
Distinctive Signs of Disease as Deterrents for the Endothelial Function: A Systematic Review

Abstract
:1. Introduction
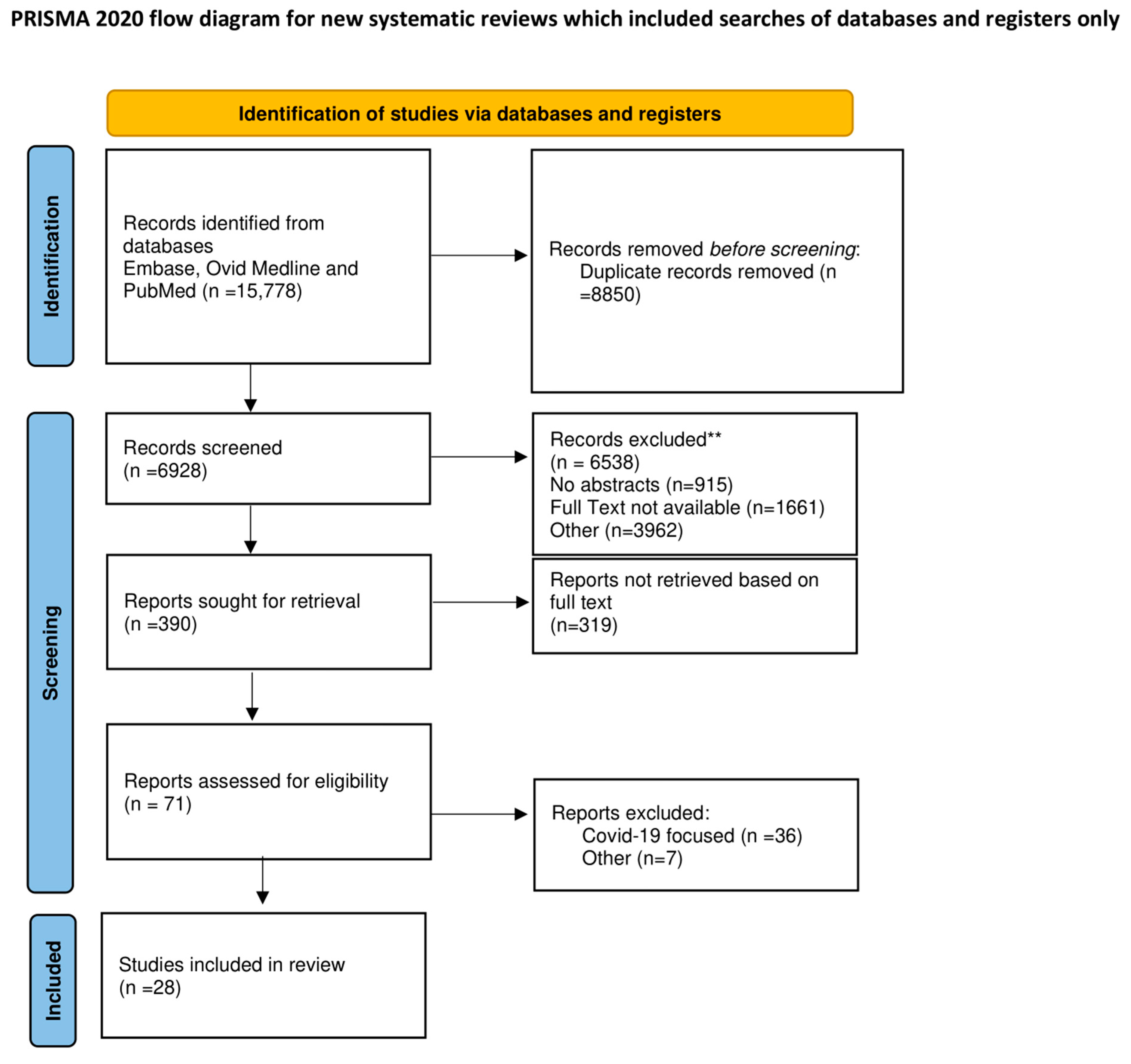
2. Methods
2.1. Search Strategy
2.2. Study Selection and Data Extraction
2.3. Endpoints and Effect Summary
3. Results
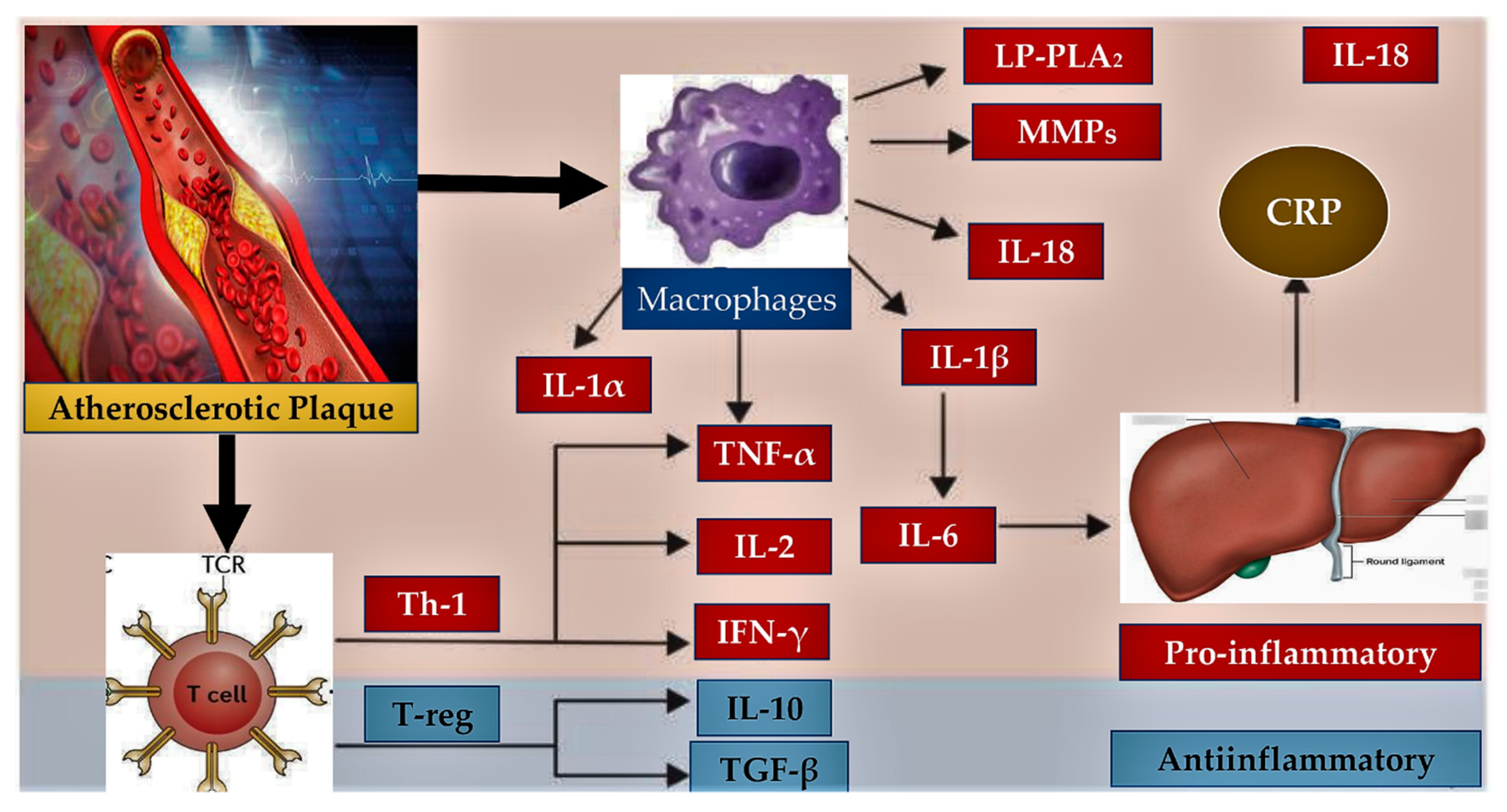
3.1. Presence of Inflammation in Endothelial Dysfunction
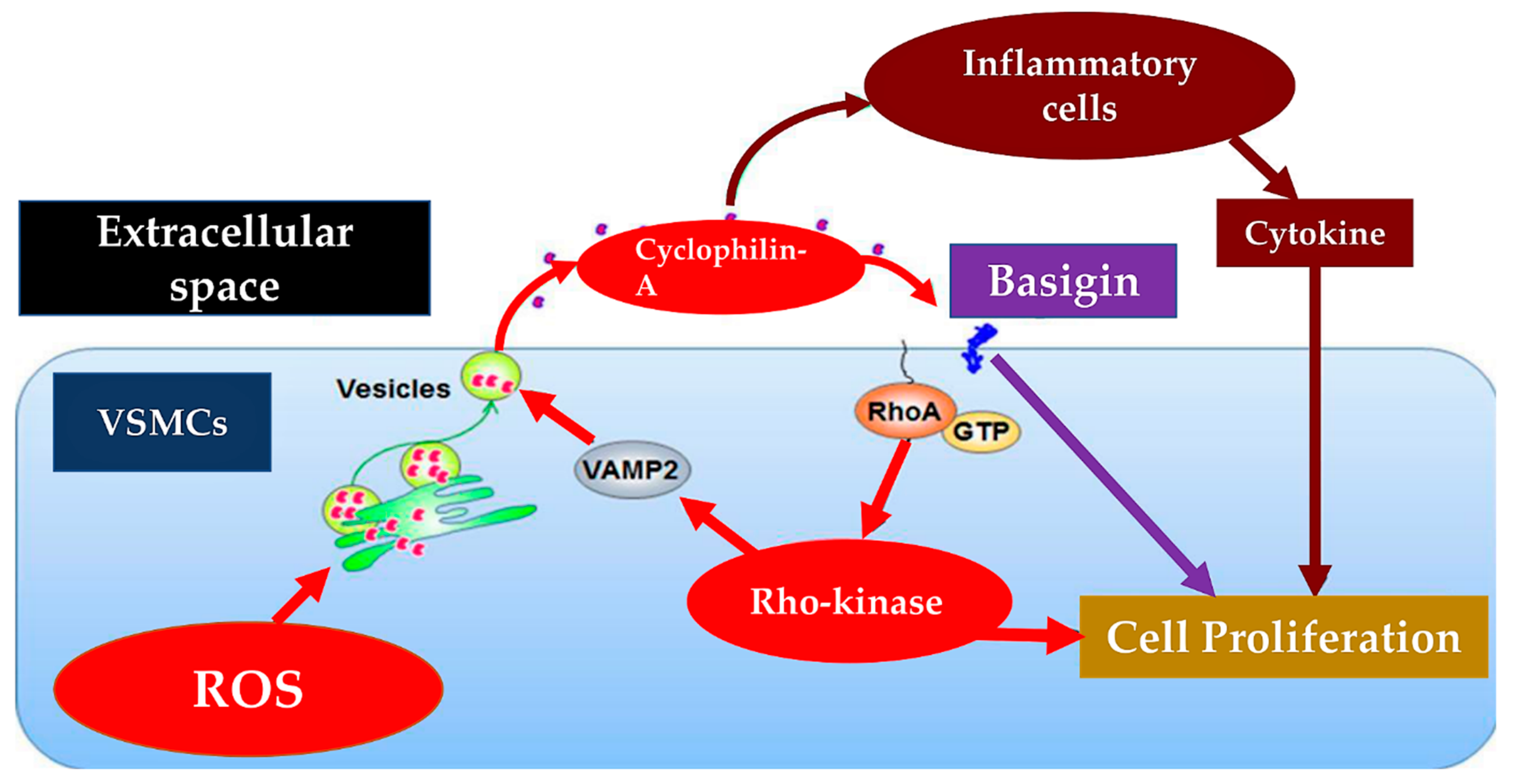
3.2. Pulmonary Hypertension Mediates Endothelial Dysfunction
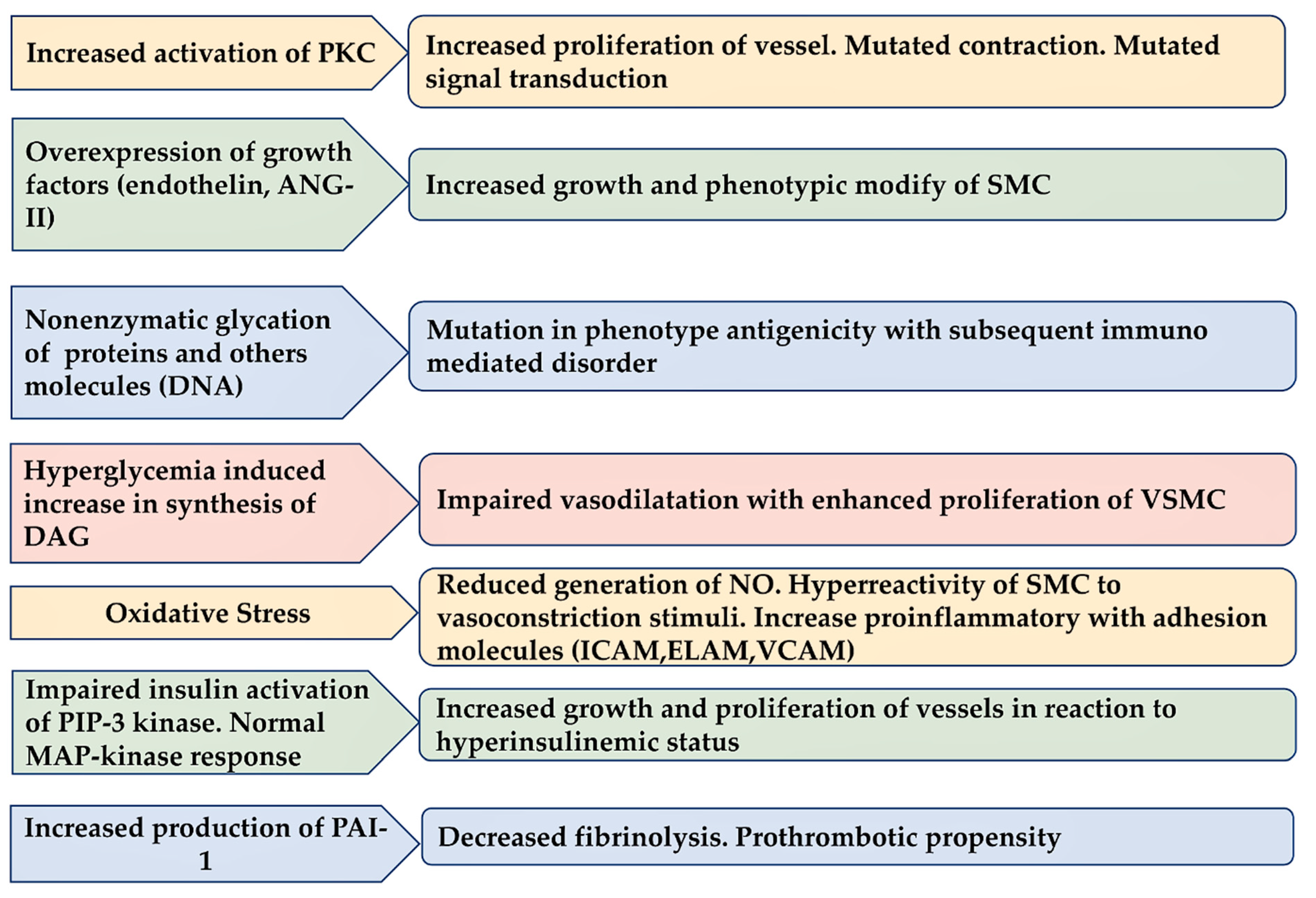
3.3. Diabetes Induce Injury in Endothelium
3.4. Endothelial Dysfunction and Fabry Disease
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nappi, F.; Fiore, A.; Masiglat, J.; Cavuoti, T.; Romandini, M.; Nappi, P.; Avtaar Singh, S.S.; Couetil, J.P. Endothelium-Derived Relaxing Factors and Endothelial Function: A Systematic Review. Biomedicines 2022, 10, 2884. [Google Scholar] [CrossRef]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H. Endothelial dysfunction and vascular disease—A 30th anniversary update. Acta Physiol. 2015, 219, 22–96. [Google Scholar] [CrossRef] [PubMed]
- Shimokawa, H. 2014 Williams Harvey lecture: Importance of coronary vasomotion abnormalities-from bench to bedside. Eur. Heart J. 2014, 35, 3180–3193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godo, S.; Takahashi, J.; Yasuda, S.; Shimokawa, H. Endothelium in Coronary Macrovascular and Microvascular Diseases. J. Cardiovasc. Pharmacol. 2021, 78 (Suppl. S6), S19–S29. [Google Scholar] [CrossRef] [PubMed]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arter. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, Y.; Kwon, T.G.; Lennon, R.J.; Lerman, L.O.; Lerman, A. Prognostic value of flow-mediated vasodilation in brachial artery and fingertip artery for cardiovascular events: A systematic review and meta-analysis. J. Am. Heart Assoc. 2015, 4, e002270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitta, Y.; Obata, J.E.; Nakamura, T.; Hirano, M.; Kodama, Y.; Fujioka, D.; Saito, Y.; Kawabata, K.; Sano, K.; Kobayashi, T.; et al. Persistent impairment of endothelial vasomotor function has a negative impact on outcome in patients with coronary artery disease. J. Am. Coll. Cardiol. 2009, 53, 323–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetti, P.O.; Pumper, G.M.; Higano, S.T.; Holmes, D.R., Jr.; Kuvin, J.T.; Lerman, A. Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. J. Am. Coll. Cardiol. 2004, 44, 2137–2141. [Google Scholar] [CrossRef] [Green Version]
- Chaulin, A.M.; Sergeev, A.K. The Role of Fine Particles (PM 2.5) in the Genesis of Atherosclerosis and Myocardial Damage: Emphasis on Clinical and Epidemiological Data, and Pathophysiological Mechanisms. Cardiol. Res. 2022, 13, 268–282. [Google Scholar] [CrossRef]
- Nataf, P.; Guettier, C.; Bourbon, A.; Nappi, F.; Lima, L.; Dorent, R.; Pavie, A.; Gandjbakhch, I. Influence of arterial allograft preparation techniques on chronic vascular rejection: A histological study. Transplant Proc. 1996, 28, 2890–2892. [Google Scholar]
- Avtaar Singh, S.S.; Nappi, F. Pathophysiology and Outcomes of Endothelium Function in Coronary Microvascular Diseases: A Systematic Review of Randomized Controlled Trials and Multicenter Study. Biomedicines 2022, 10, 3010. [Google Scholar] [CrossRef]
- Donato, A.J.; Machin, D.R.; Lesniewski, L.A. Mechanisms of Dysfunction in the Aging Vasculature and Role in Age-Related Disease. Circ Res. 2018, 123, 825–848. [Google Scholar] [CrossRef]
- Paneni, F.; Diaz Cañestro, C.; Libby, P.; Lüscher, T.F.; Camici, G.G. The aging cardiovascular system: Understanding it at the cellular and clinical levels. J. Am. Coll. Cardiol. 2017, 69, 1952–1967. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 2013, 368, 2004–2013. [Google Scholar] [CrossRef] [Green Version]
- Honda, A.; Tahara, N.; Nitta, Y.; Tahara, A.; Igata, S.; Bekki, M.; Nakamura, T.; Sugiyama, Y.; Kaida, H.; Kurata, S.; et al. Vascular inflammation evaluated by [18F]-fluorodeoxyglucose-positron emission tomography/ computed tomography is associated with endothelial dysfunction. Arter. Thromb. Vasc. Biol. 2016, 36, 1980–1988. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.K.; Kim, C.J.; Choo, E.H.; Han, E.J.; Hwang, B.H.; Kim, J.J.; Kim, S.H.; O, J.H.; Chang, K. Anti-inflammatory effect of statin is continuously working throughout use: A prospective three time point 18F-FDG PET/CT imaging study. Int. J. Cardiovasc. Imaging 2019, 35, 1745–1753. [Google Scholar] [CrossRef]
- Frauenknecht, V.; Thiel, S.; Storm, L.; Meier, N.; Arnold, M.; Schmid, J.P.; Saner, H.; Schroeder, V. Plasma levels of mannan-binding lectin (MBL)-associated serine proteases (MASPs) and MBL-associated protein in cardio—And cerebrovascular diseases. Clin. Exp. Immunol. 2013, 173, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Krogh, S.S.; Holt, C.B.; Steffensen, R.; Funck, K.L.; Høyem, P.; Laugesen, E.; Poulsen, P.L.; Thiel, S.; Hansen, T.K. Plasma levels of MASP-1, MASP-3 and MAp44 in patients with type 2 diabetes: Influence of glycaemic control, body composition and polymorphisms in the MASP1 gene. Clin. Exp. Immunol. 2017, 189, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Hertle, E.; Arts, I.C.; van der Kallen, C.J.; Feskens, E.J.; Schalkwijk, C.G.; Hoffmann-Petersen, I.T.; Thiel, S.; Stehouwer, C.D.; van Greevenbroek, M.M. Distinct longitudinal associations of MBL, MASP-1, MASP-2, MASP-3, and MAp44 with endothelial dysfunction and intima-media thickness: The cohort on diabetes and atherosclerosis maastricht (CODAM) study. Arter. Thromb. Vasc. Biol. 2016, 36, 1278–1285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertle, E.; Arts, I.C.W.; van der Kallen, C.J.H.; Feskens, E.J.M.; Schalkwijk, C.G.; Stehouwer, C.D.A.; van Greevenbroek, M.M.J. Classical Pathway of Complement Activation: Longitudinal Associations of C1q and C1-INH With Cardiovascular Outcomes: The CODAM Study (Cohort on Diabetes and Atherosclerosis Maastricht)-Brief Report. Arter. Thromb. Vasc. Biol. 2018, 38, 1242–1244. [Google Scholar] [CrossRef] [Green Version]
- Emeny, R.T.; Zierer, A.; Lacruz, M.E.; Baumert, J.; Herder, C.; Gornitzka, G.; Koenig, W.; Thorand, B.; Ladwig, K.H. KORA Investigators. Job strain-associated inflammatory burden and long-term risk of coronary events: Findings from the MONICA/KORA Augsburg case-cohort study. Psychosom. Med. 2013, 75, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Herder, C.; Kannenberg, J.M.; Carstensen-Kirberg, M.; Huth, C.; Meisinger, C.; Koenig, W.; Peters, A.; Rathmann, W.; Roden, M.; Thorand, B. Serum levels of interleukin-22, cardiometabolic risk factors and incident type 2 diabetes: KORA F4/FF4 study. Cardiovasc. Diabetol. 2017, 16, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herder, C.; Gala, T.D.L.H.; Carstensen-Kirberg, M.; Huth, C.; Zierer, A.; Wahl, S.; Sudduth-Klinger, J.; Kuulasmaa, K.; Peretz, D.; Ligthart, S.; et al. Circulating Levels of Interleukin 1-Receptor Antagonist and Risk of Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2017, 37, 1222–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. CANTOS Trial Group Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Choi, B.J.; Matsuo, Y.; Aoki, T.; Kwon, T.G.; Prasad, A.; Gulati, R.; Lennon, R.J.; Lerman, L.O.; Lerman, A. Coronary endothelial dysfunction is associated with inflammation and vasa vasorum proliferation in patients with early atherosclerosis. Arter. Thromb. Vasc. Biol. 2014, 34, 2473–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaric, B.; Obradovic, M.; Trpkovic, A.; Banach, M.; Mikhailidis, D.P.; Isenovic, E.R. Endothelial Dysfunction in Dyslipidaemia: Molecular Mechanisms and Clinical Implications. Curr. Med. Chem. 2020, 27, 1021–1040. [Google Scholar] [CrossRef] [PubMed]
- Feletou, M. The Endothelium: Part 1: Multiple Functions of the Endothelial Cells-Focus on Endothelium-Derived Vasoactive Mediators; Morgan & Claypool Life Sciences Publishers: San Rafael, CA, USA, 2011. [Google Scholar]
- Satoh, K.; Matoba, T.; Suzuki, J.; O’Dell, M.R.; Nigro, P.; Cui, Z.; Mohan, A.; Pan, S.; Li, L.; Jin, Z.G.; et al. Cyclophilin A mediates vascular remodeling by promoting inflammation and vascular smooth muscle cell proliferation. Circulation 2008, 117, 3088–3098. [Google Scholar] [CrossRef] [Green Version]
- Satoh, K.; Satoh, T.; Kikuchi, N.; Omura, J.; Kurosawa, R.; Suzuki, K.; Sugimura, K.; Aoki, T.; Nochioka, K.; Tatebe, S.; et al. Basigin mediates pulmonary hypertension by promoting inflammation and vascular smooth muscle cell proliferation. Circ. Res. 2014, 115, 738–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, C.; Sowden, M.; Berk, B.C. Extracellular Cyclophilin A, Especially Acetylated, Causes Pulmonary Hypertension by Stimulating Endothelial Apoptosis, Redox Stress, and Inflammation. Arter. Thromb. Vasc. Biol. 2017, 37, 1138–1146. [Google Scholar] [CrossRef] [Green Version]
- Xue, C.; Senchanthisai, S.; Sowden, M.; Pang, J.; White, R.J.; Berk, B.C. Endothelial-to-Mesenchymal Transition and Inflammation Play Key Roles in Cyclophilin A-Induced Pulmonary Arterial Hypertension. Hypertension 2020, 76, 1113–1123. [Google Scholar] [CrossRef]
- Rosa, A.; Butt, E.; Hopper, C.P.; Loroch, S.; Bender, M.; Schulze, H.; Sickmann, A.; Vorlova, S.; Seizer, P.; Heinzmann, D.; et al. Cyclophilin A Is Not Acetylated at Lysine-82 and Lysine-125 in Resting and Stimulated Platelets. Int. J. Mol. Sci. 2022, 23, 1469. [Google Scholar] [CrossRef]
- Liu, S.F.; Nambiar Veetil, N.; Li, Q.; Kucherenko, M.M.; Knosalla, C.; Kuebler, W.M. Pulmonary hypertension: Linking inflammation and pulmonary arterial stiffening. Front. Immunol. 2022, 13, 959209. [Google Scholar] [CrossRef]
- Rogula, S.; Pomirski, B.; Czyżak, N.; Eyileten, C.; Postuła, M.; Szarpak, Ł.; Filipiak, K.J.; Kurzyna, M.; Jaguszewski, M.; Mazurek, T.; et al. Biomarker-based approach to determine etiology and severity of pulmonary hypertension: Focus on microRNA. Front. Cardiovasc. Med. 2022, 9, 980718. [Google Scholar] [CrossRef] [PubMed]
- Kherbeck, N.; Tamby, M.C.; Bussone, G.; Dib, H.; Perros, F.; Humbert, M.; Mouthon, L. The role of inflammation and autoimmunity in the pathophysiology of pulmonary arterial hypertension. Clin. Rev. Allergy Immunol. 2013, 44, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Gomez, D.; Gladwin, M.T. Do BRD (4)S of a Feather Flock Together? How an Inflammation-Driven Epigenetic Regulator May Link Pulmonary Hypertension and Coronary Artery Disease. Arter. Thromb. Vasc. Biol. 2017, 37, 1428–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borck, P.C.; Guo, L.W.; Plutzky, J. BET Epigenetic Reader Proteins in Cardiovascular Transcriptional Programs. Circ. Res. 2020, 126, 1190–1208. [Google Scholar] [CrossRef]
- Li, M.X.; Jiang, D.Q.; Wang, Y.; Chen, Q.Z.; Ma, Y.J.; Yu, S.S.; Wang, Y. Signal Mechanisms of Vascular Remodeling in the Development of Pulmonary Arterial Hypertension. J. Cardiovasc. Pharmacol. 2016, 67, 182–190. [Google Scholar] [CrossRef]
- Meloche, J.; Pflieger, A.; Vaillancourt, M.; Paulin, R.; Potus, F.; Zervopoulos, S.; Graydon, C.; Courboulin, A.; Breuils-Bonnet, S.; Tremblay, E.; et al. Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 2014, 129, 786–797. [Google Scholar] [CrossRef] [Green Version]
- Meloche, J.; Le Guen, M.; Potus, F.; Vinck, J.; Ranchoux, B.; Johnson, I.; Antigny, F.; Tremblay, E.; Breuils-Bonnet, S.; Perros, F.; et al. miR-223 reverses experimental pulmonary arterial hypertension. Am. J. Physiol. Physiol. 2015, 309, C363–C372. [Google Scholar] [CrossRef] [Green Version]
- Archer, S.L.; Marsboom, G.; Kim, G.H.; Zhang, H.J.; Toth, P.T.; Svensson, E.C.; Dyck, J.R.; Gomberg-Maitland, M.; Thébaud, B.; Husain, A.N.; et al. Rehman. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: A basis for excessive cell proliferation and a new therapeutic target. Circulation 2010, 121, 2661–2671. [Google Scholar] [CrossRef] [Green Version]
- Yamaji-Kegan, K.; Su, Q.; Angelini, D.J.; Myers, A.C.; Cheadle, C.; Johns, R.A. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) increases lung inflammation and activates pulmonary microvascular endothelial cells via an IL-4-dependent mechanism. J. Immunol. 2010, 185, 5539–5548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, D.J.; Su, Q.; Yamaji-Kegan, K.; Fan, C.; Skinner, J.T.; Poloczek, A.; El-Haddad, H.; Cheadle, C.; Johns, R.A. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMα) in chronic hypoxia- and antigen-mediated pulmonary vascular remodeling. Respir. Res. 2013, 14, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaji-Kegan, K.; Takimoto, E.; Zhang, A.; Weiner, N.C.; Meuchel, L.W.; Berger, A.E.; Cheadle, C.; Johns, R.A. Hypoxia-induced mitogenic factor (FIZZ1/RELMα) induces endothelial cell apoptosis and subsequent interleukin-4-dependent pulmonary hypertension. Am. J. Physiol. Cell. Mol. Physiol. 2014, 306, L1090–L1103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johns, R.A.; Takimoto, E.; Meuchel, L.W.; Elsaigh, E.; Zhang, A.; Heller, N.M.; Semenza, G.L.; Yamaji-Kegan, K. Hypoxia-inducible factor 1α is a critical downstream mediator for hypoxia-induced mitogenic factor (FIZZ1/RELMα)-induced pulmonary hypertension. Arter. Thromb. Vasc. Biol. 2016, 36, 134–144. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Fan, C.; Gomez-Arroyo, J.; Van Raemdonck, K.; Meuchel, L.W.; Skinner, J.T.; Everett, A.D.; Fang, X.; Macdonald, A.A.; Yamaji-Kegan, K.; et al. HIMF (Hypoxia-Induced Mitogenic Factor) Signaling Mediates the HMGB1 (High Mobility Group Box 1)-Dependent Endothelial and Smooth Muscle Cell Crosstalk in Pulmonary Hypertension. Arter. Thromb. Vasc. Biol. 2019, 39, 2505–2519. [Google Scholar] [CrossRef]
- Félétou, M.; Köhler, R.; Vanhoutte, P.M. Endothelium-derived vasoactive factors and hypertension: Possible roles in pathogenesis and as treatment targets. Curr. Hypertens. Rep. 2010, 12, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Yaoita, N.; Shirakawa, R.; Fukumoto, Y.; Sugimura, K.; Miyata, S.; Miura, Y.; Nochioka, K.; Miura, M.; Tatebe, S.; Aoki, T.; et al. Platelets are highly activated in patients of chronic thromboembolic pulmonary hypertension. Arter. Thromb. Vasc. Biol. 2014, 34, 2486–2494. [Google Scholar] [CrossRef] [Green Version]
- Yaoita, N.; Satoh, K.; Satoh, T.; Sugimura, K.; Tatebe, S.; Yamamoto, S.; Aoki, T.; Miura, M.; Miyata, S.; Kawamura, T.; et al. Thrombin-activatable fibrinolysis inhibitor in chronic thromboembolic pulmonary hypertension. Arter. Thromb. Vasc. Biol. 2016, 36, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Loader, J.; Montero, D.; Lorenzen, C.; Watts, R.; Méziat, C.; Reboul, C.; Stewart, S.; Walther, G. Acute hyperglycemia impairs vascular function in healthy and cardiometabolic diseased subjects: Systematic review and meta-analysis. Arter. Thromb. Vasc. Biol. 2015, 35, 2060–2072. [Google Scholar] [CrossRef] [Green Version]
- Lespagnol, E.; Dauchet, L.; Pawlak-Chaouch, M.; Balestra, C.; Berthoin, S.; Feelisch, M.; Roustit, M.; Boissière, J.; Fontaine, P.; Heyman, E. Early Endothelial Dysfunction in Type 1 Diabetes Is Accompanied by an Impairment of Vascular Smooth Muscle Function: A Meta-Analysis. Front. Endocrinol. 2020, 11, 203. [Google Scholar] [CrossRef]
- Horton, W.B.; Jahn, L.A.; Hartline, L.M.; Aylor, K.W.; Patrie, J.T.; Barrett, E.J. Acute hyperglycaemia enhances both vascular endothelial function and cardiac and skeletal muscle microvascular function in healthy humans. J. Physiol. 2022, 600, 949–962. [Google Scholar] [CrossRef]
- Loader, J.; Meziat, C.; Watts, R.; Lorenzen, C.; Sigaudo-Roussel, D.; Stewart, S.; Reboul, C.; Meyer, G.; Walther, G. Effects of sugar-sweetened beverage consumption on microvascular and macrovascular function in a healthy population. Arter. Thromb. Vasc. Biol. 2017, 37, 1250–1260. [Google Scholar] [CrossRef] [Green Version]
- Tabit, C.E.; Shenouda, S.M.; Holbrook, M.; Fetterman, J.L.; Kiani, S.; Frame, A.A.; Kluge, M.A.; Held, A.; Dohadwala, M.M.; Gokce, N.; et al. Protein kinase C-β contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation 2013, 127, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Farb, M.G.; Karki, S.; Park, S.Y.; Saggese, S.M.; Carmine, B.; Hess, D.T.; Apovian, C.; Fetterman, J.L.; Bretón-Romero, R.; Hamburg, N.M.; et al. WNT5A-JNK regulation of vascular insulin resistance in human obesity. Vasc. Med. 2016, 21, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Walther, G.; Obert, P.; Dutheil, F.; Chapier, R.; Lesourd, B.; Naughton, G.; Courteix, D.; Vinet, A. Metabolic syndrome individuals with and without type 2 diabetes mellitus present generalized vascular dysfunction: Crosssectional study. Arter. Thromb. Vasc. Biol. 2015, 35, 1022–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bretón-Romero, R.; Feng, B.; Holbrook, M.; Farb, M.G.; Fetterman, J.L.; Linder, E.A.; Berk, B.D.; Masaki, N.; Weisbrod, R.M.; Inagaki, E.; et al. Endothelial dysfunction in human diabetes is mediated by Wnt5a-JNK signaling. Arter. Thromb. Vasc. Biol. 2016, 36, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.K.; Kang, Y.M.; Lee, S.E.; Lee, Y.; Seol, S.M.; Lee, W.J.; Park, J.Y.; Jung, C.H. Effect of SFRP5 (Secreted Frizzled-Related Protein 5) on the WNT5A (Wingless-Type Family Member 5A)-Induced Endothelial Dysfunction and Its Relevance With Arterial Stiffness in Human Subjects. Arter. Thromb. Vasc. Biol. 2018, 38, 1358–1367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.; Kim, J.A.; Joo, K.Y.; Choi, S.; Choi, E.N.; Shin, J.A.; Han, K.H.; Jung, S.C.; Suh, S.H. Globotriaosylceramide leads to K(Ca)3.1 channel dysfunction: A new insight into endothelial dysfunction in Fabry disease. Cardiovasc. Res. 2011, 89, 290–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, K. Globotriaosylceramide induces endothelial dysfunction in fabry disease. Arter. Thromb. Vasc. Biol. 2014, 34, 2–4. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Kim, J.A.; Na, H.Y.; Cho, S.E.; Park, S.; Jung, S.C.; Suh, S.H. Globotriaosylceramide induces lysosomal degradation of endothelial. KCa3.1 in fabry disease. Arter. Thromb. Vasc. Biol. 2014, 34, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Linhart, A.; Kampmann, C.; Zamorano, J.L.; Sunder-Plassmann, G.; Beck, M.; Mehta, A.; Elliott, P.M.; European FOS Investigators. Cardiac manifestations of Anderson-Fabry disease: Results from the international Fabry outcome survey. Eur. Heart J. 2007, 28, 1228–1235. [Google Scholar] [CrossRef] [Green Version]
- Linhart, A.; Elliott, P.M. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart 2007, 93, 528–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidemann, F.; Strotmann, J.M.; Niemann, M.; Herrmann, S.; Wilke, M.; Beer, M.; Voelker, W.; Ertl, G.; Emmert, A.; Wanner, C.; et al. Heart valve involvement in Fabry cardiomyopathy. Ultrasound Med. Biol. 2009, 35, 730–735. [Google Scholar] [CrossRef]
- Linhart, A.; Lubanda, J.C.; Palecek, T.; Bultas, J.; Karetová, D.; Ledvinová, J.; Elleder, M.; Aschermann, M. Cardiac manifestations in Fabry disease. J. Inherit. Metab. Dis. 2001, 24 (Suppl. S2), 75–83. [Google Scholar] [CrossRef] [PubMed]
- Elliott, P.M.; Kindler, H.; Shah, J.S.; Sachdev, B.; Rimoldi, O.E.; Thaman, R.; Tome, M.T.; McKenna, W.J.; Lee, P.; Camici, P.G. Coronary microvascular dysfunction in male patients with Anderson-Fabry disease and the effect of treatment with alpha galactosidase A. Heart 2006, 92, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovarnik, T.; Mintz, G.S.; Karetova, D.; Bultas, J.; Skulec, R.; Skalicka, H.; Aschermann, M.; Elleder, M.; Linhart, A. Intravascular ultrasound assessment of coronary artery involvement in Fabry disease. J. Inherit. Metab. Dis. 2008, 31, 753–776. [Google Scholar] [CrossRef]
- Nappi, F.; Avatar Singh, S.S.; Santana, O.; Mihos, C.G. Functional mitral regurgitation: An overview for surgical management framework. J. Thorac. Dis. 2018, 10, 4540–4555. [Google Scholar] [CrossRef] [PubMed]
- Nappi, F.; Nenna, A.; Spadaccio, C.; Lusini, M.; Chello, M.; Fraldi, M.; Acar, C. Predictive factors of long-term results following valve repair in ischemic mitral valve prolapse. Int. J. Cardiol. 2016, 204, 218–228. [Google Scholar] [CrossRef]
- Rama, A.; Nappi, F.; Praschker, B.G.; Gandjbakhch, I. Papillary muscle approximation for ischemic mitral valve regurgitation. J. Card. Surg. 2008, 23, 733–735. [Google Scholar] [CrossRef]
- Nappi, F.; Spadaccio, C.; Fraldi, M. Reply: Papillary Muscle Approximation Is an Anatomically Correct Repair for Ischemic Mitral Regurgitation. J. Am. Coll. Cardiol. 2016, 68, 1147–1148. [Google Scholar] [CrossRef]
- Nappi, F.; Lusini, M.; Spadaccio, C.; Nenna, A.; Covino, E.; Acar, C.; Chello, M. Papillary Muscle Approximation Versus Restrictive Annuloplasty Alone for Severe Ischemic Mitral Regurgitation. J. Am. Coll. Cardiol. 2016, 67, 2334–2346. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| First Author/Year Ref | Type of Study | Cohort | Aims | Finding |
|---|---|---|---|---|
| Honda et al. (2016) Arterioscler Thromb Vasc Biol [15] | Human Prospective Single Center (USA) | 145 pts (95 men and 50 women) | To evaluate the relationship between endothelial function and vascular inflammation by mean of flow-mediated dilation (FMD) and 18FDG-PET | Vascular inflammation in the carotid arteries evaluated independently correlates to decreased %FMD suggesting the association of vascular inflammation with endothelial dysfunction |
| Kang et al. (2019) Front Immunol [16] | Prospective Single Center (Korea) | Nine statin-naïve SA patients with inflammatory carotid plaques | To evaluate anti-inflammatory effects of statin initiation to 3 and 3 months to 1 year) by mean o 18F-FDG PET/CT | The anti-inflammatory effect of the statin continues to maintain an effect for up to 1 year. However, stable plasma LDL-C levels below the 3-month target were produced. |
| Krog et al. (2017) Clin Exp Immunol [18] | Human/Animal model Prospective Single Center (Denmark) | 100 pts with type 2 diabetes vs. 100 sex- and age-matched controls Streptozotocin-induced diabetes mouse | To study SNPs in the MASP1 gene and altered MASP-1, MASP-3 and MAp44 | Higher levels of MASP-1 levels among pts with type 2 diabetes and diabetic mice |
| Hertle et al. (2016) Arterioscler Thromb Vasc Biol [19] | Prospective Multicenter (Netherlands, Denmark) CODAM study (Cohort on Diabetes and Atherosclerosis Maastricht) | 574 pts cIMT 73 pts CVD | To study MBL-associated proteases (MASPs) and MBL-associated proteins (MAps) in complement activation and CVD | High MBL may contribute to low cIMT. MASP-1 and MASP-2 were not associated with adverse cardiovascular outcomes. MASP-3 and MAp44 crucial role in endothelial dysfunction |
| Hertle et al. (2018) Arterioscler Thromb Vasc Biol [20] | Prospective Multicenter (Netherlands Denmark) CODAM study (Cohort on Diabetes and Atherosclerosis Maastricht) | 574 pts cIMT 73 pts CVD | To determine the associations between factor C1q its regulator C1-INH and CVD | Nonlinear association between C1q and incident CVD |
| Herder et al. (2017) Cardiovasc Diabetol. [22] | Prospective Single Center (Germany) | 1107 pts KORA F4 study. | Whether higher IL-22 levels are associated with lower diabetes incidence. | High serum levels of IL-22 were inversely associated with cardiometabolic risk factors. these associations did not translate into an increased risk for type 2 diabetes |
| Herder et al. (2017) Arterioscler Thromb Vasc Biol [23] | Meta-Analysis Single Center (Germany) | 5 cohort studies IL-1RA MONICA/KORA Augsburg case-cohort study 1855 pts CVD 18,745 noncases CVD | To evaluate circulating IL-1RA and the incidence of CVD | Serum IL-1RA levels were associated with the risk of CVD after adjustment for multiple confounders. IL-1RA lead to subclinical inflammation, oxidative stress, and endothelial activation. |
| Ridker et al. (2017) NEJM J [24] | RCT Multicenter Center CANTOS Trial | 10,061 patients canakinumab (50 mg, 150 mg, and 300 mg) 3344 placebo group | To compare the therapeutic effect of a monoclonal antibody targeting interleukin-1β | Canakinumab at a dose of 150 mg every 3 months was effective as an anti-inflammatory against interleukin-1β leading to a significantly lower rate of recurrent cardiovascular events than placebo, |
| Choi et al. (2014) Arterioscler Thromb Vasc Biol [25] | Multicenter Center (USA/Korea) | 40 pts with mild coronary atherosclerosis | To study endothelial dysfunction in pts with early CAD presenting macrophages and vasa vasorum infiltrates. | Epicardial endothelial dysfunction was associated with optical coherence tomography -which identified macrophages and microchannels in mild coronary atherosclerosis. |
| First Author/Year Ref | Type of Study | Cohort | Aims | Finding |
|---|---|---|---|---|
| Satoh et al. (2008) Antioxid Redox Signal [28] | Animal Model Single Center (Japan) | CyPA knockout mice vs. Wild-type mice vs. VSMC-Tg mice | To evaluate contribute of CyPA to vascular remodeling | CyPA regulates inflammatory cell accumulation, flow-mediated vascular remodeling and intima formation. |
| Satoh et al. (2014) Circ Res [29] | Animal Model Single Center (Japan) | CyPA (±) mice vs. Bsg (±) mice | To determine the role of CyPA/Bsg signaling in the development of PH. | Increased CyPA levels in patients with PH. Cell proliferation was reduced in Bsg (±) compared with Bsg (+/+) VSMCs. |
| Xe et al.(2017) Arterioscler Thromb Vasc Biol [30] | Human/Animal Model Single Center (USA) | CyPA (±) mice vs. Human pulmonary EC | To evaluate the role of extracellular CypA in PH. To compare the effects of acetylated CypA (AcK-CypA) and CypA on EC dysfunction. | EC-derived CypA (especially AcK-CypA) favor PH due to apoptosis, inflammation, and oxidative stress |
| Rosa et al. (2022) Int J Mol Sci. [32] | Animal/Human Model Prospective Multicenter (Germany, UK,) | CyPA (±) mice vs. Human pulmonary EC | Whether K82 and K125 acetylation is required for the release of CyPA from platelets | Acetylation of CyPA no major protein modification in platelets. CyPA acetylation is not required |
| Meloche et al. (2014) Circulation [39] | Prospective Single Center (Canada) | Human PH EC vs. Healthy tissues/cells | To study PAH-PASMCs increasing during activation of poly (ADP-ribose) polymerase-1 (PARP-1) | PH development related to DNA damage/PARP-1 signaling pathway |
| Meloche et al. (2015) Am J Physiol Cell Physiol [40] | Prospective Single Center (Canada) | Human PH EC vs. Healthy tissues/cells | Wether miR-223 downregulation triggers PARP-1 overexpression | Downregulation of miR-223 in PH |
| Archer et al. (2010) Circulation [41] | Animal/ Human Model Single center (USA) | FHR (PH rat) vs. * Sprague-Dawley rat PH pts | To study the expression of SOD2 and its correlation with PH | Epigenetic SOD2 deficiency induces PH due to impairing redox signaling and creating a proliferative, apoptosis-resistant PASMC |
| Yamaji-Kegan et al. (2014) Am J Physiol Lung Cell Mol Physiol [44] | AnimalModel Single Center (USA) | IL-4/STAT6 KO mice vs. wild-type (WT) mice | To evaluate how HIMF lead to lung inflammation and vascular remodeling | IL-4 signaling exerts a substantial role in HIMF-induced lung inflammation and vascular remodeling. |
| Johns et al. (2016) Arterioscler Thromb Vasc Biol [45] | Animal Model Single Center (USA) | HIF-1α(+/−) mice vs. wild-type (HIF-1α (+/+) vs. Human resistin-like molecule-β | To evaluate hypoxia-inducible factor-1 (HIF-1) is a critical downstream signal mediator of HIMF during PH development. | HIMF can induce HIF-1, vascular endothelial growth factor-A, and interleukin-6. Mediators for hypoxic inflammation and PH pathophysiology. |
| Lin et al. (2018) Arterioscler Thromb Vasc Biol [46] | Animal/Human Model Single Center (USA) | HIMF KO mice vs. Human RELM-β | To investigate the immunomodulatory properties of HIMF signaling in PH pathogenesis | In HIMF-induced PH, HMGB1-RAGE mediates EC-smooth muscle cell crosstalk |
| First Author/Year Ref | Type of Study | Cohort | Aims | Finding |
|---|---|---|---|---|
| Loader et al. (2015) Arterioscler Thromb Vasc Biol [50] | Human Study level meta-analysis | 525 healthy pts 540 cardiometabolic pts | To compare acute hyperglycemia in EF and VSMF | In healthy and diseased subjects macrovascular but not microvascular endothelial dysfunction during acute hyperglycemia was revealed |
| Lespagnol et al. (2020) Front Endocrinol. [51] | Human Study level meta-analysis | 21 study | To evaluate early EF and VSMF alteration in type 1 diabetes. | In children and adults, VSM dysfunction with type 1 diabetes is demonstrated. Endothelial dysfunction s more pronounced in large than small vessels. |
| Horton et al. (2022) J Physiol [52] | Human RCT | Healthy young adults 6 males 7 females | To compare macrovascular and microvascular functional responses to euglycemia and hyperglycaemia | Unlike meal-promoted acute hyperglycaemia, 4 h of intravenous glucose-induced hyperglycaemia enhances brachial artery flow-mediated dilatation and evokes cardiac and skeletal muscle microvascular function without impairing aortic stiffness. |
| Loader et al. (2017) Arterioscler Thromb Vasc Biol [53] | Human RCT | Healthy young adults (12 males) 600 mL (20 oz.) of water vs. SSB | To compare EF and VSMF. | SSB infusion mediates endothelial dysfunction with increased oxidative stress and decreasing NO bioavailability after SSB infusion. |
| Tabit et al. (2013) Circulation [54] | Human Prospective comparative | 40 diabetics type 2 vs. 36 nondiabetic controls | To study the activity of PKCβ nuclear factor κB and reduced No | Altered eNOS activation, reduced insulin action and increased inflammatory activation. Increased PKCβ activity in endothelial insulin resistance. |
| Farb et al. (2016) Vasc Med [55] | Human Visceral adipose tissue arterioles | 43 obese pts | To investigate the role of WNT5A-JNK leading to insulin-mediated vasodilator responses | Up-regulation of WNT5A-JNK signaling and impaired endothelial eNOS activation |
| Walther et al. (2015) Arterioscler Thromb Vasc Biol [56] | Human RCT | 53 pts MetS without T2D vs. 25 pts T2D vs. 40 pts healthy | To compare EF and VSMF. To measure plasma glucose, insulin and inflammatory markers | MetS is associated with endothelial-dependent and endothelial-independent dysfunction, affecting both the macro- and the microvascular systems. |
| Bretón-Romero et al. (2016) Arterioscler Thromb Vasc Biol [57] | Human Prospective comparative | 42 pts T2D vs. 43 pts healthy | To evaluate whether increased activation of Wnt5a-JNK signaling contributes to impaired EF. To determine eNOS activation and NO production | Wnt5a-induced impairment of eNOS activation and NO that was reversed by Wnt5a and JNK inhibition. Noncanonical Wnt5a signaling and JNK activity contribute to vascular insulin resistance and endothelial dysfunction. |
| Cho et al. (2018) Arterioscler Thromb Vasc Biol [58] | Human/Animal model | Sprague-Dawley rat vs. Human EC | To investigate whether SFRP5 could restore WNT5A-induced endothelial dysfunction in vitro and ex vivo. | Compensatory action of SFRP5 against atherosclerosis under conditions of metabolic dysfunction. SFRPS restored WNT5A-induced reduction of NO production via eNOS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nappi, F.; Avtaar Singh, S.S. Distinctive Signs of Disease as Deterrents for the Endothelial Function: A Systematic Review. Metabolites 2023, 13, 430. https://doi.org/10.3390/metabo13030430
Nappi F, Avtaar Singh SS. Distinctive Signs of Disease as Deterrents for the Endothelial Function: A Systematic Review. Metabolites. 2023; 13(3):430. https://doi.org/10.3390/metabo13030430
Chicago/Turabian StyleNappi, Francesco, and Sanjeet Singh Avtaar Singh. 2023. "Distinctive Signs of Disease as Deterrents for the Endothelial Function: A Systematic Review" Metabolites 13, no. 3: 430. https://doi.org/10.3390/metabo13030430