
Porokeratoses—A Comprehensive Review on the Genetics and Metabolomics, Imaging Methods and Management of Common Clinical Variants

 ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
3. Disseminated Superficial Actinic Porokeratosis (DSAP)
3.1. Clinical Presentation
3.2. Dermatoscopy
3.3. Confocal Microscopy
3.4. Pathology
3.5. Genetics and Epigenetics
3.6. Treatment
4. Disseminated Superficial Porokeratosis (DSP)
4.1. Clinical Presentation
4.2. Genetics and Epigenetics
4.3. Dermatoscopy
4.4. Confocal Microscopy
4.5. Pathology
4.6. Treatment
5. Porokeratosis of Mibelli (PM)
5.1. Clinical Presentation
5.2. Genetics and Epigenetics
5.3. Dermatoscopy
5.4. Confocal Microscopy
5.5. Pathology
5.6. Treatment
6. Linear Porokeratosis (LP)
6.1. Clinical Presentation
6.2. Genetics and Epigenetics
6.3. Dermatoscopy
6.4. Confocal Microscopy
6.5. Pathology
6.6. Treatment
7. Porokeratosis Palmaris, Plantaris et Disseminata (PPPD) and Punctate Porokeratosis (PuP)
7.1. Clinical Presentation
7.2. Genetics and Epigenetics
7.3. Dermatoscopy
7.4. Confocal Microscopy
7.5. Pathology
7.6. Treatment
8. Verrucous Porokeratosis (VP)
8.1. Clinical Presentation
8.2. Genetics and Epigenetics
8.3. Dermatoscopy
8.4. Confocal Microscopy
8.5. Pathology
8.6. Treatment
9. Follicular Porokeratosis (FP)
9.1. Clinical Presentation
9.2. Genetics and Epigenetics
9.3. Dermatoscopy
9.4. Confocal Microscopy
9.5. Pathology
9.6. Treatment
10. Porokeratoma
10.1. Clinical Presentation
10.2. Genetics and Epigenetics
10.3. Dermatoscopy
10.4. Confocal Microscopy
10.5. Pathology
10.6. Treatment
11. Risk of Malignancy
12. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patterson, J.W. Weedon’s Skin Pathology, 4th ed.; Elsevier: London, UK, 2015; ISBN 978-0-7020-5183-8. [Google Scholar]
- Neumann, I. Uber Eine Noch Wenig Bekannte Hautkrankheit (Dermatitis Circumscripta Herpetiformis). Vierteljahrsschr. Dermatol. Syph. Wien. 1875, 2, 41–52. [Google Scholar]
- Respighi, E. Di Une Ipercheratosi Non Ancora Descritta. G. Ital. Dermatol. Venereol. 1893, 28, 356–386. [Google Scholar]
- Mibelli, V. Ueber Einen Fall von Porokeratosis Mit Localisation im Munde und an der Glans. Arch. Dermat. Syph. 1899, 47, 231–243. [Google Scholar] [CrossRef]
- Freyschmidt-Paul, P.; Hoffmann, R.; König, A.; Happle, R. Linear Porokeratosis Superimposed on Disseminated Superficial Actinic Porokeratosis: Report of Two Cases Exemplifying the Concept of Type 2 Segmental Manifestation of Autosomal Dominant Skin Disorders. J. Am. Acad. Dermatol. 1999, 41, 644–647. [Google Scholar] [CrossRef]
- Wallner, J.S.; Fitzpatrick, J.E.; Brice, S.L. Verrucous Porokeratosis of Mibelli on the Buttocks Mimicking Psoriasis. Cutis 2003, 72, 391–393. [Google Scholar]
- Mukhopadhyay, A.K. Simultaneous Occurrence of Disseminated Superficial, Linear and Hypertrophic Verrucous Forms of Porokeratosis in a Child. Indian J. Dermatol. Venereol. Leprol. 2004, 70, 364–366. [Google Scholar]
- Niimi, Y.; Kawana, S. Type 2 Segmental Manifestation of Disseminated Superficial Actinic Porokeratosis in a 7-Year-Old Girl. Eur. J. Dermatol. 2009, 19, 25–28. [Google Scholar] [CrossRef]
- Murase, J.; Gilliam, A.C. Disseminated Superficial Actinic Porokeratosis Co-Existing with Linear and Verrucous Porokeratosis in an Elderly Woman: Update on the Genetics and Clinical Expression of Porokeratosis. J. Am. Acad. Dermatol. 2010, 63, 886–891. [Google Scholar] [CrossRef]
- Koley, S.; Sarkar, J.; Choudhary, S.; Dhara, S.; Choudhury, M.; Bhattacharya, S. Different Morphological Variants of Hypertrophic Porokeratosis and Disseminated Lesions of Porokeratosis of Mibelli: A Rare Co-Existence. Indian J. Dermatol. Venereol. Leprol. 2011, 77, 199–202. [Google Scholar] [CrossRef]
- Guo, H.; Gao, X.-H.; Chen, H.-D.; Li, J.-H. Coexistence of Multiple Variants of Porokeratosis. Indian J. Dermatol. Venereol. Leprol. 2015, 81, 402–403. [Google Scholar] [CrossRef]
- Xu, X.; Pradhan, S.; Wang, D.; Li, W. Multiple Porokeratomas (Porokeratotic Acanthoma) Coexisting with Disseminated Superficial Porokeratosis: Clinical, Dermoscopic and Pathological Observations, and Review of Published Work. J. Dermatol. 2020, 47, 787–791. [Google Scholar] [CrossRef]
- Sotoodian, B.; Mahmood, M.N.; Salopek, T.G. Clinical and Dermoscopic Features of Pigmented Disseminated Superficial Actinic Porokeratosis: Case Report and Literature Review. J. Cutan. Med. Surg. 2018, 22, 229–231. [Google Scholar] [CrossRef]
- Zaar, O.; Polesie, S.; Navarrete-Dechent, C.; Errichetti, E.; Akay, B.N.; Jaimes, J.; Cabo, H.; Cohen Sabban, E.; Paoli, J. Dermoscopy of Porokeratosis: Results from a Multicentre Study of the International Dermoscopy Society. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, C.; Wu, F.; Ma, R.; Luan, J.; Yang, F.; Liu, W.; Wang, L.; Zhang, S.; Liu, Y.; et al. Genomic Variations of the Mevalonate Pathway in Porokeratosis. eLife 2015, 4, e06322. [Google Scholar] [CrossRef] [PubMed]
- Chernosky, M.E.; Freeman, R.G. Disseminated Superficial Actinic Porokeratosis (DSAP). Arch. Dermatol. 1967, 96, 611–624. [Google Scholar] [CrossRef]
- Kong, F.; Moreira-Lucas, T.S.; Kaminski, A.; Spelman, L. Management of Disseminated Superficial Actinic Porokeratosis and Intraepidermal Squamous Cell Carcinoma with Low-Dose Radiation Therapy. Australas. J. Dermatol. 2021, 62, 410–412. [Google Scholar] [CrossRef] [PubMed]
- Inci, R.; Zagoras, T.; Kantere, D.; Holmström, P.; Gillstedt, M.; Polesie, S.; Peltonen, S. Porokeratosis Is One of the Most Common Genodermatoses and Is Associated with an Increased Risk of Keratinocyte Cancer and Melanoma. J. Eur. Acad. Dermatol. Venereol. 2023, 37, 420–427. [Google Scholar] [CrossRef]
- Waqar, M.U.; Cohen, P.R.; Fratila, S. Disseminated Superficial Actinic Porokeratosis (DSAP): A Case Report Highlighting the Clinical, Dermatoscopic, and Pathology Features of the Condition. Cureus 2022, 14, e26923. [Google Scholar] [CrossRef]
- Redondo, P.; Sola, M.A.; Lloret, P. Porokeratosis and Povidone-Iodine: A New Clinical Diagnostic Sign. Br. J. Dermatol. 2002, 147, 383. [Google Scholar] [CrossRef]
- Katugampola, R.P.; Finlay, A.Y. Fake Sun Tan Diagnosis of Porokeratosis. J. Eur. Acad. Dermatol. Venereol. 2006, 20, 224–226. [Google Scholar] [CrossRef]
- Thomas, C.J.; Elston, D.M. Medical Pearl: Gentian Violet to Highlight the Cornoid Lamella in Disseminated Superficial Actinic Porokeratosis. J. Am. Acad. Dermatol. 2005, 52, 513–514. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Dechent, C.; Uribe, P.; Marghoob, A. Ink-Enhanced Dermoscopy Is a Useful Tool to Differentiate Acquired Solitary Plaque Porokeratosis from Other Scaly Lesions. J. Am. Acad. Dermatol. 2019, 80, e137–e138. [Google Scholar] [CrossRef] [PubMed]
- Le, C.; Bedocs, P.M. Disseminated Superficial Actinic Porokeratosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Gu, C.-Y.; Zhang, C.-F.; Chen, L.-J.; Xiang, L.-H.; Zheng, Z.-Z. Clinical Analysis and Etiology of Porokeratosis. Exp. Ther. Med. 2014, 8, 737–741. [Google Scholar] [CrossRef]
- Allen, A.L.; Glaser, D.A. Disseminated Superficial Actinic Porokeratosis Associated with Topical PUVA. J. Am. Acad. Dermatol. 2000, 43, 720–722. [Google Scholar] [CrossRef] [PubMed]
- Kawara, S.; Oiso, N.; Kawada, A. Disseminated Superficial Actinic Porokeratosis in a Patient Undergoing Treatment with Long-Term Narrowband Ultraviolet B for Psoriasis. J. Dermatol. 2011, 38, 585–587. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.; Fidai, C.; Kerr, H. A Rare Case of Bullous Eruptive Disseminated Porokeratosis. Case Reports. 2019. Available online: https://scholarlycommons.henryford.com/merf2019caserpt/124 (accessed on 4 September 2023).
- Kang, B.D.; Kye, Y.C.; Kim, S.N. Disseminated Superficial Actinic Porokeratosis with Both Typical and Prurigo Nodularis-like Lesions. J. Dermatol. 2001, 28, 81–85. [Google Scholar] [CrossRef]
- Bencini, P.L.; Tarantino, A.; Grimalt, R.; Ponticelli, C.; Caputo, R. Porokeratosis and Immunosuppression. Br. J. Dermatol. 1995, 132, 74–78. [Google Scholar] [CrossRef]
- Fields, L.L.; White, C.R.; Maziarz, R.T. Rapid Development of Disseminated Superficial Porokeratosis after Transplant Induction Therapy. Bone Marrow Transpl. 1995, 15, 993–995. [Google Scholar]
- Matsushita, S.; Kanekura, T.; Kanzaki, T. A Case of Disseminated Superficial Actinic Porokeratosis Subsequent to Renal Transplantation. J. Dermatol. 1997, 24, 110–112. [Google Scholar] [CrossRef]
- Jang, Y.-H.; Chun, S.-J.; Kang, W.H.; Lee, E.-S. Eruptive Disseminated Superficial Actinic Porokeratosis in an Immunocompetent Host: Is This Associated with Herpes Simplex Virus or Bacterial Infection? J. Am. Acad. Dermatol. 2004, 51, 1018–1019. [Google Scholar] [CrossRef]
- di Meo, N.; Fluehler, C.; Perkan, V.; Trevisan, G. Disseminated Superficial Porokeratosis and Pyoderma Gangrenosum. Dermatol. Online J. 2010, 16, 15. [Google Scholar]
- Khaled, A.; Kourda, M.; Abdelmoula, F.; M’ssedi, L.; Tougourti, M.N.; Kamoun, M.R. Late-Onset Disseminated Superficial Actinic Porokeratosis in an Elderly Woman. Dermatol. Ther. 2011, 1, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Sim, C.Y.; Shin, J.Y.; Lee, S.Y.; Park, Y.L. Disseminated Superficial Actinic Porokeratosis in a Patient with Psoriasis, after Long-Term Narrowband Ultraviolet B Phototherapy. Ann. Dermatol. 2018, 30, 211–213. [Google Scholar] [CrossRef] [PubMed]
- Breton, A.L.; Pralong, P.; Trillet-Lenoir, V.; Balme, B.; Nicolas, J.-F.; Berard, F. Disseminated Porokeratosis Transiently Healed by Cancer Chemotherapy. Eur. J. Dermatol. 2014, 24, 260–261. [Google Scholar] [CrossRef] [PubMed]
- Torchia, D.; Romanelli, P.; Schachner, L.A. Disseminated Superficial Actinic Porokeratosis Associated with Pseudoxanthoma Elasticum. Eur. J. Dermatol. 2011, 21, 600–601. [Google Scholar] [CrossRef] [PubMed]
- Monteagudo-Sánchez, B.; Ginarte, M.; Durana, C.; Labandeira, J.; de las Heras, C.; Cacharrón, J.M. Porokeratosis in a patient with dermatomyositis. Actas Dermosifiliogr. 2006, 97, 650–652. [Google Scholar] [CrossRef] [PubMed]
- Kluger, N.; Guilpain, P.; Leclerc-Mercier, S.; Emilie, S.; Guillevin, L.; Mouthon, L. Superficial porokeratosis of the lower limbs during systemic scleroderma. Presse Med. 2009, 38, 146–148. [Google Scholar] [CrossRef]
- Terranova, M.; Amato, L.; Massi, D.; Fabbri, P. Disseminated Superficial Actinic Porokeratosis in a Patient with Sjögren Syndrome. Skinmed 2003, 2, 390–391. [Google Scholar] [CrossRef]
- Yao, Z.-G.; Hua, F.; Yin, Z.-H.; Xue, Y.-J.; Hou, Y.-H.; Nie, Y.-C.; Zheng, Z.-M.; Zhao, M.-Q.; Guo, X.-H.; Ma, C.; et al. Characteristics of Glioblastomas and Immune Microenvironment in a Chinese Family with Lynch Syndrome and Concurrent Porokeratosis. Front. Oncol. 2023, 13, 1194232. [Google Scholar] [CrossRef]
- Kanitakis, J.; Arbona-Vidal, E.; Faure, M. Porokeratosis in Patients with Polycythemia Rubra Vera: A New Side Effect of Hydroxyurea? J. Eur. Acad. Dermatol. Venereol. 2012, 26, 1040–1041. [Google Scholar] [CrossRef]
- Romagnuolo, M.; Riva, D.; Alberti Violetti, S.; Di Benedetto, A.; Barberi, F.; Moltrasio, C. Disseminated Superficial Actinic Porokeratosis Following Hydroxyurea Treatment: A Case Report. Australas. J. Dermatol. 2023, 64, e72–e75. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Amor, O.; Pereiro-Ferreirós, M.; Ginarte, M.; Peteiro, C.; Toribio, J. Coexistence of Linear Porokeratosis and Disseminated Superficial Actinic Porokeratosis: A Type 2 Segmental Manifestation. Acta Derm. Venereol. 2007, 87, 363–364. [Google Scholar] [CrossRef] [PubMed]
- Gautam, R.K.; Bedi, G.K.; Schgal, V.N.; Singh, N. Simultaneous Occurrence of Disseminated Superficial Actinic Porokeratosis (DSAP), Linear, and Punctate Porokeratosis. Int. J. Dermatol. 1995, 34, 71–72. [Google Scholar] [CrossRef] [PubMed]
- Welton, W.A. Linear Porokeratosis in a Family with DSAP. Arch. Dermatol. 1972, 106, 263. [Google Scholar] [CrossRef]
- Dover, J.S.; Miller, J.A.; Levene, G.M. Linear Porokeratosis of Mibelli and DSAP. Clin. Exp. Dermatol. 1986, 11, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Löhrer, R.; Neumann-Acikel, A.; Eming, R.; Hartmann, K.; Rasokat, H.; Krieg, T.; Happle, R.; Eming, S. A Case of Linear Porokeratosis Superimposed on Disseminated Superficial Actinic Porokeratosis. Case Rep. Dermatol. 2010, 2, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Feldman, S.R.; Crosby, D.L.; Tomsick, R.S. Scaly Atrophic Lesions Both Scattered and in Linear Arrays. Disseminated Superficial Actinic Porokeratosis in a Patient with Linear Porokeratosis. Arch. Dermatol. 1991, 127, 1219–1222. [Google Scholar] [CrossRef]
- Pearson, I.C.; Cliff, S. Case 6: Plaques Extending in a Linear Pattern from Left Ankle to Hip Forming over a 2-Year Period. Diagnosis: Linear Porokeratosis with Disseminated Superficial Porokeratosis Erupting in Pregnancy. Clin. Exp. Dermatol. 2003, 28, 345–346. [Google Scholar] [CrossRef]
- Happle, R. Somatic Recombination May Explain Linear Porokeratosis Associated with Disseminated Superficial Actinic Porokeratosis. Am. J. Med. Genet. 1991, 39, 237. [Google Scholar] [CrossRef]
- Suh, D.H.; Lee, H.S.; Kim, S.D.; Cho, K.H.; Kim, K.H.; Park, K.C. Coexistence of Disseminated Superficial Porokeratosis in Childhood with Congenital Linear Porokeratosis. Pediatr. Dermatol. 2000, 17, 466–468. [Google Scholar] [CrossRef]
- Boente, M.d.C.; López-Baró, A.M.; Frontini, M.d.V.; Asial, R.A. Linear Porokeratosis Associated with Disseminated Superficial Actinic Porokeratosis: A New Example of Type II Segmental Involvement. Pediatr. Dermatol. 2003, 20, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Lee, Y.B.; Park, H.J.; Cho, B.K. Coexistence of Congenital Linear Porokeratosis and Disseminated Superficial Porokeratosis. Australas. J. Dermatol. 2012, 53, e30–e31. [Google Scholar] [CrossRef]
- Commens, C.A.; Shumack, S.P. Linear Porokeratosis in Two Families with Disseminated Superficial Actinic Porokeratosis. Pediatr. Dermatol. 1987, 4, 209–214. [Google Scholar] [CrossRef]
- Shiiya, C.; Aoki, S.; Nakabayashi, K.; Hata, K.; Amagai, M.; Kubo, A. Linear and Disseminated Porokeratosis in One Family Showing Identical and Independent Second Hits in MVD among Skin Lesions, Respectively: A Proof-of-Concept Study. Br. J. Dermatol. 2021, 184, 1209–1212. [Google Scholar] [CrossRef]
- Peng, J.-M.; Xiao, X.-M.; Chen, J.-W.; Chen, L.-F.; Cheng, B.; Ji, M.-K.; Zhang, Z.-H. Novel Mutation in MVK Gene for Co-Occurrence of Disseminated Superficial Actinic Porokeratosis with Porokeratosis Ptychotropica. J. Dermatol. 2021, 48, e137–e139. [Google Scholar] [CrossRef]
- Mei, Q.; Xing, F.; Yin, Y.; Yuan, C. Case Report: A Novel MVK Missense Mutation in the Sporadic Porokeratosis Ptychotropica in China. Clin. Cosmet. Investig. Dermatol. 2023, 16, 1325–1329. [Google Scholar] [CrossRef]
- Xu, H.-J.; Wen, G.-D. Mixed Porokeratosis with a Novel Mevalonate Kinase Gene Mutation: A Case Report. World J. Clin. Cases 2022, 10, 4528–4534. [Google Scholar] [CrossRef]
- McGuigan, K.; Shurman, D.; Campanelli, C.; Lee, J.B. Porokeratosis Ptychotropica: A Clinically Distinct Variant of Porokeratosis. J. Am. Acad. Dermatol. 2009, 60, 501–503. [Google Scholar] [CrossRef]
- Mehta, V.; Balachandran, C. Simultaneous Co-Occurrence of Porokeratosis of Mibelli with Disseminated Superficial Actinic Porokeratosis. Indian J. Dermatol. 2009, 54, 390–391. [Google Scholar] [CrossRef]
- Kubo, A.; Sasaki, T.; Suzuki, H.; Shiohama, A.; Aoki, S.; Sato, S.; Fujita, H.; Ono, N.; Umegaki-Arao, N.; Kawai, T.; et al. Clonal Expansion of Second-Hit Cells with Somatic Recombinations or C>T Transitions Form Porokeratosis in MVD or MVK Mutant Heterozygotes. J. Investig. Dermatol. 2019, 139, 2458–2466.e9. [Google Scholar] [CrossRef] [PubMed]
- Ng, D.; Brand, R. A Precancerous Skin Lesion That Is Often Misdiagnosed. Aust. J. Gen. Pract. 2019, 48, 765–768. [Google Scholar] [CrossRef]
- Zaballos, P.; Puig, S.; Malvehy, J. Dermoscopy of Disseminated Superficial Actinic Porokeratosis. Arch. Dermatol. 2004, 140, 1410. [Google Scholar] [CrossRef]
- Delfino, M.; Argenziano, G.; Nino, M. Dermoscopy for the Diagnosis of Porokeratosis. J. Eur. Acad. Dermatol. Venereol. 2004, 18, 194–195. [Google Scholar] [CrossRef]
- Chi, C.; Liu, J. Image Gallery: Porokeratosis under the Dermoscopic Furrow Ink Test and Ultraviolet Light. Br. J. Dermatol. 2017, 177, e159. [Google Scholar] [CrossRef]
- Vargas-Mora, P.; Morgado-Carrasco, D.; Fustà-Novell, X. Porokeratosis: A Review of Its Pathophysiology, Clinical Manifestations, Diagnosis, and Treatment. Actas Dermosifiliogr. 2020, 111, 545–560. [Google Scholar] [CrossRef]
- Nicola, A.; Magliano, J. Dermoscopy of Disseminated Superficial Actinic Porokeratosis. Actas Dermosifiliogr. 2017, 108, e33–e37. [Google Scholar] [CrossRef]
- Errichetti, E.; Zalaudek, I.; Kittler, H.; Apalla, Z.; Argenziano, G.; Bakos, R.; Blum, A.; Braun, R.P.; Ioannides, D.; Lacarrubba, F.; et al. Standardization of Dermoscopic Terminology and Basic Dermoscopic Parameters to Evaluate in General Dermatology (Non-Neoplastic Dermatoses): An Expert Consensus on Behalf of the International Dermoscopy Society. Br. J. Dermatol. 2020, 182, 454–467. [Google Scholar] [CrossRef]
- Hung, R.; Ahmeen, M.; Fleming, A.; Hoque, S. Itchy Lesions in Pigmented Skin. BMJ Case Rep. 2013, 2013, bcr2013201058. [Google Scholar] [CrossRef]
- Tan, T.S.P.; Tallon, B. Pigmented Porokeratosis. A Further Variant? Am. J. Dermatopathol. 2016, 38, 218–221. [Google Scholar] [CrossRef]
- Reyna-Rodríguez, I.L.; García-Lozano, J.A.; Ocampo-Candiani, J. Pigmented Disseminated Superficial Actinic Porokeratosis in Dark-Skinned Patients: Clinical, Dermoscopic, and Histopathologic Features. J. Cosmet. Dermatol. 2021, 20, 3054–3056. [Google Scholar] [CrossRef]
- Mazzeo, M.; Longo, C.; Manfreda, V.; Piana, S.; Bianchi, L.; Pellacani, G.; Pampena, R. Looking Horizontally at Disseminated Superficial Actinic Porokeratosis: Correlations between in-Vivo Reflectance Confocal Microscopy and Histopathology. Skin. Res. Technol. 2020, 26, 443–444. [Google Scholar] [CrossRef]
- Broggi, G.; Verzì, A.E.; Caltabiano, R.; Micali, G.; Lacarrubba, F. Correlation between In Vivo Reflectance Confocal Microscopy and Horizontal Histopathology in Skin Cancer: A Review. Front. Oncol. 2021, 11, 653140. [Google Scholar] [CrossRef]
- Moscarella, E.; Longo, C.; Zalaudek, I.; Argenziano, G.; Piana, S.; Lallas, A. Dermoscopy and Confocal Microscopy Clues in the Diagnosis of Psoriasis and Porokeratosis. J. Am. Acad. Dermatol. 2013, 69, e231–e233. [Google Scholar] [CrossRef]
- Ulrich, M.; Forschner, T.; Röwert-Huber, J.; González, S.; Stockfleth, E.; Sterry, W.; Astner, S. Differentiation between Actinic Keratoses and Disseminated Superficial Actinic Porokeratoses with Reflectance Confocal Microscopy. Br. J. Dermatol. 2007, 156 (Suppl. S3), 47–52. [Google Scholar] [CrossRef]
- Sertznig, P.; von Felbert, V.; Megahed, M. Porokeratosis: Present Concepts. J. Eur. Acad. Dermatol. Venereol. 2012, 26, 404–412. [Google Scholar] [CrossRef]
- Shen, C.-S.; Tabata, K.; Matsuki, M.; Goto, T.; Yokochi, T.; Yamanishi, K. Premature Apoptosis of Keratinocytes and the Dysregulation of Keratinization in Porokeratosis. Br. J. Dermatol. 2002, 147, 498–502. [Google Scholar] [CrossRef]
- Fernandez-Flores, A. Small Lesions of Porokeratosis Show a Normal Proliferation Rate with MIB-1. Acta Dermatovenerol. Alp. Pannonica Adriat. 2008, 17, 22–25. [Google Scholar]
- Champagne, C.; Moore, L.; Reule, R.; Dyer, J.A.; Rady, P.; Tyring, S.K.; North, J.P. Cornoid Lamella-Like Structures in HIV-Associated Epidermodysplasia Verruciformis: A Unique Histopathologic Finding. Am. J. Dermatopathol. 2015, 37, 929–932. [Google Scholar] [CrossRef]
- Foster, C.; Tallon, B. Porokeratosis: A Differential Diagnosis to Consider in Benign Lichenoid Keratosis. Int. J. Clin. Exp. Pathol. 2022, 15, 56–62. [Google Scholar]
- Maruyama, T.; Ito, M. Disseminated superficial actinic porokeratosis with amyloid deposition. Nihon Hifuka Gakkai Zasshi 1987, 97, 463–470. [Google Scholar]
- Yamamoto, T.; Furukawa, H.; Ohtsuka, M. Amyloid Deposition in Disseminated Superficial Porokeratosis with Inflammatory Stages. J. Dermatol. 2013, 40, 1059–1060. [Google Scholar] [CrossRef]
- Zhao, Q.; Yu, B.; Zhou, H.; Feng, C.; Zhang, X.; Zheng, Y.; Geng, S. Generalized Type 2 Segmental Disseminated Superficial Actinic Porokeratosis Coexisted with Multiple Cutaneous Squamous Cell Carcinomas: Analysis of Two Cases. Indian. J. Pathol. Microbiol. 2020, 63, 634–636. [Google Scholar] [CrossRef]
- Al-Haseni, A.; Chitgopeker, P.; Ho, J.D.; Goldberg, L.J.; Sahni, D. Amelanotic Melanoma Arising within a Lesion of Disseminated Superficial Actinic Porokeratosis: An Unusual Presentation Leading to a Novel Therapeutic Approach. Dermatol. Ther. 2018, 31, e12552. [Google Scholar] [CrossRef]
- Happle, R. Mibelli Revisited: A Case of Type 2 Segmental Porokeratosis from 1893. J. Am. Acad. Dermatol. 2010, 62, 136–138. [Google Scholar] [CrossRef]
- Xia, J.H.; Yang, Y.F.; Deng, H.; Tang, B.S.; Tang, D.S.; He, Y.G.; Xia, K.; Chen, S.X.; Li, Y.X.; Pan, Q.; et al. Identification of a Locus for Disseminated Superficial Actinic Porokeratosis at Chromosome 12q23.2-24.1. J. Investig. Dermatol. 2000, 114, 1071–1074. [Google Scholar] [CrossRef]
- Zhang, Z.H.; Niu, Z.M.; Yuan, W.T.; Zhao, J.J.; Jiang, F.X.; Zhang, J.; Chai, B.; Cui, F.; Chen, W.; Lian, C.H.; et al. A Mutation in SART3 Gene in a Chinese Pedigree with Disseminated Superficial Actinic Porokeratosis. Br. J. Dermatol. 2005, 152, 658–663. [Google Scholar] [CrossRef]
- Xia, K.; Deng, H.; Xia, J.H.; Zheng, D.; Zhang, H.L.; Lu, C.Y.; Li, C.Q.; Pan, Q.; Dai, H.P.; Yang, Y.F.; et al. A Novel Locus (DSAP2) for Disseminated Superficial Actinic Porokeratosis Maps to Chromosome 15q25.1-26.1. Br. J. Dermatol. 2002, 147, 650–654. [Google Scholar] [CrossRef]
- Zhang, Z.; Niu, Z.; Yuan, W.; Liu, W.; Xiang, L.; Zhang, J.; Chu, X.; Zhao, J.; Jiang, F.; Chai, B.; et al. Fine Mapping and Identification of a Candidate Gene SSH1 in Disseminated Superficial Actinic Porokeratosis. Hum. Mutat. 2004, 24, 438. [Google Scholar] [CrossRef]
- Zhang, Z.-H.; Huang, W.; Niu, Z.-M.; Liu, W.-D.; Xiang, L.-H.; Yuan, W.-T.; Zhao, J.-J.; Gu, C.-Y.; Chai, B.; Jiang, F.-X.; et al. Two Closely Linked Variations in Actin Cytoskeleton Pathway in a Chinese Pedigree with Disseminated Superficial Actinic Porokeratosis. J. Am. Acad. Dermatol. 2005, 52, 972–976. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, S.; Yao, Q.; Liu, X.; Wang, X.; Huang, C.; Huang, X.; Wang, P.; Yuan, M.; Liu, J.Y.; et al. Identification of a Genetic Locus for Autosomal Dominant Disseminated Superficial Actinic Porokeratosis on Chromosome 1p31.3-P31.1. Hum. Genet. 2008, 123, 507–513. [Google Scholar] [CrossRef]
- Luan, J.; Niu, Z.; Zhang, J.; Crosby, M.E.; Zhang, Z.; Chu, X.; Wang, Z.; Huang, W.; Xiang, L.; Zheng, Z. A Novel Locus for Disseminated Superficial Actinic Porokeratosis Maps to Chromosome 16q24.1-24.3. Hum. Genet. 2011, 129, 329–334. [Google Scholar] [CrossRef]
- Qian, W.; Wu, J.; Tang, H.; Zhen, Q.; Ge, H.; Gao, J.; Chen, J.; Chang, Y.; Wang, W.; Sun, L. Mutation Analysis of the MVD Gene in a Chinese Family with Disseminated Superficial Actinic Porokeratosis and a Chinese Literature Review. Indian J. Dermatol. 2021, 66, 126–131. [Google Scholar] [CrossRef]
- Zhu, T.; Tian, D.; Zhang, L.; Xu, X.; Xia, K.; Hu, Z.; Xiong, Z.; Tan, J. Novel Mutations in Mevalonate Kinase Cause Disseminated Superficial Actinic Porokeratosis. Br. J. Dermatol. 2019, 181, 304–313. [Google Scholar] [CrossRef]
- Brennenstuhl, H.; Nashawi, M.; Schröter, J.; Baronio, F.; Beedgen, L.; Gleich, F.; Jeltsch, K.; von Landenberg, C.; Martini, S.; Simon, A.; et al. Phenotypic Diversity, Disease Progression, and Pathogenicity of MVK Missense Variants in Mevalonic Aciduria. J. Inherit. Metab. Dis. 2021, 44, 1272–1287. [Google Scholar] [CrossRef]
- Miziorko, H.M. Enzymes of the Mevalonate Pathway of Isoprenoid Biosynthesis. Arch. Biochem. Biophys. 2011, 505, 131–143. [Google Scholar] [CrossRef]
- Zhang, S.-Q.; Jiang, T.; Li, M.; Zhang, X.; Ren, Y.-Q.; Wei, S.-C.; Sun, L.-D.; Cheng, H.; Li, Y.; Yin, X.-Y.; et al. Exome Sequencing Identifies MVK Mutations in Disseminated Superficial Actinic Porokeratosis. Nat. Genet. 2012, 44, 1156–1160. [Google Scholar] [CrossRef]
- Vlachou, C.; Kanelleas, A.I.; Martin-Clavijo, A.; Berth-Jones, J. Treatment of Disseminated Superficial Actinic Porokeratosis with Topical Diclofenac Gel: A Case Series. J. Eur. Acad. Dermatol. Venereol. 2008, 22, 1343–1345. [Google Scholar] [CrossRef]
- Buhaescu, I.; Izzedine, H. Mevalonate Pathway: A Review of Clinical and Therapeutical Implications. Clin. Biochem. 2007, 40, 575–584. [Google Scholar] [CrossRef]
- Cui, H.; Li, L.; Wang, W.; Shen, J.; Yue, Z.; Zheng, X.; Zuo, X.; Liang, B.; Gao, M.; Fan, X.; et al. Exome Sequencing Identifies SLC17A9 Pathogenic Gene in Two Chinese Pedigrees with Disseminated Superficial Actinic Porokeratosis. J. Med. Genet. 2014, 51, 699–704. [Google Scholar] [CrossRef]
- Kim, H.; Lee, B.H.; Do, H.-S.; Kim, G.-H.; Kang, S.; Koh, K.-N.; Im, H.J. Case Report: Mevalonic Aciduria Complicated by Acute Myeloid Leukemia After Hematopoietic Stem Cell Transplantation. Front. Immunol. 2021, 12, 782780. [Google Scholar] [CrossRef]
- Yıldız, Ç.; Gezgin Yıldırım, D.; Inci, A.; Tümer, L.; Cengiz Ergin, F.B.; Sunar Yayla, E.N.S.; Esmeray Şenol, P.; Karaçayır, N.; Eğritaş Gürkan, Ö.; Okur, I.; et al. A Possibly New Autoinflammatory Disease Due to Compound Heterozygous Phosphomevalonate Kinase Gene Mutation. Jt. Bone Spine 2023, 90, 105490. [Google Scholar] [CrossRef]
- Lang, B.M.; Peveling-Oberhag, A.; Zimmer, S.; Wegner, J.; Sohn, A.; Grabbe, S.; Staubach, P. Effective Treatment of Disseminated Superficial Actinic Porokeratosis with Chemical Peels—Customary Treatment for a Rare Disease. J. Dermatol. Treat. 2020, 31, 744–748. [Google Scholar] [CrossRef]
- Li, M.; Min, W.; Wang, J.; Wang, L.; Li, Y.; Zhou, N.; Yang, Z.; Qian, Q. Effects of Mevalonate Kinase Interference on Cell Differentiation, Apoptosis, Prenylation and Geranylgeranylation of Human Keratinocytes Are Attenuated by Farnesyl Pyrophosphate or Geranylgeranyl Pyrophosphate. Exp. Ther. Med. 2020, 19, 2861–2870. [Google Scholar] [CrossRef]
- Politiek, F.A.; Waterham, H.R. Compromised Protein Prenylation as Pathogenic Mechanism in Mevalonate Kinase Deficiency. Front. Immunol. 2021, 12, 724991. [Google Scholar] [CrossRef]
- Biswas, A. Cornoid Lamellation Revisited: Apropos of Porokeratosis with Emphasis on Unusual Clinicopathological Variants. Am. J. Dermatopathol. 2015, 37, 145–155. [Google Scholar] [CrossRef]
- Huang, M.; Zhou, B.; Gong, J.; Xing, L.; Ma, X.; Wang, F.; Wu, W.; Shen, H.; Sun, C.; Zhu, X.; et al. RNA-Splicing Factor SART3 Regulates Translesion DNA Synthesis. Nucleic Acids Res. 2018, 46, 4560–4574. [Google Scholar] [CrossRef]
- Timani, K.A.; Rezaei, S.; Whitmill, A.; Liu, Y.; He, J.J. Tip110/SART3-Mediated Regulation of NF-κB Activity by Targeting IκBα Stability Through USP15. Front. Oncol. 2022, 12, 843157. [Google Scholar] [CrossRef]
- Kurita, S.; Gunji, E.; Ohashi, K.; Mizuno, K. Actin Filaments-Stabilizing and -Bundling Activities of Cofilin-Phosphatase Slingshot-1. Genes Cells 2007, 12, 663–676. [Google Scholar] [CrossRef]
- Welch, M.D.; DePace, A.H.; Verma, S.; Iwamatsu, A.; Mitchison, T.J. The Human Arp2/3 Complex Is Composed of Evolutionarily Conserved Subunits and Is Localized to Cellular Regions of Dynamic Actin Filament Assembly. J. Cell Biol. 1997, 138, 375–384. [Google Scholar] [CrossRef]
- Schrank, B.R.; Aparicio, T.; Li, Y.; Chang, W.; Chait, B.T.; Gundersen, G.G.; Gottesman, M.E.; Gautier, J. Nuclear ARP2/3 Drives DNA Break Clustering for Homology-Directed Repair. Nature 2018, 559, 61–66. [Google Scholar] [CrossRef]
- Atzmony, L.; Choate, K.A. Second-Hit Somatic Mutations in Mevalonate Pathway Genes Underlie Porokeratosis. J. Investig. Dermatol. 2019, 139, 2409–2411. [Google Scholar] [CrossRef]
- Atzmony, L.; Lim, Y.H.; Hamilton, C.; Leventhal, J.S.; Wagner, A.; Paller, A.S.; Choate, K.A. Topical Cholesterol/Lovastatin for the Treatment of Porokeratosis: A Pathogenesis-Directed Therapy. J. Am. Acad. Dermatol. 2020, 82, 123–131. [Google Scholar] [CrossRef]
- Ugwu, N.; Choate, K.A.; Atzmony, L. Two Percent Lovastatin Ointment as a Pathogenesis-Directed Monotherapy for Porokeratosis. JAAD Case Rep. 2020, 6, 1110–1112. [Google Scholar] [CrossRef]
- Marks, S.; Varma, R.; Cantrell, W.; Chen, S.C.; Gold, M.; Muellenhoff, M.; Elewski, B. Diclofenac Sodium 3% Gel as a Potential Treatment for Disseminated Superficial Actinic Porokeratosis. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 42–45. [Google Scholar] [CrossRef]
- Darr-Foit, S.; Elsner, P. 77-year-old female with persisting erythematous and scaly plaques on the extremities and upper trunk: Preparation for the medical specialist examination: Part 20. Hautarzt 2018, 69, 165–168. [Google Scholar] [CrossRef]
- Nayeemuddin, F.A.; Wong, M.; Yell, J.; Rhodes, L.E. Topical Photodynamic Therapy in Disseminated Superficial Actinic Porokeratosis. Clin. Exp. Dermatol. 2002, 27, 703–706. [Google Scholar] [CrossRef]
- Cavicchini, S.; Tourlaki, A. Successful Treatment of Disseminated Superficial Actinic Porokeratosis with Methyl Aminolevulinate-Photodynamic Therapy. J. Dermatol. Treat. 2006, 17, 190–191. [Google Scholar] [CrossRef]
- Aird, G.A.; Sitenga, J.L.; Nguyen, A.H.; Vaudreuil, A.; Huerter, C.J. Light and Laser Treatment Modalities for Disseminated Superficial Actinic Porokeratosis: A Systematic Review. Lasers Med. Sci. 2017, 32, 945–952. [Google Scholar] [CrossRef]
- Calzavara-Pinton, P.G.; Rossi, M.T.; Aronson, E.; Sala, R.; Italian Group for Photodynamic Therapy. A Retrospective Analysis of Real-Life Practice of off-Label Photodynamic Therapy Using Methyl Aminolevulinate (MAL-PDT) in 20 Italian Dermatology Departments. Part 1: Inflammatory and Aesthetic Indications. Photochem. Photobiol. Sci. 2013, 12, 148–157. [Google Scholar] [CrossRef]
- Salas, T.; Hernandez-Gil, J.; Lopez, A.; Dorado, M.; Ruiz, J.; García, E.; Martinez, F. Two Cases of Disseminated Superficial Actinic Porokeratosis Treated with Daylight-Mediated Photodynamic Therapy. Dermatol. Ther. 2016, 29, 484–485. [Google Scholar] [CrossRef]
- Ferrer Guillén, B.; Giácaman, M.M.; Pérez Ferriols, A. Improved Effect on 2 Cases of Disseminated Superficial Actinic Porokeratosis with Daylight Photodynamic Therapy. Photodiagnosis Photodyn. Ther. 2018, 23, 365–366. [Google Scholar] [CrossRef]
- Fernández-Guarino, M.; Harto, A.; Pérez-Garcia, B.; Martin-González, M.; Urrutia, S.; Jaén, P. Photodynamic Therapy in Disseminated Superficial Actinic Porokeratosis. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 176–177. [Google Scholar] [CrossRef]
- Schmook, T.; Kraft, J.; Ulrich, C.; Stockfleth, E. Disseminated Superficial Actinic Porokeratosis: Report of 7 Patients Successfully Treated with Cryotherapy. J. Am. Acad. Dermatol. 2005, 52, 159. [Google Scholar] [CrossRef]
- Boiy, A.; de Witte, P.A.M.; Roelandts, R. Topical Treatment of Disseminated Superficial Actinic Porokeratosis with Hypericin-Photodynamic Therapy: A Case Report. Photodiagnosis Photodyn. Ther. 2010, 7, 123–125. [Google Scholar] [CrossRef]
- Ramelyte, E.; Bylaite-Bucinskiene, M.; Dummer, R.; Imhof, L. Successful Use of Grenz Rays for Disseminated Superficial Actinic Porokeratosis: Report of 8 Cases. Dermatology 2017, 233, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Ting, S.; Webster, M. Grenz Ray Therapy in Disseminated Superficial Actinic Porokeratosis: A Case Series of 17 Patients. Australas. J. Dermatol. 2022, 63, 91–94. [Google Scholar] [CrossRef]
- O’Reilly, M.; Butt, S.; Gajebasia, S.; Dawe, R. PD13: Successful Treatment of Disseminated Superficial Actinic Porokeratosis with Grenz Ray Therapy. Br. J. Dermatol. 2022, 187, 218. [Google Scholar] [CrossRef]
- Smith, J.; Narla, S.; Lyons, A.B.; Kohli, I.; Siddiqui, F.; Rao, B.K.; Penman, L.; Hamzavi, I.H. Brachytherapy for Resistant Disseminated Superficial Actinic Porokeratosis. Appl. Rad. Oncol. 2020, 9, 43–45. [Google Scholar] [CrossRef]
- Spelman, L.; Christie, D.; Kaminski, A.; Baker, C.; Supranowicz, M.; Sinclair, R. Radiotherapy, Utilizing Volumetric Modulated Arc Therapy, for Extensive Skin Field Cancerization: A Retrospective Case Series Assessing Efficacy, Safety, and Cosmetic Outcomes at 12 Months after Treatment. Case Rep. Dermatol. 2022, 14, 31–38. [Google Scholar] [CrossRef]
- Harrison, P.V.; Stollery, N. Disseminated Superficial Actinic Porokeratosis Responding to Calcipotriol. Clin. Exp. Dermatol. 1994, 19, 95. [Google Scholar] [CrossRef]
- Bakardzhiev, I.; Kavaklieva, S.; Pehlivanov, G. Successful Treatment of Disseminated Superficial Actinic Porokeratosis with Calcipotriol. Int. J. Dermatol. 2012, 51, 1139–1142. [Google Scholar] [CrossRef]
- Böhm, M.; Luger, T.A.; Bonsmann, G. Disseminated Superficial Actinic Porokeratosis: Treatment with Topical Tacalcitol. J. Am. Acad. Dermatol. 1999, 40, 479–480. [Google Scholar] [CrossRef]
- Abe, M.; Yokoyama, Y.; Ishikawa, O. Successful Treatment of Disseminated Superficial Actinic Porokeratosis with Tacalcitol Lotion. J. Dermatol. 2010, 37, 913–915. [Google Scholar] [CrossRef]
- Nakamura, Y.; Yamaguchi, M.; Nakamura, A.; Muto, M. Calcipotriol and Adapalene Therapy for Disseminated Superficial Actinic Porokeratosis. Indian J. Dermatol. Venereol. Leprol. 2014, 80, 373–374. [Google Scholar] [CrossRef]
- Tchernev, G.; Chokoeva, A.A.; Ivanova, B.; Mangarov, H.; Vidolova, N.G. Disseminated Superficial Actinic Porokeratosis (DSAP): Significant Improvement after Local Administration of Calcipotriol/Betamethasone Gel? Wien. Med. Wochenschr. 2017, 167, 85–88. [Google Scholar] [CrossRef]
- Noborio, R.; Morita, A. Split-Face Trial of CO2 Laser-Induced Ring Abrasion and High-Dose Tacalcitol in the Treatment of Disseminated Superficial Actinic Porokeratosis. J. Dermatol. 2012, 39, 879–880. [Google Scholar] [CrossRef]
- Arun, B.; Pearson, J.; Chalmers, R. Disseminated Superficial Actinic Porokeratosis Treated Effectively with Topical Imiquimod 5% Cream. Clin. Exp. Dermatol. 2011, 36, 509–511. [Google Scholar] [CrossRef] [PubMed]
- Riad, H.; Mansour, K.; Sada, H.A.; Shaika, S.A.; Ansari, H.A.; Mohannadi, H.A. Disseminated Superficial Actinic Porokeratosis on the Face Treated with Imiquimod 5% Cream. Case Rep. Dermatol. 2013, 5, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Anderson, I.; Routt, E.T.; Jim On, S.C. Disseminated Superficial Actinic Porokeratosis Treated with Ingenol Mebutate Gel 0.05. Cutis 2017, 99, E36–E39. [Google Scholar] [PubMed]
- Nahm, W.K.; Donohue, K.G.; Danahy, J.F.; Badiavas, E.; Falanga, V. Systemic 5-Fluorouracil Producing an Inflammatory Response in Porokeratosis. J. Eur. Acad. Dermatol. Venereol. 2003, 17, 190–192. [Google Scholar] [CrossRef]
- Teixeira, S.P.; de Nascimento, M.M.; Bagatin, E.; Hassun, K.M.; Talarico, S.; Michalany, N. The Use of Fluor-Hydroxy Pulse Peel in Actinic Porokeratosis. Dermatol. Surg. 2005, 31, 1145–1148. [Google Scholar] [CrossRef]
- Mendonça, R.F.; Salem, L.A.N.; Alves, R.O.; Hong, B.; Lellis, R.F.; Crocco, E.I. Tratamento da poroqueratose actínica superficial disseminada com laser 1340-nm Nd:YAP. Surg. Cosmet. Dermatol. 2019, 11, 65–67. [Google Scholar] [CrossRef]
- Narbutt, J.; Słowik-Rylska, M.; Sysa-Jędrzejowska, A.; Słowik-Kwiatkowska, I.; Lesiak, A. Disseminated Superficial Actinic Porokeratosis. Two Case Reports. Adv. Dermatol. Allergol. 2010, 27, 435–439. [Google Scholar]
- Ross, N.A.; Rosenbaum, L.E.; Saedi, N.; Arndt, K.A.; Dover, J.S. Disseminated Superficial Actinic Porokeratosis Improved with Fractional 1927-Nm Laser Treatments. J. Cosmet. Laser Ther. 2016, 18, 53–55. [Google Scholar] [CrossRef]
- Borroni, R.G.; Poddighe, D.; Zecca, M.; Brazzelli, V. Efficacy of Acitretin for Porokeratosis in a Child with Chronic Cutaneous Graft versus Host Disease. Pediatr. Dermatol. 2013, 30, 148–150. [Google Scholar] [CrossRef]
- Park, B.J.; Oh, E.H.; Kim, J.E.; Ko, J.Y.; Ro, Y.S. Treatment of Disseminated Superficial Actinic Porokeratosis with Oral Alitretinoin. J. Eur. Acad. Dermatol. Venereol. 2017, 31, e505–e507. [Google Scholar] [CrossRef]
- Kariniemi, A.L.; Stubb, S.; Lassus, A. Treatment of Disseminated Superficial Actinic Porokeratosis with a New Aromatic Retinoid (Ro 10-9359). Br. J. Dermatol. 1980, 102, 213–214. [Google Scholar] [CrossRef]
- Knobler, R.M.; Neumann, R.A. Exacerbation of Porokeratosis during Etretinate Therapy. Acta Derm. Venereol. 1990, 70, 319–322. [Google Scholar] [CrossRef]
- Carmichael, A.J.; Tan, C.Y. Digitate Keratoses—A Complication of Etretinate Used in the Treatment of Disseminated Superficial Actinic Porokeratosis. Clin. Exp. Dermatol. 1990, 15, 370–371. [Google Scholar] [CrossRef]
- Schwarz, T.; Seiser, A.; Gschnait, F. Disseminated Superficial “Actinic” Porokeratosis. J. Am. Acad. Dermatol. 1984, 11, 724–730. [Google Scholar] [CrossRef]
- Vergara, G.; Bañuls, J.; Botella, R.; Silvestre, J.F.; Belinchón, I.; Betlloch, I. Porokeratosis of the Lower Lip. Eur. J. Dermatol. 2002, 12, 500–502. [Google Scholar]
- Tan, L.S.; Chong, W.-S. Porokeratosis in Singapore: An Asian Perspective. Australas. J. Dermatol. 2012, 53, e40–e44. [Google Scholar] [CrossRef]
- Lolis, M.S.; Marmur, E.S. Treatment of Disseminated Superficial Actinic Porokeratosis (DSAP) with the Q-Switched Ruby Laser. J. Cosmet. Laser Ther. 2008, 10, 124–127. [Google Scholar] [CrossRef]
- Itoh, M.; Nakagawa, H. Successful Treatment of Disseminated Superficial Actinic Porokeratosis with Q-Switched Ruby Laser. J. Dermatol. 2007, 34, 816–820. [Google Scholar] [CrossRef]
- Rosenblum, J.; Roenigk, H.H. Erbium Laser for the Treatment of Disseminated Superficial Actinic Porokeratosis: A Case Report. Dermatol. Surg. 2013, 39, 1543–1545. [Google Scholar] [CrossRef]
- Hou, P.; Miao, Y.; Hou, S.; Cheng, S.; Hu, Z. Application of Q-Switch Alexandrite Laser Combined with Fractional CO2 Laser in Treating Disseminated Superficial Actinic Porokeratosis: Report of Two Cases. Int. J. Clin. Exp. Med. 2017, 10, 14532–14535. [Google Scholar]
- Kim, H.S.; Baek, J.H.; Park, Y.M.; Kim, H.O.; Lee, J.Y. Photodynamic Therapy Combined with CO2 Laser Vaporization on Disseminated Superficial Actinic Porokeratosis: A Report of 2 Cases on the Face. Ann. Dermatol. 2011, 23, S211–S213. [Google Scholar] [CrossRef]
- Chrastil, B.; Glaich, A.S.; Goldberg, L.H.; Friedman, P.M. Fractional Photothermolysis: A Novel Treatment for Disseminated Superficial Actinic Porokeratosis. Arch. Dermatol. 2007, 143, 1447–1462. [Google Scholar] [CrossRef]
- Marcuzzi, A.; Zanin, V.; Piscianz, E.; Tricarico, P.M.; Vuch, J.; Girardelli, M.; Monasta, L.; Bianco, A.M.; Crovella, S. Lovastatin-Induced Apoptosis Is Modulated by Geranylgeraniol in a Neuroblastoma Cell Line. Int. J. Dev. Neurosci. 2012, 30, 451–456. [Google Scholar] [CrossRef]
- Marcuzzi, A.; Tricarico, P.M.; Piscianz, E.; Kleiner, G.; Vecchi Brumatti, L.; Crovella, S. Lovastatin Induces Apoptosis through the Mitochondrial Pathway in an Undifferentiated SH-SY5Y Neuroblastoma Cell Line. Cell Death Dis. 2013, 4, e585. [Google Scholar] [CrossRef]
- Byth, L.A.; Byth, J. Topical Simvastatin-Cholesterol for Disseminated Superficial Actinic Porokeratosis: An Open-Label, Split-Body Clinical Trial. Australas. J. Dermatol. 2021, 62, 310–313. [Google Scholar] [CrossRef]
- Tomsitz, D.; Biedermann, T. Successful Treatment of Disseminated Superficial Actinic Porokeratosis with Topical 2% Cholesterol/ 2% Lovastatin Cream: A Case Series with 7 Patients. J. Eur. Acad. Dermatol. Venereol. 2022, 36, e52–e54. [Google Scholar] [CrossRef]
- Leow, Y.H.; Soon, Y.H.; Tham, S.N. A Report of 31 Cases of Porokeratosis at the National Skin Centre. Ann. Acad. Med. Singap. 1996, 25, 837–841. [Google Scholar]
- Santa Lucia, G.; Snyder, A.; Lateef, A.; Drohan, A.; Gregoski, M.J.; Barton, V.; Elston, D.M. Safety and Efficacy of Topical Lovastatin Plus Cholesterol Cream vs Topical Lovastatin Cream Alone for the Treatment of Disseminated Superficial Actinic Porokeratosis: A Randomized Clinical Trial. JAMA Dermatol. 2023, 159, 488–495. [Google Scholar] [CrossRef]
- Andrews, G.C. Porokeratosis (Mibelli) Disseminated and Superficial Type. Arch. Dermatol. Syphilol. 1937, 36, 1111. [Google Scholar]
- Kim, S.W.; Min, S.U.; Won, C.H.; Cho, S. Disseminated Superficial Porokeratosisin a Patient with Gastric Cancer. Ann. Dermatol. 2008, 20, 193–196. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Liu, F.; Huang, C.; Han, S.; Lv, Y.; Liu, C.-J.; Zhang, S.; Qin, Y.; Ling, L.; et al. Loss-of-Function Mutation in PMVK Causes Autosomal Dominant Disseminated Superficial Porokeratosis. Sci. Rep. 2016, 6, 24226. [Google Scholar] [CrossRef] [PubMed]
- Rosón, E.; García-Doval, I.; De La Torre, C.; Losada, A.; Rodríguez, T.; Ocampo, C.; Cruces, M. Disseminated Superficial Porokeratosis with Mucosal Involvement. Acta Derm. Venereol. 2001, 81, 64–65. [Google Scholar] [CrossRef]
- Kanak, K.; Jaiswal, A.K.; Reddy, P. Disseminated Superficial and Warty Type of Porokeratosis: A Rare Coexistence. Indian J. Dermatol. 2011, 56, 576–577. [Google Scholar] [CrossRef]
- Bhatia, R.; Gupta, V.; Khanna, N. Oral Involvement in Disseminated Superficial Porokeratosis. Indian J. Dermatol. Venereol. Leprol. 2017, 83, 244–246. [Google Scholar] [CrossRef]
- Kanekura, T.; Yoshii, N. Eruptive Pruritic Papular Porokeratosis: A Pruritic Variant of Porokeratosis. J. Dermatol. 2006, 33, 813–816. [Google Scholar] [CrossRef]
- Makino, E.; Inaoki, M.; Fujimoto, W. Inflammatory Stage of Disseminated Superficial Porokeratosis. J. Dermatol. 2005, 32, 890–893. [Google Scholar] [CrossRef]
- Sakhiya, J.J.; Sakhiya, D.J.; Patel, M.R.; Daruwala, F.R. Case Report on Rare Clinical Variant of Porokeratosis: Disseminated Superficial Porokeratosis. J. Cutan. Aesthet. Surg. 2020, 13, 145–148. [Google Scholar] [CrossRef]
- Navarro, V.; Pinazo, I.; Martínez, E.; Monteagudo, C.; Jordá, E. Facial Superficial Porokeratosis. Dermatology 2000, 201, 361. [Google Scholar] [CrossRef]
- Kanzaki, T.; Miwa, N.; Kobayashi, T.; Ogawa, S. Eruptive Pruritic Papular Porokeratosis. J. Dermatol. 1992, 19, 109–112. [Google Scholar] [CrossRef]
- Stork, J.; Kodetová, D. Disseminated Superficial Porokeratosis: An Eruptive Pruritic Papular Variant. Dermatology 1997, 195, 304–305. [Google Scholar] [CrossRef] [PubMed]
- Goulding, J.M.R.; Teoh, J.K.; Carr, R.A.; Humphreys, F.; Gee, B.C. Eruptive Disseminated Superficial Porokeratosis with Rapid Resolution: A Drug-Induced Phenomenon? Clin. Exp. Dermatol. 2009, 34, 895–897. [Google Scholar] [CrossRef] [PubMed]
- Patrizi, A.; Virdi, A.; Misciali, C.; Bardazzi, F. Eruptive Pruritic Papular Porokeratosis in a Caucasian Woman: A Transient Inflammatory Stage of Porokeratosis. G. Ital. Dermatol. Venereol. 2017, 152, 529–531. [Google Scholar] [CrossRef] [PubMed]
- Morgado-Carrasco, D.; Feola, H.; Fustà-Novell, X. Eruptive Pruritic Papular Porokeratosis or Inflammatory Form of Disseminated Superficial Porokeratosis: A New Case and Review of the Literature. Dermatol. Online J. 2020, 26, 7. [Google Scholar] [CrossRef]
- Soni, R.; Phiske, M.; Kanade, P.; John, J.; Joshi, R.; Shylaja, S. Eruptive Pruritic Papular Porokeratosis: A Rare Variant of Porokeratosis. Indian J. Dermatol. 2021, 66, 212–214. [Google Scholar] [CrossRef]
- Zhang, W.-L.; Huang, D.; Zhang, W.; Wu, Y.-D.; Feng, S.-Y.; Jiang, Y.-Q.; Li, C.-R. Eruptive Pruritic Papular Porokeratosis. Postepy Dermatol. Alergol. 2021, 38, 167–169. [Google Scholar] [CrossRef]
- Wakatabi, K.; Kakurai, M.; Yamada, T.; Umemoto, N.; Demitsu, T.; Yoneda, K. Inflammatory Disseminated Superficial Porokeratosis with an Unusual Clinical Feature of the Pruritic, Erythematous Papules Preceding Annular Brownish Pigmentation. J. Dermatol. 2012, 39, 946–948. [Google Scholar] [CrossRef]
- Shoimer, I.; Robertson, L.H.; Storwick, G.; Haber, R.M. Eruptive Disseminated Porokeratosis: A New Classification System. J. Am. Acad. Dermatol. 2014, 71, 398–400. [Google Scholar] [CrossRef] [PubMed]
- Happle, R. Loss of Heterozygosity in Human Skin. J. Am. Acad. Dermatol. 1999, 41, 143–164. [Google Scholar] [CrossRef] [PubMed]
- Korviriyakamol, T.; Kattipathananpong, P.; Chunhasewee, C.; Wessagowit, V.; Kootiratrakarn, T. Co-Existence of Porokeratosis Variants Concurrent with Bowen’s Disease: Two Rare Cases Report. J. Med. Assoc. Thai 2014, 97, 356–359. [Google Scholar]
- Palleschi, G.M.; Torchia, D. Porokeratosis of Mibelli and Superficial Disseminated Porokeratosis. J. Cutan. Pathol. 2008, 35, 253–255. [Google Scholar] [CrossRef]
- Raychaudhury, T.; Valsamma, D.P.C. Giant Porokeratosis. IJDVL 2011, 77, 601. [Google Scholar] [CrossRef]
- Kluger, N.; Dereure, O.; Guilhou, J.-J.; Guillot, B. Genital Porokeratosis: Treatment with Diclofenac Topical Gel. J. Dermatol. Treat. 2007, 18, 188–190. [Google Scholar] [CrossRef]
- Chun, S.I.; Lee, J.S.; Kim, N.S.; Park, K.D. Disseminated Epidermolytic Acanthoma with Disseminated Superficial Porokeratosis and Verruca Vulgaris in an Immunosuppressed Patient. J. Dermatol. 1995, 22, 690–692. [Google Scholar] [CrossRef]
- Vasudevan, B.; Chatterjee, M.; Grewal, R.; Rana, V.; Lodha, N. A Case of Disseminated Superficial Porokeratosis Associated with Giant Porokeratosis in Pregnancy. Indian J. Dermatol. 2014, 59, 492–494. [Google Scholar] [CrossRef]
- Ibbotson, S.H. Disseminated Superficial Porokeratosis: What Is the Association with Ultraviolet Radiation? Clin. Exp. Dermatol. 1996, 21, 48–50. [Google Scholar] [CrossRef]
- Kanitakis, J.; Misery, L.; Nicolas, J.F.; Lyonnet, S.; Chouvet, B.; Haftek, M.; Faure, M.; Claudy, A.; Thivolet, J. Disseminated Superficial Porokeratosis in a Patient with AIDS. Br. J. Dermatol. 1994, 131, 284–289. [Google Scholar] [CrossRef]
- Romaní, J.; Pujol, R.M.; Casanova, J.M.; de Moragas, J.M. Disseminated Superficial Porokeratosis Developing after Electron-Beam Total Skin Irradiation for Mycosis Fungoides. Clin. Exp. Dermatol. 1996, 21, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Dumas, M.; Corre, F.; Payancé, A.; Guedj, N.; Durand, F.; Descamps, V.; Le Bozec, P. Eruptive disseminated superficial porokeratosis associated with acute hepatitis E. Ann. Dermatol. Venereol. 2019, 146, 655–658. [Google Scholar] [CrossRef] [PubMed]
- Luelmo-Aguilar, J.; Gonzalez-Castro, U.; Mieras-Barcelo, C.; Castells-Rodellas, A. Disseminated Porokeratosis and Myelodysplastic Syndrome. Dermatology 1992, 184, 289. [Google Scholar] [CrossRef] [PubMed]
- Levin, R.M.; Heymann, W.R. Superficial Disseminate Porokeratosis in a Patient with Myelodysplastic Syndrome. Int. J. Dermatol. 1999, 38, 138–139. [Google Scholar] [CrossRef]
- Diluvio, L.; Campione, E.; Paterno, E.J.; Hagman, J.H.; Anemona, L.; Orlandi, A.; Chimenti, S. Acute Onset Disseminated Superficial Porokeratosis Heralding Diffuse Large B-Cell Lymphoma. Eur. J. Dermatol. 2008, 18, 349–350. [Google Scholar] [CrossRef]
- Benmously Mlika, R.; Kenani, N.; Badri, T.; Ben Romdhane, S.; Debbiche, A.; Souissi, A.; Ben Ayed, M.; Mokhtar, I.; Fenniche, S. Localized Genital Porokeratosis in a Female Patient with Multiple Myeloma. J. Eur. Acad. Dermatol. Venereol. 2009, 23, 584–585. [Google Scholar] [CrossRef]
- Rossiello, L.; Lupoli, A.; Ruggiero, F.; Boscaino, A.; Cozzi, R. Acquired Disseminated Superficial Porokeratosis in a Patient Affected by Chronic Lymphocitic Leukemia. G. Ital. Dermatol. Venereol. 2017, 152, 533–534. [Google Scholar] [CrossRef]
- Chokoeva, A.A.; Wollina, U.; Lotti, T.; Maximov, G.K.; Lozev, I.; Tchernev, G. Disseminated Porokeratosis with Idiopathic Thrombocytopenia—Case Report and Literature Review of Porokeratosis and Related Disorders. Open Access Maced. J. Med. Sci. 2018, 6, 139–142. [Google Scholar] [CrossRef]
- Irie, K. A Case of Disseminated Superficial Porokeratosis in a Patient with Chronic Graft-versus-Host Disease. Dermatol. Online J. 2020, 26, 22. [Google Scholar] [CrossRef]
- Rio, B.; Magana, C.; Le Tourneau, A.; Bachmeyer, C.; Lévy, V.; Hamont, N.; Diebold, J.; Zittoun, R. Disseminated Superficial Porokeratosis after Autologous Bone Marrow Transplantation. Bone Marrow Transpl. 1997, 19, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Knoell, K.A.; Patterson, J.W.; Wilson, B.B. Sudden Onset of Disseminated Porokeratosis of Mibelli in a Renal Transplant Patient. J. Am. Acad. Dermatol. 1999, 41, 830–832. [Google Scholar] [CrossRef]
- Kanitakis, J.; Euvrard, S.; Claudy, A. Porokeratosis in Organ Transplant Recipients. J. Am. Acad. Dermatol. 2001, 44, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Feuerman, E.J.; Sandbank, M. Disseminated Superficial Porokeratosis in Patients with Pemphigus Vulgaris Treated with Steroids. Acta Derm. Venereol. Suppl. 1979, 59, 59–61. [Google Scholar]
- Bednarek, R.; Ezra, N.; Toubin, Y.; Linos, K.; Mousdicas, N. Eruptive Disseminated Porokeratosis Associated with Corticosteroid-Induced Immunosuppression. Clin. Exp. Dermatol. 2015, 40, 753–756. [Google Scholar] [CrossRef]
- Schena, D.; Papagrigoraki, A.; Frigo, A.; Girolomoni, G. Eruptive Disseminated Porokeratosis Associated with Internal Malignancies: A Case Report. Cutis 2010, 85, 156–159. [Google Scholar]
- Jung, J.-Y.; Yeon, J.-H.; Ryu, H.-S.; Youn, S.-W.; Park, K.-C.; Huh, C.-H. Disseminated Superficial Porokeratosis Developed by Immunosuppression Due to Rheumatoid Arthritis Treatment. J. Dermatol. 2009, 36, 466–467. [Google Scholar] [CrossRef]
- Stewart, L.; Howat, A.; Coulson, I. Disseminated Superficial Porokeratosis Secondary to Immunosuppression Induced by Etanercept for Extensive Psoriasis. Arch. Dermatol. 2010, 146, 1193–1194. [Google Scholar] [CrossRef]
- Mangas, C.; Espeli, V.; Blum, R. A Case of Eruptive Disseminated Porokeratosis in a Cancer Patient after Trastuzumab and Exemestane Treatment: Cancer Related or Drug Induced Phenomenon? Actas Dermosifiliogr. 2018, 109, 559–560. [Google Scholar] [CrossRef]
- Cinotti, E.; Fiorani, D.; Provvidenziale, L.; Miracco, C.; Calamai, V.; Danielli, R.; Rubegni, P. Eruptive Porokeratosis under Nivolumab Adjuvant Treatment for Melanoma. Int. J. Dermatol. 2019, 58, e138–e140. [Google Scholar] [CrossRef] [PubMed]
- Maredia, H.; Simkin, D.; Soni, A.; Loss, M.J. Eruptive Porokeratosis during Pembrolizumab Treatment of Invasive Cutaneous Squamous Cell Carcinoma. Int. J. Dermatol. 2020, 59, e141–e142. [Google Scholar] [CrossRef] [PubMed]
- Klager, S.; Khalil, M.; Shulman, K.; Sami, N. Abatacept-Induced Disseminated Superficial Porokeratosis. J. Clin. Rheumatol. 2021, 27, S634–S635. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.D.; Mufti, A.; Sachdeva, M.; Rahat, S.; Lansang, R.P.; Yeung, J. Drugs Associated with Development of Porokeratosis: A Systematic Review. Dermatol. Ther. 2021, 34, e14560. [Google Scholar] [CrossRef] [PubMed]
- Kroiss, M.M.; Stolz, W.; Hohenleutner, U.; Landthaler, M. Disseminated Superficial Porokeratosis Induced by Furosemide. Acta Derm. Venereol. 2000, 80, 52–53. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, O.; Duarte, A.F.; Baudrier, T.; Mota, A.; Azevedo, F. Development of Disseminated Superficial Porokeratosis in a Patient with Complicated Acute Pancreatitis. Dermatol. Online J. 2011, 17, 5. [Google Scholar] [CrossRef] [PubMed]
- Kono, T.; Kobayashi, H.; Ishii, M.; Nishiguchi, S.; Taniguchi, S. Synchronous Development of Disseminated Superficial Porokeratosis and Hepatitis C Virus-Related Hepatocellular Carcinoma. J. Am. Acad. Dermatol. 2000, 43, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Mundi, J.P.; Cerullo, L.; Cotliar, J. Porokeratosis in a Patient with Hepatitis of Unclear Etiology. J. Drugs Dermatol. 2010, 9, 258–260. [Google Scholar]
- Park, B.S.; Moon, S.E.; Kim, J.A. Disseminated Superficial Porokeratosis in a Patient with Chronic Liver Disease. J. Dermatol. 1997, 24, 485–487. [Google Scholar] [CrossRef]
- Rao, A.G.; Lakshmi, T.S.S.; Haritha, S. Disseminated Superficial Porokeratosis. Indian J. Dermatol. Venereol. Leprol. 2002, 68, 284–285. [Google Scholar]
- Shimizu, S.; Takashima, Y.; Hotta, M.; Ito, E.; Moriuchi, R. Inflammatory Disseminated Superficial Porokeratosis Successfully Controlled with a Combination of Topical Diclofenac Gel and Systemic Etretinate. J. Eur. Acad. Dermatol. Venereol. 2018, 32, e201–e202. [Google Scholar] [CrossRef]
- Lee, S.Y.; Lee, K.H.; Ishii, N.; Hashimoto, T.; Kim, J.H.; Oh, C.H.; Park, K. Rare Case of Bullous Pemphigoid Occurring on Atrophic Centers of Disseminated Superficial Porokeratosis Lesions. J. Dermatol. 2021, 48, e530–e531. [Google Scholar] [CrossRef]
- Sen, B.B.; Ekiz, Ö.; Rifaioglu, E.N.; Sen, T.; Atik, E.; Dogramaci, A.Ç. Localized Bullous Pemphigoid Occurring on Surgical Scars. Indian J. Dermatol. Venereol. Leprol. 2013, 79, 554. [Google Scholar] [CrossRef]
- Chun, S.H.; Kim, B.Y.; Kim, C.M.; Park, J.B.; Ryu, H.J. A Case of Wolf’s Isotopic Response Presenting as Bullous Pemphigoid. Ann. Dermatol. 2017, 29, 499–500. [Google Scholar] [CrossRef]
- Pietkiewicz, P.; Gornowicz-Porowska, J.; Dmochowska, M.B.; Dmochowski, M. Malignancy in Relation to Autoimmune Blistering Dermatoses: Molecular and Clinical Aspects. In Highlights in Skin Cancer; IntechOpen: London, UK, 2013; ISBN 978-953-51-1073-6. [Google Scholar]
- Murata, Y.; Kumano, K.; Takai, T. Type 2 Segmental Manifestation of Disseminated Superficial Porokeratosis Showing a Systematized Pattern of Involvement and Pronounced Cancer Proneness. Eur. J. Dermatol. 2001, 11, 191–194. [Google Scholar]
- Lee, W.J.; Kim, C.H.; Park, G.H.; Won, C.H.; Chang, S.E.; Lee, M.W.; Choi, J.H.; Moon, K.C. Disseminated Superficial Porokeratosis in a Patient with Esophageal Cancer. J. Dermatol. 2010, 37, 747–748. [Google Scholar] [CrossRef]
- Choi, K.H.; Kim, T.Y. A Case of Inflammatory Disseminated Superficial Porokeratosis in a Colon Cancer Patient. Ann. Dermatol. 2009, 21, 150–153. [Google Scholar] [CrossRef]
- Jiang, L.-Y.; Guo, Z.; Kong, Y.-L.; Luan, H.; Yu, J.-X.; Wang, K.-Y. Rectal Cancer Concurrent with Disseminated Superficial Porokeratosis in Three Brothers. J. Dermatol. 2015, 42, 756–757. [Google Scholar] [CrossRef]
- Lee, H.-W.; Oh, S.-H.; Choi, J.-C.; Chang, S.-E.; Lee, M.-W.; Choi, J.-H.; Moon, K.-C.; Koh, J.-K. Disseminated Superficial Porokeratosis in a Patient with Cholangiocarcinoma. J. Am. Acad. Dermatol. 2006, 54, S56–S58. [Google Scholar] [CrossRef]
- Torres, T.; Velho, G.C.; Selores, M. Disseminated Superficial Porokeratosis in a Patient with Cholangiocarcinoma: A Paraneoplastic Manifestation? An. Bras. Dermatol. 2010, 85, 229–231. [Google Scholar] [CrossRef]
- Cannavó, S.P.; Borgia, F.; Adamo, B.; Guarneri, B. Simultaneous Development and Parallel Course of Disseminated Superficial Porokeratosis and Ovarian Cancer: Coincidental Association or True Paraneoplastic Syndrome? J. Am. Acad. Dermatol. 2008, 58, 657–660. [Google Scholar] [CrossRef]
- Hui, H.-Z.; Wang, Y.-J.; Cheng, J.-R.; Mao, H.; Guo, H.-X.; Shi, B.-J. A Novel Missense Mutation in the MVK Gene Is Associated with Disseminated Superficial Porokeratosis. Int. J. Dermatol. 2023, 62, e223–e225. [Google Scholar] [CrossRef]
- Wei, S.; Yang, S.; Lin, D.; Li, M.; Zhang, X.; Bu, L.; Zheng, G.; Hu, L.; Kong, X.; Zhang, X. A Novel Locus for Disseminated Superficial Porokeratosis Maps to Chromosome 18p11.3. J. Investig. Dermatol. 2004, 123, 872–875. [Google Scholar] [CrossRef]
- Cao, H.M.; Wang, Z.Y.; Zhang, G.W.; Liu, C.F.; Pan, C.M.; Zhao, S.X.; Song, Z.Y.; Song, H.D.; Zhang, L. Identification of a Locus (DSP2) for Disseminated Superficial Porokeratosis at Chromosome 12q21.2-24.21. Clin. Exp. Dermatol. 2012, 37, 672–676. [Google Scholar] [CrossRef]
- Awatani, K.; Hashimoto, T.; Satoh, T. Eruptive Pruritic Papular Porokeratosis Accompanied by Eosinophilic and Basophilic Infiltrate with Upregulation of Epidermal CCL26/Eotaxin-3 and Thymic Stromal Lymphopoietin. J. Dermatol. 2021, 48, e382–e383. [Google Scholar] [CrossRef]
- Gao, Z.; Sun, Y. Dermoscopy Assisting the Diagnosis of Eruptive Disseminated Porokeratosis. J. Dermatol. 2021, 48, e460–e461. [Google Scholar] [CrossRef]
- Tokat, F.; Sezer, E.; Erdemoglu, Y.; Cetin, E.D.; Durmaz, E.O. Pruriginous Follicular Porokeratosis. J. Dtsch. Dermatol. Ges. 2017, 15, 845–847. [Google Scholar] [CrossRef]
- Pietkiewicz, P.; Navarrete-Dechent, C.; Salwowska, N.; Cantisani, C.; Goldust, M.; Errichetti, E. Ultraviolet-Induced Fluorescence Dermatoscopy Reveals Fluorescent Clues in Pitted Keratolysis. Dermatol. Pract. Concept. 2023, 13, e2023242. [Google Scholar] [CrossRef]
- Pietkiewicz, P.; Navarrete-Dechent, C.; Mayisoğlu, H.; Jolly, G.; Kutlu, Ö.; Errichetti, E. Pink-Red Fluorescence Observed in Ultraviolet-Induced Fluorescence Dermoscopy of Psoriatic Plaques. Dermatol. Pract. Concept. 2023, 13, e2023243. [Google Scholar] [CrossRef]
- Pietkiewicz, P.; Navarrete-Dechent, C.; Goldust, M.; Korecka, K.; Todorovska, V.; Errichetti, E. Differentiating Fordyce Spots from Their Common Simulators Using Ultraviolet-Induced Fluorescence Dermatoscopy—Retrospective Study. Diagnostics 2023, 13, 985. [Google Scholar] [CrossRef]
- Al-Nasiri, M.; Navarrete-Dechent, C.; Korecka, K.; Salwowska, N.; Goldust, M.; Pietkiewicz, P. Ultraviolet-Induced Fluorescence Dermatoscopy of Trichobacteriosis Axillaris Reveals Peripilar Yellow-Green Luminescent Concretions. Dermatol. Pract. Concept. 2023, 13, e2023169. [Google Scholar] [CrossRef]
- Togawa, Y.; Yamamoto, Y.; Matsue, H. Clinical study on the comparison of dermoscopic images using two wavelengths of near-ultraviolet-visible light. JEADV Clin. Pract. 2023, 1–5. [Google Scholar] [CrossRef]
- Ramakrishnan, R.; Vignesh, T.A.; Durai, P.C.T.; Narasimhan, M. A Rare Case of Disseminated Superficial Porokeratosis-Case Report. J. Fam. Med. Prim. Care 2022, 11, 1195–1197. [Google Scholar] [CrossRef]
- Das, A.; Vasudevan, B.; Talwar, A. Porokeratosis: An Enigma Beginning to Unravel. Indian J. Dermatol. Venereol. Leprol. 2022, 88, 291–299. [Google Scholar] [CrossRef]
- Jurecka, W.; Neumann, R.A.; Knobler, R.M. Porokeratoses: Immunohistochemical, Light and Electron Microscopic Evaluation. J. Am. Acad. Dermatol. 1991, 24, 96–101. [Google Scholar] [CrossRef]
- Akino, S.; Okano, T.; Takeuchi, S.; Ariizumi, Y.; Kadono, T.; Miyagaki, T. Complete Pruritus Relief by Oren-Gedoku-to in Eruptive Pruritic Papular Porokeratosis. J. Dermatol. 2021, 48, e378–e379. [Google Scholar] [CrossRef]
- Tanaka, M.; Terui, T.; Kudo, K.; Tagami, H. Inflammatory Disseminated Superficial Porokeratosis Followed by Regression. Br. J. Dermatol. 1995, 132, 153–155. [Google Scholar] [CrossRef]
- Ito, M.; Fujiwara, H.; Maruyama, T.; Oguro, K.; Ishihara, O.; Sato, Y. Morphogenesis of the Cornoid Lamella: Histochemical, Immunohistochemical, and Ultrastructural Study of Porokeratosis. J. Cutan. Pathol. 1991, 18, 247–256. [Google Scholar] [CrossRef]
- Matsuta, M.; Kon, S.; Sasaki, K.; Matsuta, M. Immunohistochemical Detection of p21WAF1/CIP1 and P53 Proteins in Formalin-Fixed Paraffin-Embedded Tissue Sections of Squamous Cell Carcinoma of the Skin. J. Dermatol. Sci. 1997, 14, 233–239. [Google Scholar] [CrossRef]
- Manganoni, A.M.; Facchetti, F.; Gavazzoni, R. Involvement of Epidermal Langerhans Cells in Porokeratosis of Immunosuppressed Renal Transplant Recipients. J. Am. Acad. Dermatol. 1989, 21, 799–801. [Google Scholar] [CrossRef]
- Stefanato, C.M.; Youssef, E.A.; Cerio, R.; Kobza-Black, A.; Greaves, M.W. Atypical Nekam’s Disease—Keratosis Lichenoides Chronica Associated with Porokeratotic Histology and Amyloidosis. Clin. Exp. Dermatol. 1993, 18, 274–276. [Google Scholar] [CrossRef] [PubMed]
- Piamphongsant, T.; Sittapairoachana, D. Localized Cutaneous Amyloidosis in Disseminated Superficial Actinic Porokeratosis. J. Cutan. Pathol. 1974, 1, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.P.; Balme, B.; Gho, A.; Perrot, H. Porokératose Disséminée Superficielle Avec Amylose Dermique. Ann. Dermatol. Venereol. 1992, 119, 651–654. [Google Scholar] [PubMed]
- Yasuda, K.; Ikeda, M.; Ikeda, M.; Kodama, H. Disseminated Superficial Porokeratosis with Amyloid Deposition. J. Dermatol. 1996, 23, 111–115. [Google Scholar] [CrossRef]
- Amantea, A.; Giuliano, M.C.; Balus, L. Disseminated Superficial Porokeratosis with Dermal Amyloid Deposits: Case Report and Immunohistochemical Study of Amyloid. Am. J. Dermatopathol. 1998, 20, 86–88. [Google Scholar] [CrossRef]
- Demitsu, T.; Okada, O. Disseminated Superficial Porokeratosis with Dermal Amyloid Deposition. J. Dermatol. 1999, 26, 405–406. [Google Scholar] [CrossRef]
- Kim, J.H.; Yim, H.; Kang, W.H. Secondary Cutaneous Amyloidosis in Disseminated Superficial Porokeratosis: A Case Report. J. Korean Med. Sci. 2000, 15, 478–481. [Google Scholar] [CrossRef]
- Garcia-F-Villalta, M.J.; Daudén, E.; Ruiz-Genao, D.; Fraga, J.; García-Díez, A. Dermal Amyloid Deposits in Disseminated Superficial Porokeratosis. Acta Derm. Venereol. 2004, 84, 173–174. [Google Scholar] [CrossRef]
- Ginarte, M.; León, A.; Toribio, J. Disseminated Superficial Porokeratosis with Amyloid Deposits. Eur. J. Dermatol. 2005, 15, 298–300. [Google Scholar]
- Carlesimo, M.; Rossi, A.; Fidanza, L.; Narcisi, A.; La Pietra, M.; Mari, E.; Cacchi, C.; Camplone, G. Disseminated Superficial Porokeratosis with Dermal Amyloid Deposits. Case Rep. Dermatol. 2009, 1, 35–38. [Google Scholar] [CrossRef]
- Inazawa, M.; Satoh, T.; Yokozeki, H. Hyperkeratotic Variant of Inflammatory Disseminated Superficial Porokeratosis with Lichenoid Reaction and Extensive Amyloid Deposition. Int. J. Dermatol. 2014, 53, e94–e95. [Google Scholar] [CrossRef] [PubMed]
- Husein, H.-E.; Inmaculada, R.-M.; Vicente, C.-A.; Eduardo, S.-G. Disseminated Superficial Porokeratosis with Dermal Amyloid Deposits in an Elderly Man: A Rare Entity. J. Dtsch. Dermatol. Ges. 2015, 13, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.T. Treatment of Lichen Amyloidosis (LA) and Disseminated Superficial Porokeratosis (DSP) with Frequency-Doubled Q-Switched Nd:YAG Laser. Dermatol. Surg. 2000, 26, 958–962. [Google Scholar] [CrossRef]
- Tee, S.-I.; Chong, W.-S. Eruptive Pruritic Papular Porokeratosis. Indian J. Dermatol. Venereol. Leprol. 2012, 78, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Klein, N.; Enk, A.; Hartschuh, W. Inflammatory stage of disseminated superficial porokeratosis in a 71-year old patient. Hautarzt 2009, 60, 420–423. [Google Scholar] [CrossRef]
- Mu, X.; Li, W.; Zhang, M.; Yang, C.; Yang, X.; Li, D.; Ding, Y. Successful Treatment of Eruptive Pruritic Papular Porokeratosis in the Elderly with Tofacitinib: A Case Report. Clin. Cosmet. Investig. Dermatol. 2023, 16, 1741–1747. [Google Scholar] [CrossRef]
- Jourdan, M.; Bachmeyer, C.; Duriez, P.; Francès, C. Disseminated superficial porokeratosis in a black woman. Ann. Dermatol. Venereol. 2014, 141, 52–54. [Google Scholar] [CrossRef]
- Pruitt, L.G.; Hsia, L.-L.B.; Burke, W.A. Disseminated Superficial Porokeratosis Involving the Groin and Genitalia in a 72-Year-Old Immunocompetent Man. JAAD Case Rep. 2015, 1, 277–279. [Google Scholar] [CrossRef]
- Cha, S.H.; Park, H.J.; Lee, J.Y.; Cho, B.K. Atypical Porokeratosis Developing Following Bone Marrow Transplantation in a Patient with Myelodysplastic Syndrome. Ann. Dermatol. 2010, 22, 206–208. [Google Scholar] [CrossRef]
- Shelley, W.B.; Shelley, E.D. Disseminated Superficial Porokeratosis: Rapid Therapeutic Response to 5-Fluorouracil. Cutis 1983, 32, 139–140. [Google Scholar]
- Elisia, I.; Nakamura, H.; Lam, V.; Hofs, E.; Cederberg, R.; Cait, J.; Hughes, M.R.; Lee, L.; Jia, W.; Adomat, H.H.; et al. DMSO Represses Inflammatory Cytokine Production from Human Blood Cells and Reduces Autoimmune Arthritis. PLoS ONE 2016, 11, e0152538. [Google Scholar] [CrossRef]
- Saki, N.; Ahramiyanpour, N.; Heiran, A.; Alipour, S.; Parvizi, M.M. Efficacy of Topical Dimethyl Sulfoxide (DMSO) 50% Solution vs Tretinoin 0.5% Cream in Treatment of Patients with Primary Macular Amyloidosis: A Split-Side Single-Blinded Randomized Clinical Trial. Dermatol. Ther. 2020, 33, e13305. [Google Scholar] [CrossRef]
- Padda, I.S.; Bhatt, R.; Parmar, M. Tofacitinib. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Chen, Y.; Xian, Y.-F.; Loo, S.; Lai, Z.; Chan, W.Y.; Liu, L.; Lin, Z.-X. Huang-Lian-Jie-Du Extract Ameliorates Atopic Dermatitis-like Skin Lesions Induced by 2,4-Dinitrobenzene in Mice via Suppression of MAPKs and NF-κB Pathways. J. Ethnopharmacol. 2020, 249, 112367. [Google Scholar] [CrossRef]
- Takagi, R.; Kawano, M.; Nakagome, K.; Hashimoto, K.; Higashi, T.; Ohbuchi, K.; Kaneko, A.; Matsushita, S. Wogonin Attenuates Ovalbumin Antigen-Induced Neutrophilic Airway Inflammation by Inhibiting Th17 Differentiation. Int. J. Inflam. 2014, 2014, 571508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, H.; Shao, S.; Shen, Y.; Xiao, F.; Sun, J.; Piao, S.; Zhao, D.; Li, G.; Yan, M. Compound Traditional Chinese Medicine Dermatitis Ointment Ameliorates Inflammatory Responses and Dysregulation of Itch-Related Molecules in Atopic Dermatitis. Chin. Med. 2022, 17, 3. [Google Scholar] [CrossRef]
- Schamroth, J.M.; Zlotogorski, A.; Gilead, L. Porokeratosis of Mibelli. Overview and Review of the Literature. Acta Derm. Venereol. 1997, 77, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.R.; Santos, L.D.N.; Tagliarini, F.A.N.M.; Lira, M.L. de A. Porokeratosis of Mibelli—Literature Review and a Case Report. An. Bras. Dermatol. 2013, 88, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Nabai, H.; Mehregan, A.H. Porokeratosis of Mibelli. A Report of Two Unusual Cases. Dermatologica 1979, 159, 325–331. [Google Scholar] [CrossRef]
- Schramm, P.; Bork, K. Naeviform porokeratosis—Only a morphologic variant of porokeratosis of Mibelli (author’s transl). Z. Hautkr 1982, 57, 963–970. [Google Scholar]
- Guillot, P.; Taieb, A.; Fontan, I.; Bilhou-Nabera, C.; Viard, E.; Renaud, P.; Maleville, J. Linear porokeratosis of Mibelli in monozygotic twin girls. Ann. Dermatol. Venereol. 1991, 118, 519–524. [Google Scholar]
- Götz, A.; Kopera, D.; Wach, F.; Hohenleutner, U.; Landthaler, M. Porokeratosis Mibelli gigantea: Case report and literature review. Hautarzt 1999, 50, 435–438. [Google Scholar] [CrossRef] [PubMed]
- Schaller, M.; Korting, H.C.; Kollmann, M.; Kind, P. The Hyperkeratotic Variant of Porokeratosis Mibelli Is a Distinct Entity: Clinical and Ultrastructural Evidence. Dermatology 1996, 192, 255–258. [Google Scholar] [CrossRef]
- Wagner, G.; Meyer, V.; Sachse, M.M. Verrucous variant of porokeratosis of Mibelli as a differential diagnosis of psoriasis vulgaris. Hautarzt 2016, 67, 244–248. [Google Scholar] [CrossRef]
- de Wet, J.; Swart, M.; Jordaan, H.F.; Schneider, J.W.; Mulder, S.; Visser, W.I. An Unusual Case of Generalized Hyperkeratotic and Verrucous Porokeratosis. JAAD Case Rep. 2020, 6, 925–930. [Google Scholar] [CrossRef]
- Yu, H.-J.; Park, K.-T.; Oh, D.-H.; Kim, J.-S.; Park, Y.-W. A Case of the Hyperkeratotic Variant of Porokeratosis Mibelli. J. Dermatol. 2006, 33, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Uenishi, T.; Teramura, K.; Kitamura, M.; Fujii, N.; Nakanishi, G.; Tanaka, T.; Uehara, M. Hyperkeratotic Variant of Porokeratosis Mibelli with Dermal Amyloid Deposits. J. Dermatol. 2010, 37, 475–479. [Google Scholar] [CrossRef]
- Kuno, Y.; Sato, K.; Tsuji, T. Porokeratosis of Mibelli Associated with Dermal Amyloid Deposits. Br. J. Dermatol. 1999, 141, 949–950. [Google Scholar] [CrossRef] [PubMed]
- Ghorpade, A. Localized Actinic Nasal Porokeratosis. Clin. Exp. Dermatol. 2010, 35, 211–212. [Google Scholar] [CrossRef]
- Perlis, C.; Robinson-Bostom, L.; Telang, G.H.; DiGiovanna, J. A Thick Lichenified Plaque on the Ventral Penile Shaft. Penile Porokeratosis of Mibelli. Arch. Dermatol. 2006, 142, 1221–1226. [Google Scholar] [CrossRef]
- Neri, I.; Marzaduri, S.; Passarini, B.; Patrizi, A. Genital Porokeratosis of Mibelli. Genitourin. Med. 1995, 71, 410–411. [Google Scholar] [CrossRef]
- Miranda, S.M.B.; De Miranda, J.N.R.; De Souza Filho, J.B. Facial Porokeratosis Characterized by Destructive Lesions. Int. J. Dermatol. 2004, 43, 913–914. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Bel, P.; Sanmartín-Jimenez, O.; Sorni-Bröker, G.; Guillén-Barona, C. Labial Porokeratosis. Am. J. Dermatopathol. 2010, 32, 638. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, G.; Ganguly, S.; Khare, S. Atrophic Lingual Plaque in Father-Son Duo. Indian Dermatol. Online J. 2022, 13, 672. [Google Scholar] [CrossRef] [PubMed]
- Yong, A.S.W.; Singh, M.; Goulding, J.M.R.; Swale, V.J. Follicular Porokeratosis of Mibelli on the Buttocks. Clin. Exp. Dermatol. 2009, 34, 43–45. [Google Scholar] [CrossRef] [PubMed]
- Robati, R.M.; Rahmati-Roodsari, M.; Ayatollahi, A.; Hejazi, S. Facial and Bilateral Acral Porokeratosis with Nail Dystrophy: A Case Report. Dermatol. Online J. 2011, 17, 5. [Google Scholar] [CrossRef]
- Nenoff, P.; Arnold, T.; Nenning, H.; Hindermann, W. Centrifugal plaques with central atrophy and peripheral scale on the scalp. Hautarzt 2011, 62, 544–547. [Google Scholar] [CrossRef]
- Buzina, D.S.; Rajič, S.; Radoš, J.; Marinović, B.; Lipozenčić, J. Focal Porokeratosis of Nuchae: Case Report. Acta Dermatovenerol. Croat. 2010, 18, 257–260. [Google Scholar]
- Odeyinde, S.; Belcher, H. Isolated Single Digit Porokeratosis of Mibelli: An Unusual Case. Dermatol. Online J. 2012, 18, 13. [Google Scholar] [CrossRef]
- Pawar, M. Onychodystrophy Due to Porokeratosis of Mibelli: A Rare Association. Acta Dermatovenerol. Alp. Pannonica Adriat. 2017, 26, 51–52. [Google Scholar] [CrossRef]
- Yendo, T.M.; Gabbi, T.V.B.; Nico, M.M.S. Porokeratosis of the Nail Unit: Case Series and Review. Skin. Appendage Disord. 2021, 7, 489–492. [Google Scholar] [CrossRef]
- Karthikeyan, K.; Thappa, D.M.; Udayashankar, C. Porokeratosis of Mibelli with Nail Dystrophy. J. Dermatol. 2003, 30, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Roh, M.R.; Lee, J.H.; Lee, K.H. Pterygium Unguis Formation in Porokeratosis of Mibelli. Br. J. Dermatol. 2007, 156, 1384–1385. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, G.; Devan, P.; Keerthi, S.; Karthikeyan, K. Acral Porokeratosis Associated with Anonychia. Indian J. Dermatol. Venereol. Leprol. 2018, 84, 81–82. [Google Scholar] [CrossRef]
- Handjani, F.; Shahbaz, S.; Aslani, F.S.; Gheisari, F.; Mozaffarian, K.; Kasraee, B. Porokeratosis of Mibelli with Mutilation: A Case Report. Cutis 2010, 86, 77–80. [Google Scholar] [PubMed]
- Alexis, A.F.; Busam, K.; Myskowski, P.L. Porokeratosis of Mibelli Following Bone Marrow Transplantation. Int. J. Dermatol. 2006, 45, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Mizukawa, Y.; Shiohara, T. Onset of Porokeratosis of Mibelli in Organ Transplant Recipients: Lack of a Search for Transmissible Agents in These Patients. J. Am. Acad. Dermatol. 2001, 44, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.P.; Smoller, B.R. Porokeratosis in Immunosuppressed and Nonimmunosuppressed Patients. Int. J. Dermatol. 1992, 31, 781–782. [Google Scholar] [CrossRef]
- Han, Y.W.; Kim, Y.J.; Kim, H.O.; Park, Y.M. Clinical Study of Porokeratosis Associated with Immunosuppressive Therapy in Renal Transplant Recipients. Ann. Dermatol. 2008, 20, 167–171. [Google Scholar] [CrossRef]
- Yazkan, F.; Turk, B.G.; Dereli, T.; Kazandi, A.C. Porokeratosis of Mibelli Induced by Topical Corticosteroid. J. Cutan. Pathol. 2006, 33, 516–518. [Google Scholar] [CrossRef]
- Protopsaltis, J.; Katsantonis, J.C.; Kokkoris, S.; Agapitos, E.; Lavranos, G.; Korantzopoulos, P.; Giannoulis, G. Isolated Primary Cardiac Amyloidosis Associated with Porokeratosis of Mibelli. Int. J. Cardiol. 2008, 126, e22–e24. [Google Scholar] [CrossRef]
- Yilmaz, M.; Erdoğan, B.; Özaslan, M.; Varnali, E.; Kavak, A.; Sakiz, D. Coexistence of Multiple Porokeratoma and Porokeratosis of Mibelli. Ann. Dermatol. Venereol. 2022, 149, 214–215. [Google Scholar] [CrossRef]
- Dippel, E.; Haas, N.; Czarnetzki, B.M. Porokeratosis of Mibelli Associated with Active Chronic Hepatitis and Vitiligo. Acta Derm. Venereol. 1994, 74, 463–464. [Google Scholar] [CrossRef]
- Ma, Y.; Li, C.; Wu, J.; Cui, P.; Lin, L.; Feng, S. Coexistence of Porokeratosis Ptychotropica with Porokeratosis of Mibelli in a Chinese Man. Postepy Dermatol. Alergol. 2015, 32, 307–309. [Google Scholar] [CrossRef]
- Eng, A.M.; Kolton, B. Generalized Eruptive Porokeratosis of Mibelli with Associated Psoriasis. J. Cutan. Pathol. 1975, 2, 203–213. [Google Scholar] [CrossRef]
- Beer, W.E.; Smith, N.P. Hyperkeratotic Porokeratosis (Mibelli) with Psoriasis—Response to an Aromatic Retinoid. Clin. Exp. Dermatol. 1984, 9, 509–513. [Google Scholar] [CrossRef]
- De Simone, C.; Paradisi, A.; Massi, G.; Proietti, I.; Capponi, A.; Amerio, P.L.; Capizzi, R. Giant Verrucous Porokeratosis of Mibelli Mimicking Psoriasis in a Patient with Psoriasis. J. Am. Acad. Dermatol. 2007, 57, 665–668. [Google Scholar] [CrossRef]
- Ehsani, A.H.; Shakoei, S.; Ranjbar, M. Giant Porokeratosis of Mibelli with Squamous Cell Carcinoma. Indian J. Dermatol. Venereol. Leprol. 2014, 80, 96. [Google Scholar] [CrossRef]
- Zhang, F.; Bai, W.; Sun, S.; Li, N.; Zhang, X. Squamous Cell Carcinoma Arising from Giant Porokeratosis and Rare Postoperative Recurrence and Metastasis: A Case Report. Medicine 2020, 99, e18697. [Google Scholar] [CrossRef]
- Hanumanthayya, K.; Magavi, S.; Tophakhane, R.; Rathod, R. Coexistence of Disseminated Superficial and Giant Porokeratosis of Mibelli with Squamous Cell Carcinoma. Indian J. Dermatol. Venereol. Leprol. 2003, 69, 296–297. [Google Scholar]
- Bhunia, D.; Ghosh, S.; Rudra, O.; Biswas, S.K.; Agarwal, M.; Ghosh, A. Keratoacanthoma Arising over Margin of Porokeratosis of Mibelli: A New Association? Indian J. Dermatol. 2016, 61, 107–109. [Google Scholar] [CrossRef]
- Saritha, M.; Kumari, R.; Thappa, D.M.; Rajesh, N.G.; Verma, S.K. Benign Giant Cutaneous Horn Formed by Giant Porokeratosis of Mibelli with Dysplasia. IJDVL 2013, 79, 433. [Google Scholar] [CrossRef]
- Breneman, D.L.; Breneman, J.C. Cutaneous T-Cell Lymphoma Mimicking Porokeratosis of Mibelli. J. Am. Acad. Dermatol. 1993, 29, 1046–1048. [Google Scholar] [CrossRef]
- Elfatoiki, F.Z.; Soussi, W.; Chiheb, S.; Jabri, L.; Benchikhi, H. Cutaneous Sarcoidosis Simulating Porokeratosis of Mibelli. Pan Afr. Med. J. 2015, 20, 195. [Google Scholar] [PubMed]
- Zeng, K.; Zhang, Q.-G.; Li, L.; Duan, Y.; Liang, Y.-H. Splicing Mutation in MVK Is a Cause of Porokeratosis of Mibelli. Arch. Dermatol. Res. 2014, 306, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Song, N.J.; Luan, J.; Zhang, Z.H. Updating and Identifying a Novel Mutation in the PMVK Gene in Classic Porokeratosis of Mibelli. Clin. Exp. Dermatol. 2017, 42, 910–911. [Google Scholar] [CrossRef] [PubMed]
- Occella, C.; Bleidl, D.; Nozza, P.; Mascelli, S.; Raso, A.; Gimelli, G.; Gimelli, S.; Tassano, E. Identification of an Interstitial 18p11.32-P11.31 Duplication Including the EMILIN2 Gene in a Family with Porokeratosis of Mibelli. PLoS ONE 2013, 8, e61311. [Google Scholar] [CrossRef] [PubMed]
- Hivnor, C.; Williams, N.; Singh, F.; VanVoorhees, A.; Dzubow, L.; Baldwin, D.; Seykora, J. Gene Expression Profiling of Porokeratosis Demonstrates Similarities with Psoriasis. J. Cutan. Pathol. 2004, 31, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Sonthalia, S.; Lallas, A. Dermoscopy of Porokeratosis of Mibelli. Indian Dermatol. Online J. 2017, 8, 304–305. [Google Scholar] [CrossRef]
- Pizzichetta, M.A.; Canzonieri, V.; Massone, C.; Soyer, H.P. Clinical and Dermoscopic Features of Porokeratosis of Mibelli. Arch. Dermatol. 2009, 145, 91–92. [Google Scholar] [CrossRef]
- Uhara, H.; Kamijo, F.; Okuyama, R.; Saida, T. Open Pores with Plugs in Porokeratosis Clearly Visualized with the Dermoscopic Furrow Ink Test: Report of 3 Cases. Arch. Dermatol. 2011, 147, 866–868. [Google Scholar] [CrossRef]
- Ahlgrimm-Siess, V.; Koller, S.; El Shabrawi-Caelen, L.; Hofmann-Wellenhof, R.; Kerl, H. New Diagnostic Methods in Dermatopathology: In Vivo Reflectance Confocal Microscopy. J. Dtsch. Dermatol. Ges. 2008, 6, 591–592. [Google Scholar] [CrossRef]
- Marghescu, S.; Anton-Lamprecht, I.; Melz-Rothfuss, B. Disseminated Bilateral Hyperkeratotic Variant of Porokeratosis Mibelli. Arch. Dermatol. Res. 1987, 279, S38–S47. [Google Scholar] [CrossRef]
- Sato, A.; Masu, S.; Seiji, M. Electron Microscopic Studies of Porokeratosis Mibelli—Civatte Bodies and Amyloid Deposits in the Dermis. J. Dermatol. 1980, 7, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Böhm, W.; Bersch, A. Hyperkeratotic Form of Porokeratosis Mibelli. Dermatologica 1977, 155, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Giuliodori, K.; Campanati, A.; Ganzetti, G.; Conocchiari, L.; Cataldi, I.; Simonetti, O.; Giangiacomi, M.; Offidani, A. The Successful Off-Label Use of Photodynamic Therapy for Classic Porokeratosis of Mibelli: Case Report. Dermatol. Ther. 2011, 24, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez Paredes, E.; Bella Navarro, R.; Montesinos Villaescusa, E.; Jordá Cuevas, E. Porokeratosis of Mibelli: A New Indication for Photodynamic Therapy? Actas Dermosifiliogr. 2013, 104, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Levitt, J.; Emer, J.J.; Emanuel, P.O. Treatment of Porokeratosis of Mibelli with Combined Use of Photodynamic Therapy and Fluorouracil Cream. Arch. Dermatol. 2010, 146, 371–373. [Google Scholar] [CrossRef]
- Chowdhury, M.M.; Inaloz, H.S.; Holt, P.J. A Scaly Macule on the Bridge of the Nose of a 15-Year-Old Boy. Pediatr. Dermatol. 2000, 17, 149–150. [Google Scholar] [CrossRef]
- Dereli, T.; Ozyurt, S.; Ozturk, G. Porokeratosis of Mibelli: Successful Treatment with Cryosurgery. J. Dermatol. 2004, 31, 223–227. [Google Scholar] [CrossRef]
- Levell, N.J.; Bewley, A.P.; Levene, G.M. Porokeratosis of Mibelli on the Penis, Scrotum and Natal Cleft. Clin. Exp. Dermatol. 1994, 19, 77–78. [Google Scholar] [CrossRef]
- Harrison, S.; Sinclair, R. Porokeratosis of Mibelli: Successful Treatment with Topical 5% Imiquimod Cream. Australas. J. Dermatol. 2003, 44, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Shahmoradi, Z.; Sadeghiyan, H.; Pourazizi, M.; Saber, M.; Abtahi-Naeini, B. Recalcitrant Digital Porokeratosis of Mibelli: A Successful Surgical Treatment. N. Am. J. Med. Sci. 2015, 7, 295–296. [Google Scholar] [CrossRef] [PubMed]
- McCullough, T.L.; Lesher, J.L. Porokeratosis of Mibelli: Rapid Recurrence of a Large Lesion after Carbon Dioxide Laser Treatment. Pediatr. Dermatol. 1994, 11, 267–270. [Google Scholar] [CrossRef]
- Rabbin, P.E.; Baldwin, H.E. Treatment of Porokeratosis of Mibelli with CO2 Laser Vaporization versus Surgical Excision with Split-Thickness Skin Graft. A Comparison. J. Dermatol. Surg. Oncol. 1993, 19, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Ottoni, L.d.Q.; Kakizaki, P.; Pinheiro, R.R.; Sittart, J.A.d.S.; Valente, N.Y.S. Porokeratosis of Mibelli in an HIV-Positive Patient. An. Bras. Dermatol. 2016, 91, 131–133. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Nie, X.; Yan, L.; Liu, Y.; Zhou, J.; Tong, W.; Tao, J. Treatment of Classic Porokeratosis of Mibelli with Q-Switched Ruby Laser. Eur. J. Dermatol. 2012, 22, 125–126. [Google Scholar] [CrossRef]
- Spencer, J.M.; Katz, B.E. Successful Treatment of Porokeratosis of Mibelli with Diamond Fraise Dermabrasion. Arch. Dermatol. 1992, 128, 1187–1188. [Google Scholar] [CrossRef]
- Venkatarajan, S.; LeLeux, T.M.; Yang, D.; Rosen, T.; Orengo, I. Porokeratosis of Mibelli: Successful Treatment with 5 Percent Topical Imiquimod and Topical 5 Percent 5-Fluorouracil. Dermatol. Online J. 2010, 16, 10. [Google Scholar] [CrossRef]
- McDonald, S.G.; Peterka, E.S. Porokeratosis (Mibelli): Treatment with Topical 5-Fluorouracil. J. Am. Acad. Dermatol. 1983, 8, 107–110. [Google Scholar] [CrossRef]
- Zasada, M.; Budzisz, E. Retinoids: Active Molecules Influencing Skin Structure Formation in Cosmetic and Dermatological Treatments. Postepy Dermatol. Alergol. 2019, 36, 392–397. [Google Scholar] [CrossRef]
- Ratka, P.; Słoboda, T.; Szubstarski, F.; Dudzik, W. Favorable outcome of the treatment of Mibelli’s porokeratosis linearis using occlusive dressings with retinoic acid. Przegl Dermatol. 1988, 75, 149–153. [Google Scholar]
- Laureano, A.; Macias, V.C.; Pacheco, A. Poroqueratose de Mibelli—Um Caso Clínico. Revista SPDV 2012, 70, 209–212. [Google Scholar] [CrossRef]
- Torne Escasany, R.; Villodres Ramos, E.; Umbert Millet, P.; Guix, M. Generalized Mibelli’s porokeratosis in transformation to spinocellular carcinoma. Clinical, histologic and ultrastructural study. Favorable response to RO-109359. Med. Cutan. Ibero Lat. Am. 1983, 11, 323–328. [Google Scholar] [PubMed]
- Bundino, S.; Zina, A.M. Disseminated Porokeratosis Mibelli Treated with RO 10-9359. A Study of Two Cases with Ultrastructural Remarks. Dermatologica 1980, 160, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Hacham-Zadeh, S.; Holubar, K. Etretinate in the Treatment of Disseminated Porokeratosis of Mibelli. Int. J. Dermatol. 1985, 24, 258–260. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.P.; Voorhees, J.J. Etretinate Improves Localized Porokeratosis of Mibelli. Int. J. Dermatol. 1985, 24, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Verma, K.K.; Singh, O.P. Dexamethasone Pulse Treatment in Disseminated Porokeratosis of Mibelli. J. Dermatol. Sci. 1994, 7, 71–72. [Google Scholar] [CrossRef]
- Gracia-Cazaña, T.; Vera-Álvarez, J.; García-Patos, V.; Gilaberte, Y. Imiquimod and Photodynamic Therapy Are Useful in the Treatment of Porokeratosis in Children with Bone Marrow Transplantation. Pediatr. Dermatol. 2015, 32, e291–e293. [Google Scholar] [CrossRef]
- Montes-De-Oca-Sánchez, G.; Tirado-Sánchez, A.; García-Ramírez, V. Porokeratosis of Mibelli of the Axillae: Treatment with Topical Imiquimod. J. Dermatolog Treat. 2006, 17, 319–320. [Google Scholar] [CrossRef]
- Erbagci, Z.; Tuncel, A.A.; Erbagci, I. Successful Treatment of Porokeratosis with Topical Imiquimod in 2 Immunosuppressed Cases. J. Drugs Dermatol. 2006, 5, 668–671. [Google Scholar]
- Jain, S. Successful Treatment of Porokeratosis of Mibelli with Imiquimod 5% Cream. Clin. Exp. Dermatol. 2006, 31, 302–303. [Google Scholar] [CrossRef]
- Esser, A.C.; Pittelkow, M.R.; Randle, H.W. Human Papillomavirus Isolated from Transplant-Associated Porokeratoses of Mibelli Responsive to Topical 5% Imiquimod Cream. Dermatol. Surg. 2006, 32, 858–861. [Google Scholar] [CrossRef]
- Gajic, B.; Tang, K.; Whitfeld, M. Porokeratosis of Mibelli: Involution and Resolution with 5% Imiquimod Cream. Australas. J. Dermatol. 2011, 52, 301–303. [Google Scholar] [CrossRef]
- Ataseven, A.; Öztürk, P.; Dilek, N.; Küçükosmanoğlu, I. Localized Actinic Nasal Porokeratosis: A Case Report. Acta Dermatovenerol. Alp. Pannonica Adriat. 2012, 21, 79–80. [Google Scholar]
- Kindem, S.; Serra-Guillén, C.; Sorní, G.; Guillén, C.; Sanmartín, O. Treatment of Porokeratosis of Mibelli with Ingenol Mebutate: A Possible New Therapeutic Option. JAMA Dermatol. 2015, 151, 85–86. [Google Scholar] [CrossRef]
- Goldner, R. Zosteriform Porokeratosis of Mibelli. Arch. Dermatol. 1971, 104, 425–426. [Google Scholar] [CrossRef]
- Rahbari, H.; Cordero, A.A.; Mehregan, A.H. Linear Porokeratosis. A Distinctive Clinical Variant of Porokeratosis of Mibelli. Arch. Dermatol. 1974, 109, 526–528. [Google Scholar] [CrossRef]
- Sommerlad, M.; Lock, A.; Moir, G.; McGregor, J.; Bull, R.; Cerio, R.; Harwood, C. Linear Porokeratosis with Multiple Squamous Cell Carcinomas Successfully Treated by Electrochemotherapy. Br. J. Dermatol. 2016, 175, 1342–1345. [Google Scholar] [CrossRef]
- Agrawal, S.N.; Pawar, P.C.; Dhillan, P.V. Linear Porokeratosis: An Unusual Presentation. Indian J. Dermatol. 2014, 59, 318. [Google Scholar] [CrossRef]
- Bogaert, M.A.; Hogan, D.J. Linear Porokeratosis in a 74-Year-Old Woman. J. Am. Acad. Dermatol. 1991, 25, 338. [Google Scholar] [CrossRef]
- Fisher, C.A.; LeBoit, P.E.; Frieden, I.J. Linear Porokeratosis Presenting as Erosions in the Newborn Period. Pediatr. Dermatol. 1995, 12, 318–322. [Google Scholar] [CrossRef]
- Nayak, M.K.; Dhanta, A.; Hazarika, N.; Kumar, A. Linear Systematized Porokeratosis—A Rare Case and Dermoscopic Clues to Diagnosis. Indian Dermatol. Online J. 2020, 12, 465–466. [Google Scholar] [CrossRef]
- Yang, J.; Du, Y.-Q.; Fang, X.-Y.; Li, B.; Xi, Z.-Q.; Feng, W.-L. Linear Porokeratosis of the Foot with Dermoscopic Manifestations: A Case Report. World J. Clin. Cases 2022, 10, 11585–11589. [Google Scholar] [CrossRef]
- Hashimoto, T.; Moriyama, Y.; Satoh, T. Linear Porokeratosis with Severe Itch Accompanied by Lesional Upregulation of Interleukin 31, Thymic Stromal Lymphopoietin, and Periostin. Eur. J. Dermatol. 2021, 31, 570–572. [Google Scholar] [CrossRef]
- Kienast, A.K.; Hoeger, P.H. Penile Linear Porokeratosis in a Child: A Case Report. Pediatr. Dermatol. 2009, 26, 216–217. [Google Scholar] [CrossRef]
- Yadav, P.; Sanke, S.; Mendiratta, V.; Chander, R. Generalized Linear Porokeratosis in a Bilateral Distribution: An Unusual Presentation. Skinmed 2022, 20, 220–223. [Google Scholar]
- Kudligi, C.; Bhagwat, P.V.; Giriyan, S.S.; Eshwarrao, M.S. Unilateral systematized linear porokeratosis: A report of a rare case. Indian J. Dermatol. 2011, 56, 452–453. [Google Scholar] [CrossRef]
- Garg, T.; Ramchander; Varghese, B.; Barara, M.; Nangia, A. Generalized Linear Porokeratosis: A Rare Entity with Excellent Response to Acitretin. Dermatol. Online J. 2011, 17, 3. [Google Scholar] [CrossRef]
- Escanilla-Figueroa, C.; Jimeno-Ortega, I.; Fuenzalida-Wong, H.; Chávez-Rojas, F. Generalized Linear Porokeratosis. An. Bras. Dermatol. 2018, 93, 477–478. [Google Scholar] [CrossRef]
- McMillan, G.L.; Krull, E.A.; Mikhail, G.R. Linear Porokeratosis with Giant Cornoid Lamella. Arch. Dermatol. 1976, 112, 515–516. [Google Scholar] [CrossRef]
- Kono, M.; Yokoyama, N.; Ogawa, Y.; Takama, H.; Sugiura, K.; Akiyama, M. Unilateral Generalized Linear Porokeratosis with Nail Dystrophy. J. Dermatol. 2016, 43, 286–287. [Google Scholar] [CrossRef]
- Tseng, S.S.; Levit, E.K.; Ilarda, I.; Garzon, M.C.; Grossman, M.E. Linear Porokeratosis with Underlying Bony Abnormalities. Cutis 2002, 69, 309–312. [Google Scholar]
- Chen, H.-H.; Liao, Y.-H. Onychodystrophy in Congenital Linear Porokeratosis. Br. J. Dermatol. 2002, 147, 1272–1273. [Google Scholar] [CrossRef]
- Dervis, E.; Demirkesen, C. Generalized Linear Porokeratosis. Int. J. Dermatol. 2006, 45, 1077–1079. [Google Scholar] [CrossRef]
- Kohara, Y.; Takeo, T.; Oshima, Y.; Akita, Y.; Tamada, Y.; Watanabe, D. Linear Porokeratosis with Nail Dystrophy. Eur. J. Dermatol. 2011, 21, 625–626. [Google Scholar] [CrossRef]
- Blue, E.; Abbott, J.; Bowen, A.; Cipriano, S.D. Linear Porokeratosis with Bone Abnormalities Treated with Compounded Topical 2% Cholesterol/2% Lovastatin Ointment. Pediatr. Dermatol. 2021, 38, 242–245. [Google Scholar] [CrossRef]
- Wei, B.; Liu, M.; Qu, L.; Xiao, T.; Chen, H.-D.; He, C. Congenital Linear Porokeratosis with Pseudoainhum. Eur. J. Dermatol. 2010, 20, 817–818. [Google Scholar] [CrossRef]
- Witkowski, J.A.; Parish, L.C. Linear Porokeratosis Presenting as Mosaic Plantar Warts. Int. J. Dermatol. 1982, 21, 40–41. [Google Scholar] [CrossRef]
- Hunt, S.J.; Sharra, W.G.; Abell, E. Linear and Punctate Porokeratosis Associated with End-Stage Liver Disease. J. Am. Acad. Dermatol. 1991, 25, 937–939. [Google Scholar] [CrossRef]
- Tappel, A.C.; Tiwari, N.; Zlotoff, B. Linear Porokeratosis Associated with Bardet-Biedl Syndrome: A Case Report. Pediatr. Dermatol. 2019, 36, 346–348. [Google Scholar] [CrossRef]
- Verma, S.B. A Rare Case of Porokeratosis Ptychotropica and Coexistent Linear Porokeratosis in a 10-Year-Old Boy. Clin. Exp. Dermatol. 2009, 34, e501–e502. [Google Scholar] [CrossRef]
- Schwarz, N.; Stadie, V.; Kreft, B.; Happle, R.; Marsch, W.C.; Fiedler, E. Systematized Linear Porokeratosis: Concept of Type 2 Segmental Manifestation Implies an Increased Cancer Risk. J. Dermatol. 2012, 39, 953–954. [Google Scholar] [CrossRef]
- Sasson, M.; Krain, A.D. Porokeratosis and Cutaneous Malignancy. A Review. Dermatol. Surg. 1996, 22, 339–342. [Google Scholar] [CrossRef]
- Happle, R. Cancer Proneness of Linear Porokeratosis May Be Explained by Allelic Loss. Dermatology 1997, 195, 20–25. [Google Scholar] [CrossRef]
- Saleva-Stateva, M.; Hess, M.; Technau-Hafsi, K.; Weibel, L.; Badea, M.-A.; Boente, M.D.C.; Theiler, M.; Fiandrino, M.J.; Hoeger, P.; Zimmer, A.; et al. Molecular Characterization and Natural History of Linear Porokeratosis: A Case Series. J. Am. Acad. Dermatol. 2021, 85, 1603–1606. [Google Scholar] [CrossRef]
- Atzmony, L.; Khan, H.M.; Lim, Y.H.; Paller, A.S.; Levinsohn, J.L.; Holland, K.E.; Mirza, F.N.; Yin, E.; Ko, C.J.; Leventhal, J.S.; et al. Second-Hit, Postzygotic PMVK and MVD Mutations in Linear Porokeratosis. JAMA Dermatol. 2019, 155, 548–555. [Google Scholar] [CrossRef]
- Paller, A.S.; van Steensel, M.A.M.; Rodriguez-Martín, M.; Sorrell, J.; Heath, C.; Crumrine, D.; van Geel, M.; Cabrera, A.N.; Elias, P.M. Pathogenesis-Based Therapy Reverses Cutaneous Abnormalities in an Inherited Disorder of Distal Cholesterol Metabolism. J. Investig. Dermatol. 2011, 131, 2242–2248. [Google Scholar] [CrossRef]
- Li, D.; Technau-Hafsi, K.; Giehl, K.; Hoeger, P.H.; Has, C. Targeted Anti-IL-17 Therapy for Linear Porokeratosis. Br. J. Dermatol. 2023, 189, 630–631. [Google Scholar] [CrossRef]
- Errichetti, E.; Francesco, V.D.; Pegolo, E.; Stinco, G. Dermoscopy in Assisting the Diagnosis of Linear Porokeratosis. J. Dermatol. 2017, 44, e248–e249. [Google Scholar] [CrossRef]
- Turkmen, M.; Gerceker Turk, B.; Kilinc Karaarslan, I.; Kandiloglu, G.; Ozdemir, F. Dermoscopic Features of Linear Porokeratosis: Different Aspects in Its Development. J. Dermatol. Res. Ther. 2019, 5, 63. [Google Scholar] [CrossRef]
- Kovacikova Curkova, A.; Hegyi, J.; Kozub, P.; Szep, Z.; D’Erme, A.M.; Simaljakova, M. A Case of Linear Porokeratosis Treated with Photodynamic Therapy with Confocal Microscopy Surveillance. Dermatol. Ther. 2014, 27, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Vaishnani, J.B.; Bosamiya, S.S.; Sapariya, B.J.; Udhreja, P.R. Linear Porokeratosis with Follicular Involvement. Indian J. Dermatol. 2011, 56, 460–461. [Google Scholar] [CrossRef] [PubMed]
- Minami-Hori, M.; Ishida-Yamamoto, A.; Iizuka, H. Cornoid Lamellae Associated with Follicular Infundibulum and Acrosyringium in Porokeratosis. J. Dermatol. 2009, 36, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.K.; Puri, K.J.P.S.; Goyal, T.; Chahal, K.S. Linear Porokeratosis. Dermatol. Online J. 2007, 13, 15. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y.; Yuasa, T.; Shimizu, M. Linear Porokeratosis. J. Dermatol. 1993, 20, 489–492. [Google Scholar] [CrossRef] [PubMed]
- Barnett, J.H. Linear Porokeratosis: Treatment with the Carbon Dioxide Laser. J. Am. Acad. Dermatol. 1986, 14, 902–904. [Google Scholar] [CrossRef]
- Saleva-Stateva, M.; Weibel, L.; Theiler, M.; Balabanova, M.; Boente, M.C.; Has, C. Lack of Effect of Topical Statins in Linear Porokeratosis. J. Eur. Acad. Dermatol. Venereol. 2021, 35, e26–e28. [Google Scholar] [CrossRef]
- Alster, T.S.; Nanni, C.A. Successful Treatment of Porokeratosis with 585 Nm Pulsed Dye Laser Irradiation. Cutis 1999, 63, 265–266. [Google Scholar]
- Cohen, P.R.; Held, J.L.; Katz, B.E. Linear Porokeratosis: Successful Treatment with Diamond Fraise Dermabrasion. J. Am. Acad. Dermatol. 1990, 23, 975–977. [Google Scholar] [CrossRef]
- Bhushan, M.; Craven, N.M.; Beck, M.H.; Chalmers, R.J. Linear Porokeratosis of Mibelli: Successful Treatment with Cryotherapy. Br. J. Dermatol. 1999, 141, 389. [Google Scholar] [CrossRef]
- Gonçalves, S.V.C.B.; Valente, N.Y.S.; de Oliveira Junior, J.V.; Paiva, D.L.M. Case for Diagnosis. An. Bras. Dermatol. 2012, 87, 489–490. [Google Scholar] [CrossRef] [PubMed]
- Casale, F.; Tchanque-Fossuo, C.N.; Durkin, J.R. Successful Clearance of Linear Porokeratosis with Aminolevulinic Acid and Pulsed Dye Laser. Dermatol. Surg. 2021, 47, 1300–1301. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Colmenero, C.; Ruiz-Villaverde, R.; Martínez-García, E.; Aneiros-Fernández, J.; Tercedor-Sánchez, J. Photoletter to the Editor: Response of Linear Porokeratosis to Photodynamic Therapy in an 11-Year-Old Girl. J. Dermatol. Case Rep. 2015, 9, 118–119. [Google Scholar] [CrossRef] [PubMed]
- García-Navarro, X.; Garcés, J.R.; Baselga, E.; Alomar, A. Linear Porokeratosis: Excellent Response to Photodynamic Therapy. Arch. Dermatol. 2009, 145, 526–527. [Google Scholar] [CrossRef]
- Weidner, T.; Illing, T.; Miguel, D.; Elsner, P. Treatment of Porokeratosis: A Systematic Review. Am. J. Clin. Dermatol. 2017, 18, 435–449. [Google Scholar] [CrossRef]
- Nazarian, R.S.; Hosseinipour, M.; Amin, B.; Cohen, S.R. Linear Porokeratosis Presenting in Adulthood: A Diagnostic Challenge: A Case Report. SAGE Open Med. Case Rep. 2020, 8, 2050313X20919613. [Google Scholar] [CrossRef]
- Alakeel, A.; Dawari, S.; Alhumidi, A.; Alekrish, K. Combining Isotretinoin and Topical Cholesterol/Atorvastatin in the Treatment of Linear Porokeratosis: A Case Report. Cureus 2023, 15, e38873. [Google Scholar] [CrossRef]
- Hong, J.-B.; Hsiao, C.-H.; Chu, C.-Y. Systematized Linear Porokeratosis: A Rare Variant of Diffuse Porokeratosis with Good Response to Systemic Acitretin. J. Am. Acad. Dermatol. 2009, 60, 713–715. [Google Scholar] [CrossRef]
- Pehamberger, H.; Esca, S.A.; Holubar, K. Treatment of hyperkeratotic dermatoses with an oral aromatic retinoid (Ro 10-9359). Z. Hautkr 1980, 55, 68–78. [Google Scholar]
- Goldman, G.D.; Milstone, L.M. Generalized Linear Porokeratosis Treated with Etretinate. Arch. Dermatol. 1995, 131, 496–497. [Google Scholar] [CrossRef]
- Parks, A.C.; Conner, K.J.; Armstrong, C.A. Long-Term Clearance of Linear Porokeratosis with Tacrolimus, 0.1%, Ointment. JAMA Dermatol. 2014, 150, 194–196. [Google Scholar] [CrossRef]
- Kim, C. Linear Porokeratosis. Dermatol. Online J. 2005, 11, 22. [Google Scholar] [CrossRef]
- Grover, C.; Goel, A.; Nanda, S.; Khurana, N.; Reddy, B.S.N. A Case of Extensive Linear Porokeratosis with Evaluation of Topical Tretinoin versus 5-Flourouracil as Treatment Modalities. J. Dermatol. 2005, 32, 1000–1004. [Google Scholar] [CrossRef]
- Diep, D.; Janitz, T.; Kannan, K.S.; Crane, A.; Aluri, B.; Wright, K.; Baker, W. Bilateral Linear Porokeratosis Treated with Topical Cholesterol 2%/Lovastatin 2%. Cureus 2022, 14, e27540. [Google Scholar] [CrossRef]
- Ahn, S.-J.; Lee, H.-J.; Chang, S.-E.; Lee, M.-W.; Choi, J.-H.; Moon, K.-C.; Koh, J.-K. Case of Linear Porokeratosis: Successful Treatment with Topical 5% Imiquimod Cream. J. Dermatol. 2007, 34, 146–147. [Google Scholar] [CrossRef]
- Buhle, A.C.; Fagan, K.K.; Johnson, N.M.; Grider, D.J. Treating Linear Porokeratosis with Topical Lovastatin/Cholesterol Cream. Dermatol. Online J. 2022, 28, 12. [Google Scholar] [CrossRef]
- Guss, S.B.; Osbourn, R.A.; Lutzner, M.A. Porokeratosis Plantaris, Palmaris, et Disseminata. A Third Type of Porokeratosis. Arch. Dermatol. 1971, 104, 366–373. [Google Scholar] [CrossRef]
- Irisawa, R.; Yamazaki, M.; Yamamoto, T.; Tsuboi, R. A Case of Porokeratosis Plantaris Palmaris et Disseminata and Literature Review. Dermatol. Online J. 2012, 18, 5. [Google Scholar] [CrossRef]
- Jedlowski, P.M.; Rainwater, G.; Paek, S.Y. Punctate Porokeratosis-Pruritic and Hyperkeratotic Papules on the Palms and Feet. Bayl. Univ. Med. Cent. Proc. 2020, 33, 415–416. [Google Scholar] [CrossRef]
- Lanka, P.; Lanka, L.R.; Manivachagam, D. Punctate Porokeratosis Palmaris et Plantaris. Indian J. Dermatol. 2015, 60, 284–286. [Google Scholar] [CrossRef]
- Roberts, L.C.; DeVillez, R.L. Congenital Unilateral Punctate Porokeratosis. Am. J. Dermatopathol. 1984, 6, 57–61. [Google Scholar] [CrossRef]
- Udare, S.; Hemmady, K. Clinical and Dermatoscopic Features of Porokeratosis Palmaris et Plantaris. Indian Dermatol. Online J. 2016, 7, 290–292. [Google Scholar] [CrossRef]
- Kanitakis, J. Porokeratoses: An Update of Clinical, Aetiopathogenic and Therapeutic Features. Eur. J. Dermatol. 2014, 24, 533–544. [Google Scholar] [CrossRef]
- Shaw, J.C.; White, C.R. Porokeratosis Plantaris Palmaris et Disseminata. J. Am. Acad. Dermatol. 1984, 11, 454–460. [Google Scholar] [CrossRef]
- Jensen, J.-M.; Egberts, F.; Proksch, E.; Hauschild, A. Disseminated Porokeratosis Palmaris and Plantaris Treated with Imiquimod Cream to Prevent Malignancy. Acta Derm. Venereol. 2005, 85, 550–551. [Google Scholar] [CrossRef]
- Patrizi, A.; Passarini, B.; Minghetti, G.; Masina, M. Porokeratosis Palmaris et Plantaris Disseminata: An Unusual Clinical Presentation. J. Am. Acad. Dermatol. 1989, 21, 415–418. [Google Scholar] [CrossRef]
- Hartman, R.; Mandal, R.; Sanchez, M.; Stein, J.A. Porokeratosis Plantaris, Palmaris, et Disseminata. Dermatol. Online J. 2010, 16, 22. [Google Scholar] [CrossRef]
- Hayashi, K.; Iinuma, S.; Ishida-Yamamoto, A. Porokeratosis Plantaris Palmaris et Disseminata with Pruritic Inflammatory Changes. JEADV Clin. Pract. 2023, 2, 366–368. [Google Scholar] [CrossRef]
- Sengupta, S.; Das, J.K.; Gangopadhyay, A. Multicentric Squamous Cell Carcinoma over Lesions of Porokeratosis Palmaris et Plantaris Disseminata and Giant Porokeratosis. Indian J. Dermatol. Venereol. Leprol. 2005, 71, 414–416. [Google Scholar] [CrossRef]
- Ruocco, V.; Satriano, R.A.; Florio, M.; Pettinato, G.; Pisani, M. Porokeratosis palmaris et plantaris disseminata with squamous cell carcinoma. G. Ital. Dermatol. Venereol. 1990, 125, 53–58. [Google Scholar]
- Seishima, M.; Izumi, T.; Oyama, Z.; Maeda, M. Squamous Cell Carcinoma Arising from Lesions of Porokeratosis Palmaris et Plantaris Disseminata. Eur. J. Dermatol. 2000, 10, 478–480. [Google Scholar] [PubMed]
- Marschalkó, M.; Somlai, B. Porokeratosis Plantaris, Palmaris, et Disseminata. Arch. Dermatol. 1986, 122, 890–891. [Google Scholar] [CrossRef] [PubMed]
- Brown, F.C. Punctate Keratoderma. Arch. Dermatol. 1971, 104, 682–683. [Google Scholar] [CrossRef]
- Herman, P.S. Punctate Porokeratotic Keratoderma. Dermatologica 1973, 147, 206–213. [Google Scholar] [CrossRef]
- Rahbari, H.; Cordero, A.A.; Mehregan, A.H. Punctate Porokeratosis. A Clinical Variant of Porokeratosis of Mibelli. J. Cutan. Pathol. 1977, 4, 338–341. [Google Scholar] [CrossRef]
- Himmelstein, R.; Lynfield, Y.L. Punctate Porokeratosis. Arch. Dermatol. 1984, 120, 263–264. [Google Scholar] [CrossRef]
- Ryan, C.; Bennett, R.; Davis, M.; Yan, S.; Aaron, D.M. A Rare Case of Punctate Porokeratosis Treated with Topical Lovastatin/Cholesterol. JAAD Case Rep. 2023, 35, 57–59. [Google Scholar] [CrossRef]
- Teixeira, V.B.; Reis, J.P.; Vieira, R.; Tellechea, Ó.; Figueiredo, A. Unilateral Punctate Porokeratosis—Case Report. An. Bras. Dermatol. 2013, 88, 441–446. [Google Scholar] [CrossRef]
- Touraud, J.P.; Dalac, S.; Collet, E.; Tanter, Y.; Justrabo, E.; Dutronc, Y.; Lambert, D. Punctate Porokeratosis in a Renal Transplant Recipient. Clin. Exp. Dermatol. 2003, 28, 329–330. [Google Scholar] [CrossRef]
- Sakas, E.L.; Gentry, R.H. Porokeratosis Punctata Palmaris et Plantaris (Punctate Porokeratosis). Case Report and Literature Review. J. Am. Acad. Dermatol. 1985, 13, 908–912. [Google Scholar] [CrossRef]
- Lucke, T.W.; Fallowfield, M.; Kemmett, D. A Sporadic Case of Porokeratosis Plantaris Palmaris et Disseminata. Br. J. Dermatol. 1998, 138, 556–557. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Yang, S.; Li, M.; Song, Y.X.; Zhang, X.Q.; Bu, L.; Zheng, G.Y.; Kong, X.Y.; Zhang, X.J. Identification of a Locus for Porokeratosis Palmaris et Plantaris Disseminata to a 6.9-cM Region at Chromosome 12q24.1-24.2. Br. J. Dermatol. 2003, 149, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Jägle, S.; Juratli, H.A.; Hickman, G.; Süssmuth, K.; Boente, M.C.; Kopp, J.; Kirchmeier, P.; Zimmer, A.; Happle, R.; Bourrat, E.; et al. Porokeratosis Plantaris, Palmaris et Disseminata Caused by Congenital Pathogenic Variants in the MVD Gene and Loss of Heterozygosity in Affected Skin. Acta Derm. Venereol. 2021, 101, adv00397. [Google Scholar] [CrossRef] [PubMed]
- Topin-Ruiz, S.; Debarre, J.-M.; Blanchard, E.; Kettani, S.; Valmier, P.-J.; Martin, L.; Le Corre, Y. Circumscribed palmar hypokeratosis (CPM): The diagnostic value of dermoscopy. Ann. Dermatol. Venereol. 2017, 144, 197–202. [Google Scholar] [CrossRef]
- Skroza, N.; Proietti, I.; Bernardini, N.; Nicolucci, F.; Tolino, E.; La Viola, G.; Orsini, D.; Zuber, S.; Potenza, C. Acitretin for Treatment of Familial Porokeratosis Palmaris et Plantaris Disseminate. Eur. J. Dermatol. 2012, 22, 699–700. [Google Scholar] [CrossRef]
- McCallister, R.E.; Estes, S.A.; Yarbrough, C.L. Porokeratosis Plantaris, Palmaris, et Disseminata. Report of a Case and Treatment with Isotretinoin. J. Am. Acad. Dermatol. 1985, 13, 598–603. [Google Scholar] [CrossRef]
- Helfman, R.J.; Poulos, E.G. Reticulated Porokeratosis. A Unique Variant of Porokeratosis. Arch. Dermatol. 1985, 121, 1542–1543. [Google Scholar] [CrossRef]
- Lucker, G.P.; Happle, R.; Steijlen, P.M. An Unusual Case of Porokeratosis Involving the Natal Cleft: Porokeratosis Ptychotropica? Br. J. Dermatol. 1995, 132, 150–151. [Google Scholar] [CrossRef]
- Stone, N.; Ratnavel, R.; Wilkinson, J.D. Bilateral Perianal Inflammatory Verrucous Porokeratosis (Porokeratosis Ptychotropica). Br. J. Dermatol. 1999, 140, 553–555. [Google Scholar] [CrossRef]
- Takiguchi, R.H.; White, K.P.; White, C.R.; Simpson, E.L. Verrucous Porokeratosis of the Gluteal Cleft (Porokeratosis Ptychotropica): A Rare Disorder Easily Misdiagnosed. J. Cutan. Pathol. 2010, 37, 802–807. [Google Scholar] [CrossRef]
- Rodríguez-Tejero, A.; Bueno-Rodríguez, A.; Montero-Vílchez, T.; Arias-Santiago, S.; Molina-Leyva, A. Genitogluteal Porokeratosis: An Unusual Presentation Treated Successfully with the Novel Combination of Imiquimod 5% Cream and Photodynamic Therapy. Dermatol. Ther. 2020, 33, e13656. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-J.; Chou, Y.-C.; Chen, C.-H.; Kuo, T.-T.; Hong, H.-S. Genital Porokeratosis: A Series of 10 Patients and Review of the Literature. Br. J. Dermatol. 2006, 155, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Shrestha, S.; Aryal, R.; Homagain, S.; Tiwari, S.B.; Rayamajhi, B.; Parajuli, S.; Paudel, U. Porokeratosis of Gluteal Region: A Case Report. SAGE Open Med. Case Rep. 2022, 10, 2050313X221139559. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-L.; Liu, Y.-H.; Chen, W. Genitogluteal Porokeratosis. J. Eur. Acad. Dermatol. Venereol. 2006, 20, 899–900. [Google Scholar] [CrossRef] [PubMed]
- Bari, O.; Vazirnia, A.; Cohen, P.R.; Romero, L.S. Genitogluteal Porokeratosis in an HIV-Positive Man: A Case Report and Review of the Literature on Genital Porokeratosis. Dermatol. Online J. 2018, 24, 7. [Google Scholar] [CrossRef]
- Wanat, K.A.; Gormley, R.H.; Bennett, D.D.; Kovarik, C.L. Genitogluteal Porokeratosis Involving the Scrotum: An Unusual Presentation of an Uncommon Disease. J. Cutan. Pathol. 2012, 39, 72–74. [Google Scholar] [CrossRef]
- Foran, T.K.; Day, T.; Bradford, J.; Scurry, J. Genitogluteal Porokeratosis in a Well Woman. J. Low. Genit. Tract. Dis. 2017, 21, e8–e9. [Google Scholar] [CrossRef]
- Joshi, R.; Minni, K. Genitogluteal Porokeratosis: A Clinical Review. Clin. Cosmet. Investig. Dermatol. 2018, 11, 219–229. [Google Scholar] [CrossRef]
- Ryoo, Y.-W.; Kim, Y.; Yun, J.-M.; Kim, S.-A. Porokeratosis Ptychotropica: A Case Report. J. Yeungnam Med. Sci. 2022, 40, 423–425. [Google Scholar] [CrossRef]
- Scheiba, N.; Enk, A.; Proske, S.; Hartschuh, W. Porokeratosis Ptychotropica: Successful Treatment with the Dermatome. Dermatol. Surg. 2010, 36, 257–260. [Google Scholar] [CrossRef]
- Malek, J.; Chedraoui, A.; Kibbi, A.G.; Ghosn, S. Genitogluteal Porokeratosis: 10 Years to Make the Diagnosis! Am. J. Dermatopathol. 2009, 31, 604–606. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.S.; Lee, S. Genitogluteal Porokeratosis: An Unusual Clinical Presentation. Australas. J. Dermatol. 2012, 53, e36–e39. [Google Scholar] [CrossRef] [PubMed]
- Hazarika, D.; Pawar, M. Hyperkeratotic Porokeratosis Ptychotropica with Satellite Lesions: A Rare Presentation of an Unusual Variant of Porokeratosis. Acta Dermatovenerol. Alp. Pannonica Adriat. 2017, 26, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Bari, O.; Calame, A.; Marietti-Shepherd, S.; Barrio, V.R. Pediatric Penile Porokeratosis: A Case Report. Pediatr. Dermatol. 2018, 35, e103–e104. [Google Scholar] [CrossRef]
- Thomas, C.; Ogboli, M.I.; Carr, R.A.; Charles-Holmes, R. Hypertrophic Perianal Porokeratosis in Association with Superficial Actinic Porokeratosis of the Leg. Clin. Exp. Dermatol. 2003, 28, 676–677. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Das, J.K.; Gangopadhyay, A. Porokeratosis Confined to the Genital Area: A Report of Three Cases. Indian J. Dermatol. Venereol. Leprol. 2008, 74, 80. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.B.; Im, D.D.; Jockle, G.; Rosenshein, N.B. Vulvar Porokeratosis: Case Report and Review of the Literature. Int. J. Gynecol. Pathol. 1999, 18, 169–173. [Google Scholar] [CrossRef]
- Kogut, M.; Schiller, M.; Hadaschik, E.; Enk, A.; Haenssle, H.A. Porokeratosis Ptychotropica Involving the Glans Penis: A Unique Case of This Rare Condition. J. Dtsch. Dermatol. Ges. 2016, 14, 181–183. [Google Scholar] [CrossRef]
- Qian, Y.-T.; Vano-Galvan, S.; Liu, J.-W.; Liu, W.; Ma, D.-L. Porokeratosis Ptychotropica on the Penis and Scrotum: A Case Report. Acta Derm. Venereol. 2019, 99, 246–247. [Google Scholar] [CrossRef]
- Yeo, J.; Winhoven, S.; Tallon, B. Porokeratosis Ptychotropica: A Rare and Evolving Variant of Porokeratosis. J. Cutan. Pathol. 2013, 40, 1042–1047. [Google Scholar] [CrossRef]
- Lacarrubba, F.; Musumeci, M.L.; Verzì, A.E.; Poma, C.; Caltabiano, R.; Micali, G. Porokeratosis Ptychotropica: Dermoscopy, Reflectance Confocal Microscopy, and Histopathological Correlation. Indian J. Dermatol. 2021, 66, 540–542. [Google Scholar] [CrossRef]
- Terrell, J.R.; Urban, J.R.; Fung, M.A.; Tartar, D.M.; Kiuru, M. Pink Verrucous Plaque in a Man with Systemic Mastocytosis. Dermatol. Online J. 2019, 25, 12. [Google Scholar] [CrossRef]
- Liu, W.; Liu, J.-W.; Ma, D.-L. Porokeratosis Ptychotropica. JAMA Dermatol. 2019, 155, 845. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.; Schwentker, A.R.; Barron, D.R.; Lucky, A.W. Clinical Course of Porokeratosis Ptychotropica over 7 Years in an Otherwise Healthy Child. Pediatr. Dermatol. 2020, 37, 248–250. [Google Scholar] [CrossRef] [PubMed]
- Laino, L.; Pala, S.; Innocenzi, D.; Accappaticcio, G.; Van Steensel, M.A.M. Genital Porokeratosis. Eur. J. Dermatol. 2004, 14, 190–192. [Google Scholar]
- Ahmed, A.; Hivnor, C. A Case of Genital Porokeratosis and Review of Literature. Indian J. Dermatol. 2015, 60, 217. [Google Scholar] [CrossRef] [PubMed]
- Tebet, A.C.F.; de Oliveira, T.G.; de Oliveira, A.R.F.M.; Moriya, F.S.; de Oliveira Filho, J.; Cucé, L.C. Porokeratosis Ptychotropica. An. Bras. Dermatol. 2016, 91, 134–136. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Peng, S.; Mao, T.; Li, X.; Ye, W.; Fang, M. An Unusual Case Report of Porokeratosis Ptychotropica on the Buttocks. Medicine 2022, 101, e32074. [Google Scholar] [CrossRef]
- Tallon, B.; Blumental, G.; Bhawan, J. Porokeratosis Ptychotropica: A Lesser-Known Variant. Clin. Exp. Dermatol. 2009, 34, e895–e897. [Google Scholar] [CrossRef]
- Pitney, L.; Weedon, D.; Pitney, M. Porokeratosis Ptychotropica: A Rare Variant with Discrete Lesions. Australas. J. Dermatol. 2015, 56, e28–e29. [Google Scholar] [CrossRef]
- Fustà-Novell, X.; Podlipnik, S.; Combalia, A.; Morgado-Carrasco, D.; Ferrando, J.; Mascaró, J.M.; Aguilera, P. Porokeratosis Ptychotropica Responding to Photodynamic Therapy: An Alternative Treatment for a Refractory Disease. Photodermatol. Photoimmunol. Photomed. 2017, 33, 271–274. [Google Scholar] [CrossRef]
- Albanell-Fernández, M.; Luque-Luna, M.; López-Cabezas, C.; Alamon-Reig, F.; Espinosa-Villaseñor, N.; Barboza-Guadagnini, L.; Mascaró, J.M. Treatment of Porokeratosis Ptychotropica with a Topical Combination of Cholesterol and Simvastatin. JAMA Dermatol. 2023, 159, 458–460. [Google Scholar] [CrossRef]
- Mazori, D.R.; Shvartsbeyn, M.; Meehan, S.A.; Tarsis, S.L. Transformation of Porokeratosis Ptychotropica into Invasive Squamous Cell Carcinoma. Int. J. Dermatol. 2017, 56, 679–680. [Google Scholar] [CrossRef]
- Ferreira, F.R.; Lessa, P.P.; Alvarenga, M.L. de Genitogluteal Porokeratosis—Case Report. An. Bras. Dermatol. 2013, 88, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Hoang, N.; Harper, H.E.; Jibbe, A.; Siscos, S.M.; Cargnel, A.L.; Kaplan, D.L. Porokeratosis Ptychotropica: A Rare Variant That Is Commonly Misdiagnosed. Dermatol. Online J. 2020, 26, 15. [Google Scholar] [CrossRef]
- Kulhari, M.; Khan, H.Q.; Amin, S.S.; Afrose, R. A Rare Case of Genital Porokeratosis Associated with Epididymo-Orchitis. Indian J. Sex. Transm. Dis. 2022, 43, 220–221. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, J. Image Gallery: Verrucous Porokeratosis with Characteristic Histopathological and Dermoscopic Features. Br. J. Dermatol. 2017, 176, e38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S. Porokeratosis Ptychotropica: A Rare Manifestation and Dermoscopic Feature. Indian J. Dermatol. 2022, 67, 744–746. [Google Scholar] [CrossRef]
- Cabete, J.; Fidalgo, A.; Lencastre, A.; Diamantino, F.; João, A. Porokeratosis Ptychotropica of the Scrotum: Dermoscopic Evaluation of an Atypical Presentation. An. Bras. Dermatol. 2015, 90, 191–193. [Google Scholar] [CrossRef]
- Veasey, J.V.; Dalapicola, M.C.; Lellis, R.F.; Campaner, A.B.; Manzione, T.d.S.; Rodrigues, M.C.d.F.S. Porokeratosis Ptychotropica: A Rare Manifestation with Typical Histological Exam. An. Bras. Dermatol. 2016, 91, 496–498. [Google Scholar] [CrossRef]
- D’souza, P.; Dhali, T.K.; Arora, S.; Gupta, H.; Khanna, U. Porokeratosis Ptychotropica: A Rare Variant of Porokeratosis. Dermatol. Online J. 2014, 20, 12. [Google Scholar] [CrossRef]
- Lee, D.K.; Oh, S.H.; Chang, S.E.; Lee, M.W.; Choi, J.H.; Moon, K.C.; Koh, J.K. A Rare Variant of Porokeratosis: Porokeratosis Ptychotropica. J. Am. Acad. Dermatol. 2006, 55, S120–S122. [Google Scholar] [CrossRef] [PubMed]
- Jee, M.S.; Chang, S.E.; Suh, H.S.; Choi, J.H.; Sung, K.J.; Moon, K.C.; Koh, J.K. Porokeratosis Ptychotropica Associated with Dermal Amyloid Deposits. Clin. Exp. Dermatol. 2003, 28, 450–452. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Liu, X.-Y.; Li, B.; Gao, X.-H.; Chen, H.-D.; Li, J.-H. Porokeratosis on the Scrotum: Two Cases. Dermatol. Ther. 2015, 28, 83–85. [Google Scholar] [CrossRef] [PubMed]
- Valdivielso-Ramos, M. Genital porokeratosis. Actas Dermosifiliogr. 2008, 99, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Tangoren, I.A.; Weinberg, J.M.; Ioffreda, M.; Werth, V.P.; James, W.D. Penile Porokeratosis of Mibelli. J. Am. Acad. Dermatol. 1997, 36, 479–481. [Google Scholar] [CrossRef]
- Ghahartars, M.; Zahraei, S.A.H.; Sari Aslani, F.; Hadibarhaghtalab, M.; Parvizi, M.M. A 58-Year-Old Man with Porokeratosis Ptychotropica: A Successful Treatment with Alone Cryotherapy and Literature Review. Dermatol. Ther. 2021, 34, e14732. [Google Scholar] [CrossRef]
- Fernández Ballesteros, M.D.; Gómez Moyano, E.; Ayala Blanca, M.; Simonsen, S. A Verrucous Plaque on the Intergluteal Cleft. Indian J. Dermatol. Venereol. Leprol. 2018, 84, 641–642. [Google Scholar] [CrossRef]
- Merkle, T.; Hohenleutner, U.; Braun-Falco, O.; Landthaler, M. Reticulate Porokeratosis--Successful Treatment with CO2-Laser Vaporization. Clin. Exp. Dermatol. 1992, 17, 178–181. [Google Scholar] [CrossRef]
- Contreras-Ruiz, J.; Toussaint-Caire, S.; Torres-Camacho, P.; Villa-Castro, V.B. Porokeratosis Ptychotropica: A Diagnostic and Therapeutic Challenge. J. Eur. Acad. Dermatol. Venereol. 2018, 32, e114–e115. [Google Scholar] [CrossRef]
- Kawakami, Y.; Mitsui, S. A Case of Porokeratosis Ptychotropica: Successful Treatment with Topical 5% Imiquimod Cream. Clin. Exp. Dermatol. 2017, 42, 839–841. [Google Scholar] [CrossRef]
- Burch, H.W.; Leclerq, A.H.; Maize, J.C. Pruritic Natal Cleft Plaque. Porokeratosis Ptychotropica. Arch. Dermatol. 2010, 146, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Asawanonda, P.; Noppakun, N.; Huiprasert, P. Seborrheic Keratosis-like Porokeratosis: A Case Report. Dermatol. Online J. 2005, 11, 18. [Google Scholar] [PubMed]
- Markantoni, V.; Platsidaki, E.; Kouris, A.; Aroni, K.; Kontochristopoulos, G. Disseminated Verrucous Porokeratosis Successfully Treated with 5-FU Followed by Oral Isotretinoin—A Case Report. J. Dermat. Cosmetol. 2018, 2, 46–47. [Google Scholar] [CrossRef]
- Yu, Y.-F.; Wu, Y.-H. Verrucous Porokeratosis (Porokeratosis Ptychotropica) with Dermal Amyloid Deposits. Dermatol. Sin. 2013, 31, 145–148. [Google Scholar] [CrossRef]
- Pereira, N.; Teixeira, V.; Cordeiro, M.R.; Tellechea, O. A Rarely Diagnosed Disorder of the Gluteal Cleft. Clin. Exp. Dermatol. 2013, 38, 430–432. [Google Scholar] [CrossRef] [PubMed]
- Neumann, E. Some Unusual Findings in Porokeratosis Mibelli. Acta Derm. Venereol. 1980, 60, 355–359. [Google Scholar] [CrossRef]
- Tallon, B.; Emanuel, P. Follicular Porokeratosis, a Porokeratosis Variant. Am. J. Dermatopathol. 2017, 39, e107–e109. [Google Scholar] [CrossRef]
- de Almeida, H.L.; Guarenti, I.M.; de Castro, L.; Rocha, N.M. Follicular Involvement in Porokeratosis. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 109–111. [Google Scholar] [CrossRef]
- Zhao, M.; Sanusi, T.; Zhao, Y.; Huang, C.; Chen, S. Porokeratosis with Follicular Involvement: Report of Three Cases and Review of Literatures. Int. J. Clin. Exp. Pathol. 2015, 8, 4248–4252. [Google Scholar]
- Pongpudpunth, M.; Farber, J.; Mahalingam, M. Follicular Porokeratosis: Distinct Clinical Entity or Histologic Variant? J. Cutan. Pathol. 2009, 36, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Sud, A.; Shipman, A.R.; Odeke, M.; Varma, K.; Read-Jones, M.; Carr, R.A. Follicular Porokeratosis: Four New Cases. Clin. Exp. Dermatol. 2017, 42, 881–886. [Google Scholar] [CrossRef] [PubMed]
- Young, P.M.; Leavens, J.; Gaspard, S.; Kim, G.; Armstrong, A.W. An Unusual Spiculated Presentation of Follicular Porokeratosis. Dermatol. Online J. 2019, 25, 7. [Google Scholar] [CrossRef]
- Sun, R.; Chen, H.; Lian, S.; Zhu, W. Follicular Porokeratosis: A Case Study and Literature Review. Eur. J. Dermatol. 2017, 27, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Bembry, R.; Louie, E.; Overholser, E.; Tirado, M. Exclusive Facial Porokeratosis with Follicular Involvement in an African American Female. J. Cutan. Pathol. 2023, 50, 383–384. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Wood, B.A.; Harvey, N.T. Follicular Porokeratosis of the Nose: Two Further Cases of an Emerging Variant of Porokeratosis. Pathology 2015, 47, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Trikha, R.; Wile, A.; King, J.; Ward, K.H.M.; Brodell, R.T. Punctate Follicular Porokeratosis: Clinical and Pathologic Features. Am. J. Dermatopathol. 2015, 37, e134–e136. [Google Scholar] [CrossRef]
- Chua, I.S.Y.; Lee, J.S.; Chiam, L.Y. Pruritic Papules on the Nose in a 25-Year-Old Female. Indian J. Dermatol. 2014, 59, 106. [Google Scholar] [CrossRef]
- Wang, N.S.; Gruson, L.M.; Kamino, H. Facial Follicular Porokeratosis: A Case Report. Am. J. Dermatopathol. 2010, 32, 720–722. [Google Scholar] [CrossRef]
- Rifaioğlu, E.N.; Özyalvaçlı, G. Follicular Porokeratosis at Alae Nasi; A Case Report and Short Review of Literature. Indian J. Dermatol. 2014, 59, 398–400. [Google Scholar] [CrossRef]
- Rocha-Sousa, V.L.L.; Costa, J.B.; de Aquino Paulo-Filho, T.; Rocha, K.B.F.; da Trindade-Neto, P.B. Follicular Porokeratosis on the Face. Am. J. Dermatopathol. 2011, 33, 636–638. [Google Scholar] [CrossRef]
- Gómez-Zubiaur, A.; Medina-Expósito, I.; Fernández-Flores, A.; Trasobares-Marugán, L. Follicular Porokeratosis of the Scalp: First Description of Clinical and Trichoscopic Features. Int. J. Trichology 2022, 14, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.; Handa, S.; Chatterjee, D.; Vinay, K.; Mahajan, R. Disseminated Filiform Hyperkeratosis—A Variant of Porokeratosis? J. Eur. Acad. Dermatol. Venereol. 2018, 32, e419–e421. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Choi, E.H. Exclusive Facial Porokeratosis: Histopathologically Showing Follicular Cornoid Lamellae. J. Dermatol. 2011, 38, 1072–1075. [Google Scholar] [CrossRef]
- Walsh, S.N.; Hurt, M.A.; Santa Cruz, D.J. Porokeratoma. Am. J. Surg. Pathol. 2007, 31, 1897–1901. [Google Scholar] [CrossRef] [PubMed]
- Batalla, A.; Rosón, E.; De la Torre, C. Porokeratoma: A Different Entity or a Variant of Verrucous (Hyperkeratotic) Porokeratosis? Indian J. Dermatol. 2013, 58, 158. [Google Scholar] [CrossRef] [PubMed]
- Sherban, A.K.; Fuller, C.G.; Griffin, T.; Lee, J.B. Porokeratoma: A Distinct Variant of Porokeratosis. J. Cutan. Pathol. 2022, 49, 998–1003. [Google Scholar] [CrossRef]
- Kanitakis, J.; Fournier, N.; Jullien, D. Follicular Porokeratoma: Report of a New Variant of Porokeratoma (Porokeratotic Acanthoma) and Literature Review. Am. J. Dermatopathol. 2022, 44, 748–752. [Google Scholar] [CrossRef]
- Caseiro Silverio, P.; Pham, X.-C.; Kaya, G. Porokeratoma: A Possible Association with Human Papillomavirus Infection. Dermatopathology 2015, 2, 43–45. [Google Scholar] [CrossRef]
- Kanitakis, J.; Rival-Tringali, A.L.; Chouvet, B.; Vignot, E.; Claudy, A.; Faure, M. Porokeratoma (Porokeratotic Acanthoma): Immunohistological Study of a New Case. J. Cutan. Pathol. 2009, 36, 804–807. [Google Scholar] [CrossRef]
- Li, S.S.; Compton, L.A.; Nemer, K.M. Porokeratoma Treated with Topical 5% 5-Fluorouracil Cream. Int. J. Womens Dermatol. 2021, 7, 830–831. [Google Scholar] [CrossRef]
- Tan, Q.; Tan, C. Porokeratotic Acanthoma. J. Dtsch. Dermatol. Ges. 2015, 13, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Xiong, S.; Tian, X. Porokeratoma in the Nipple: A Case Report. Australas. J. Dermatol. 2021, 62, e325–e326. [Google Scholar] [CrossRef] [PubMed]
- Vigne, P. Porokératose de Mibelli. Trois Cas Familiaux. Transformation Neoplasique Chez l’un d’eux. Ann. Derm. Syph. 1942, 8, 5–28. [Google Scholar]
- Friedman, B.; Golubets, K.; Ho, J.; Patton, T. Linear Porokeratosis Associated with Multiple Squamous Cell Carcinomas. Cutis 2017, 100, E11–E14. [Google Scholar] [PubMed]
- Lopes, L.N.; Gouveia, A.I.; Soares-Almeida, L.; Sacramento-Marques, M.; Filipe, P. Porokeratosis and Malignant Melanoma: A Causal or Incidental Association? Indian Dermatol. Online J. 2015, 6, 451–452. [Google Scholar] [CrossRef]
- Novice, T.; Nakamura, M.; Helfrich, Y. The Malignancy Potential of Porokeratosis: A Single-Center Retrospective Study. Cureus 2021, 13, e13083. [Google Scholar] [CrossRef] [PubMed]
- Maubec, E.; Duvillard, P.; Margulis, A.; Bachollet, B.; Degois, G.; Avril, M.-F. Common Skin Cancers in Porokeratosis. Br. J. Dermatol. 2005, 152, 1389–1391. [Google Scholar] [CrossRef]
- Schierbeck, J.; Vestergaard, T.; Bygum, A. Skin Cancer Associated Genodermatoses: A Literature Review. Acta Derm. Venereol. 2019, 99, 360–369. [Google Scholar] [CrossRef]
- Goerttler, E.A.; Jung, E.G. Porokeratosis [Correction of Parakeratosis] Mibelli and Skin Carcinoma: A Critical Review. Humangenetik 1975, 26, 291–296. [Google Scholar]
- Otsuka, F.; Someya, T.; Ishibashi, Y. Porokeratosis and Malignant Skin Tumors. J. Cancer Res. Clin. Oncol. 1991, 117, 55–60. [Google Scholar] [CrossRef]
- Riyaz, N. Porokeratosis and Malignancy: Incidental or Causal Association? Indian Dermatol. Online J. 2015, 6, 452–453. [Google Scholar] [CrossRef]
- Kanitakis, J.; Euvrard, S.; Faure, M.; Claudy, A. Porokeratosis and Immunosuppression. Eur. J. Dermatol. 1998, 8, 459–465. [Google Scholar]
- Böhm, N.; Sandritter, W. DNA in Human Tumors: A Cytophotometric Study. Curr. Top. Pathol. 1975, 60, 151–219. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, F.; Shima, A.; Ishibashi, Y. Porokeratosis as a Premalignant Condition of the Skin. Cytologic Demonstration of Abnormal DNA Ploidy in Cells of the Epidermis. Cancer 1989, 63, 891–896. [Google Scholar] [CrossRef]
- Imakado, S.; Otsuka, F.; Ishibashi, Y.; Ohara, K. Abnormal DNA Ploidy in Cells of the Epidermis in a Case of Porokeratosis. Arch. Dermatol. 1988, 124, 331–332. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, F.; Chi, H.I.; Shima, A.; Ishibashi, Y. Cytological Demonstration of Abnormal DNA Ploidy in the Epidermis of Porokeratosis. Arch. Dermatol. Res. 1988, 280, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Beers, B.; Jaszcz, W.; Sheetz, K.; Hogan, D.J.; Lynch, P.J. Porokeratosis Palmaris et Plantaris Disseminata. Report of a Case with Abnormal DNA Ploidy in Lesional Epidermis. Arch. Dermatol. 1992, 128, 236–239. [Google Scholar] [CrossRef]
- Leão, R.; Apolónio, J.D.; Lee, D.; Figueiredo, A.; Tabori, U.; Castelo-Branco, P. Mechanisms of Human Telomerase Reverse Transcriptase (hTERT) Regulation: Clinical Impacts in Cancer. J. Biomed. Sci. 2018, 25, 22. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-R.; Min, S.K.; Cho, H.D.; Kim, K.H.; Shin, H.S.; Park, Y.E. Expression Profiles of P63, P53, Survivin, and hTERT in Skin Tumors. J. Cutan. Pathol. 2004, 31, 544–549. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a Signature of in Vivo Senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- Sakiz, D.; Turkmenoglu, T.T.; Kabukcuoglu, F. The Expression of P63 and P53 in Keratoacanthoma and Intraepidermal and Invasive Neoplasms of the Skin. Pathol. Res. Pract. 2009, 205, 589–594. [Google Scholar] [CrossRef]
- Arranz-Salas, I.; Sanz-Trelles, A.; Ojeda, D.B. P53 Alterations in Porokeratosis. J. Cutan. Pathol. 2003, 30, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Urano, Y.; Sasaki, S.; Ninomiya, Y.; Oura, H.; Arase, S. Immunohistochemical Detection of P53 Tumor Suppressor Protein in Porokeratosis. J. Dermatol. 1996, 23, 365–368. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Park, E.J.; Seo, Y.J.; Cho, H.S.; Kim, C.W.; Kim, K.J.; Park, H.R. Immunohistochemical Study of Cyclooxygenase-2 and P53 Expression in Skin Tumors. J. Dermatol. 2006, 33, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, Y.; Urano, Y.; Yoshimoto, K.; Iwahana, H.; Sasaki, S.; Arase, S.; Itakura, M. P53 Gene Mutation Analysis in Porokeratosis and Porokeratosis-Associated Squamous Cell Carcinoma. J. Dermatol. Sci. 1997, 14, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Pietkiewicz, P.; Gornowicz-Porowska, J.; Bowszyc-Dmochowska, M.; Jagielska, J.; Helak-Łapaj, C.; Kaczmarek, E.; Dmochowski, M. Discordant Expression of Desmoglein 2 and 3 at the mRNA and Protein Levels in Nodular and Superficial Basal Cell Carcinoma Revealed by Immunohistochemistry and Fluorescent in Situ Hybridization. Clin. Exp. Dermatol. 2014, 39, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Mowla, S.N.; Lam, E.W.-F.; Jat, P.S. Cellular Senescence and Aging: The Role of B-MYB. Aging Cell 2014, 13, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Scola, N.; Skrygan, M.; Wieland, U.; Kreuter, A.; Gambichler, T. Altered Gene Expression in Squamous Cell Carcinoma Arising from Congenital Unilateral Linear Porokeratosis. Clin. Exp. Dermatol. 2012, 37, 781–785. [Google Scholar] [CrossRef]
- Uryu, M.; Furue, M. p16INK4a Expression in Porokeratosis. Ann. Dermatol. 2017, 29, 373–376. [Google Scholar] [CrossRef]
- Jaiswal, P.K.; Goel, A.; Mittal, R.D. Survivin: A Molecular Biomarker in Cancer. Indian J. Med. Res. 2015, 141, 389–397. [Google Scholar] [CrossRef] [PubMed]



















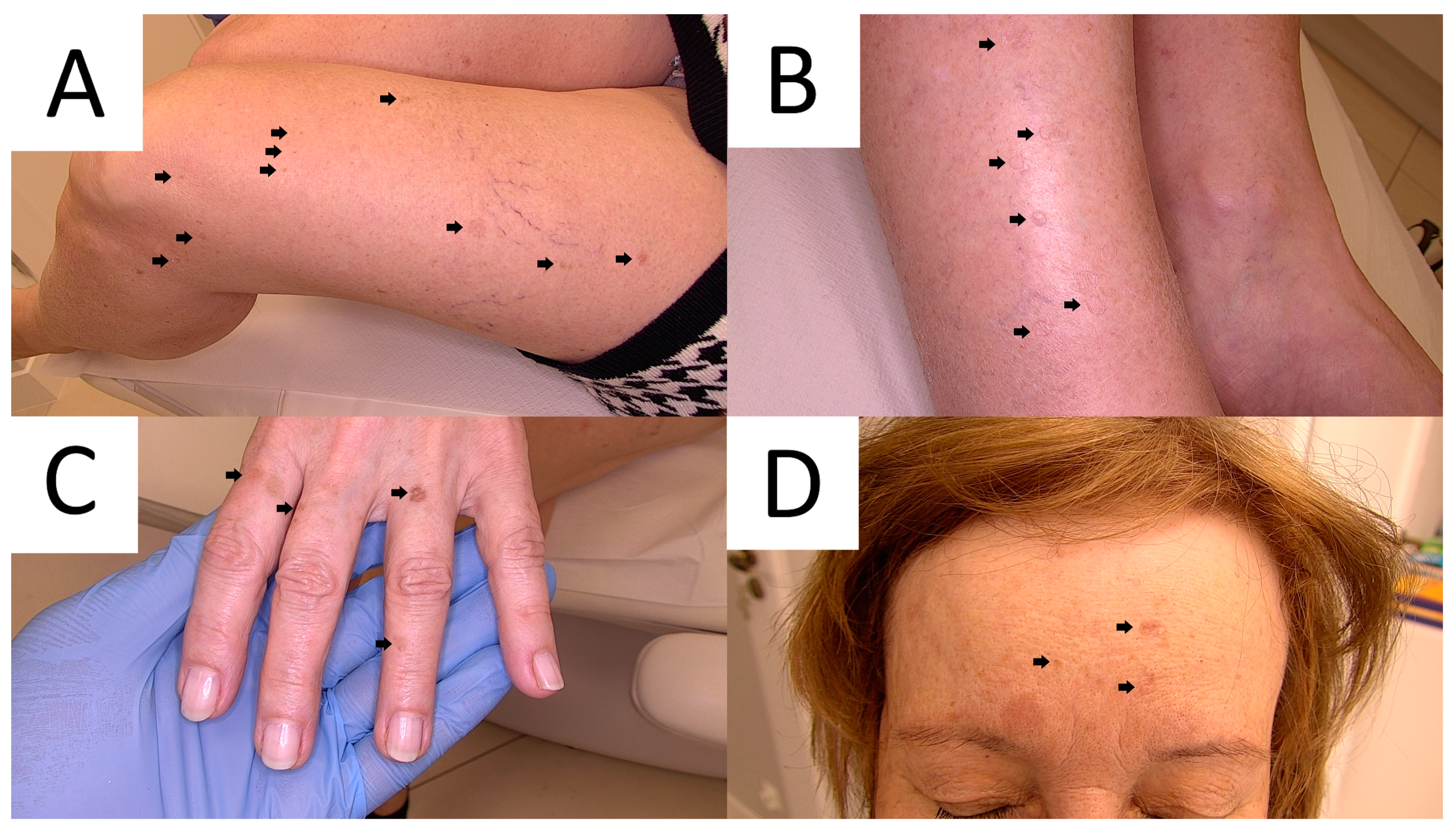{kind=link}
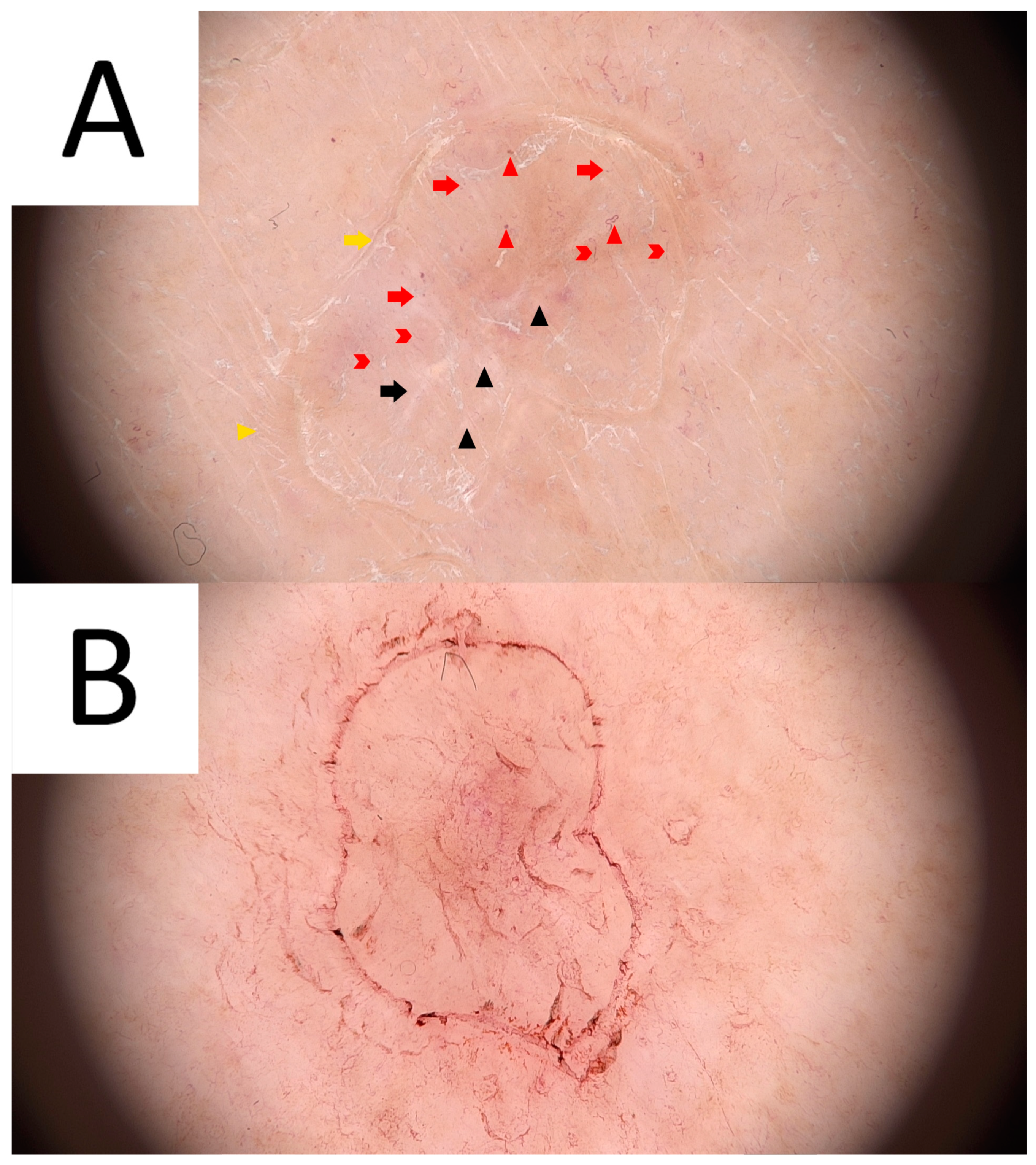{kind=link}
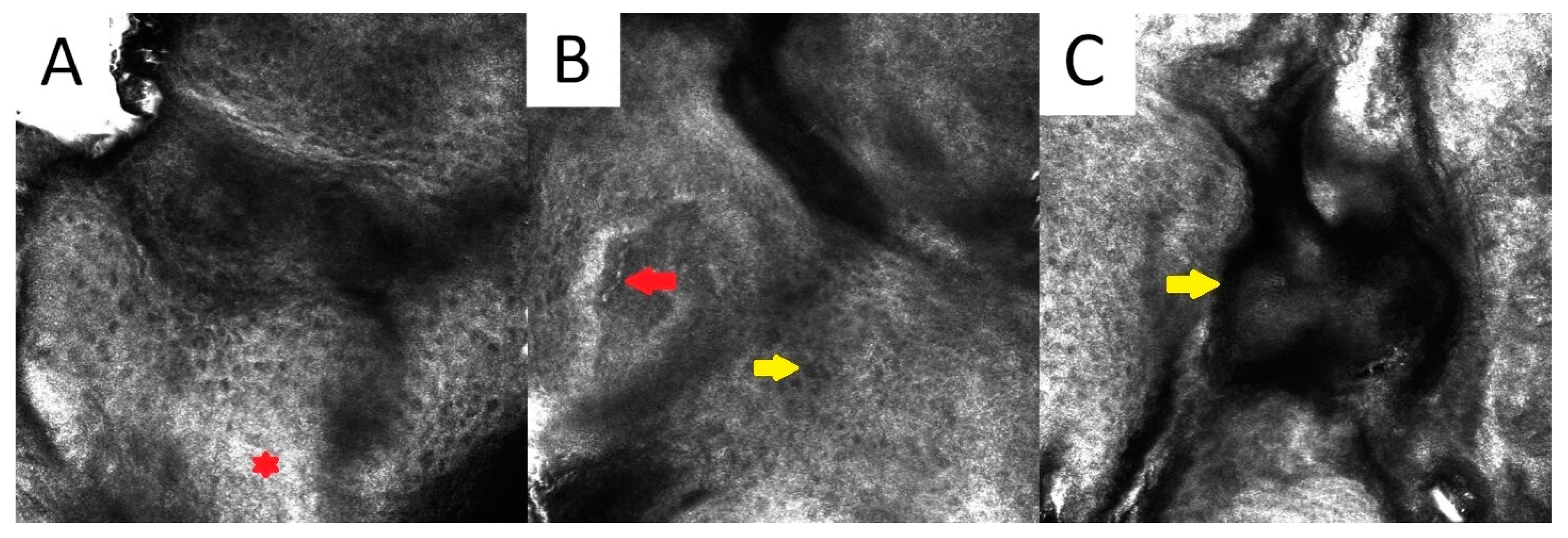{kind=link}
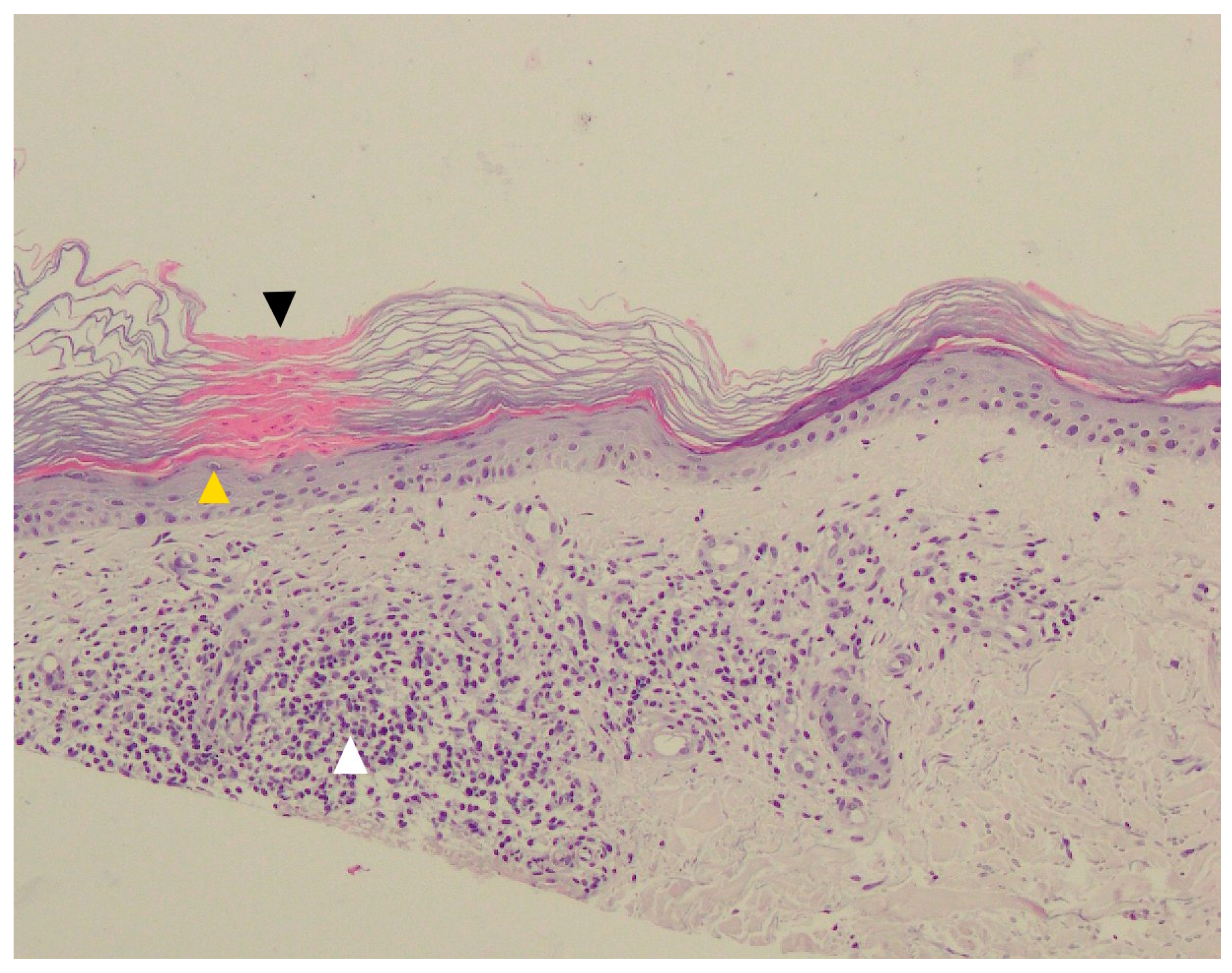{kind=link}
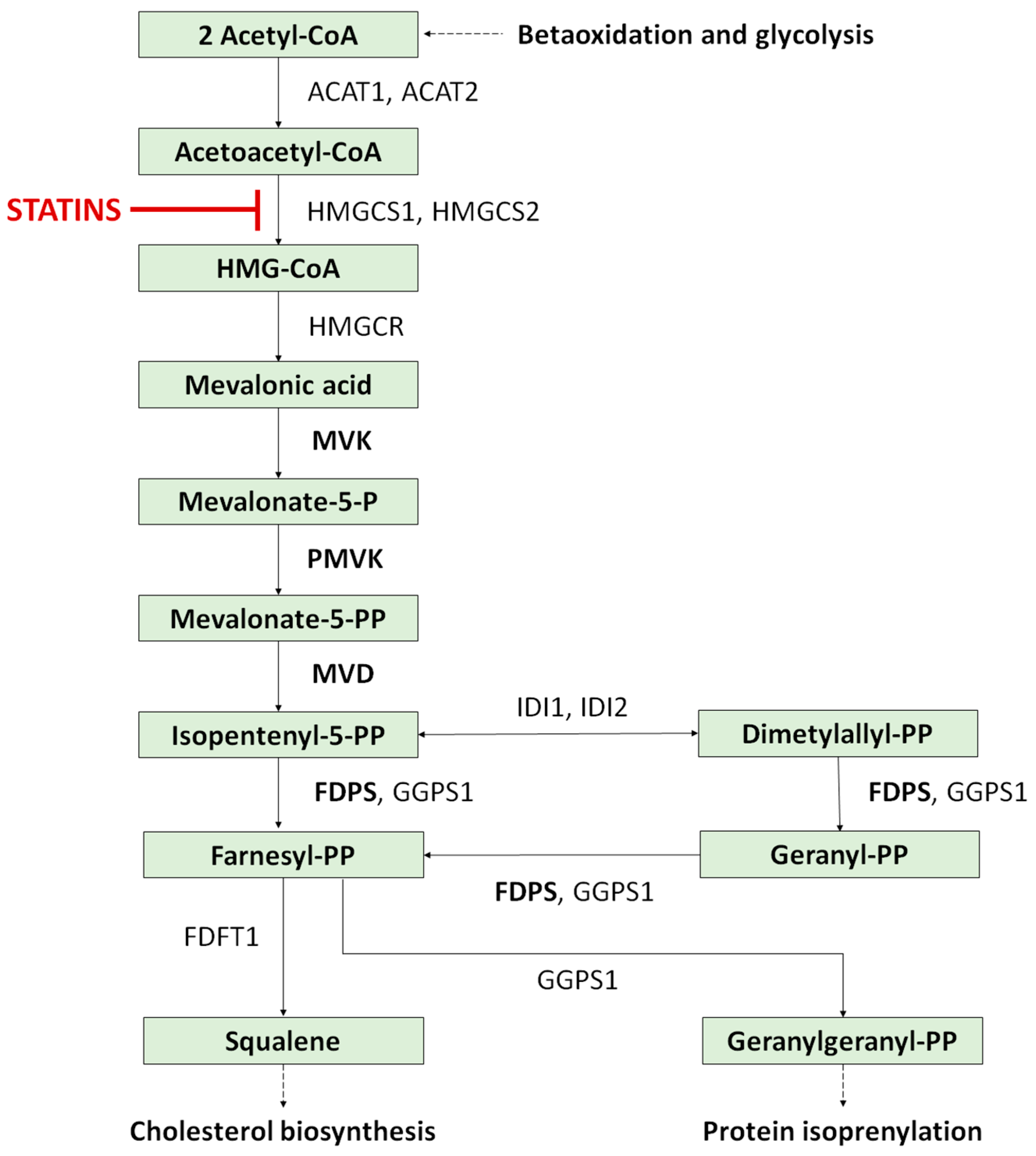{kind=link}
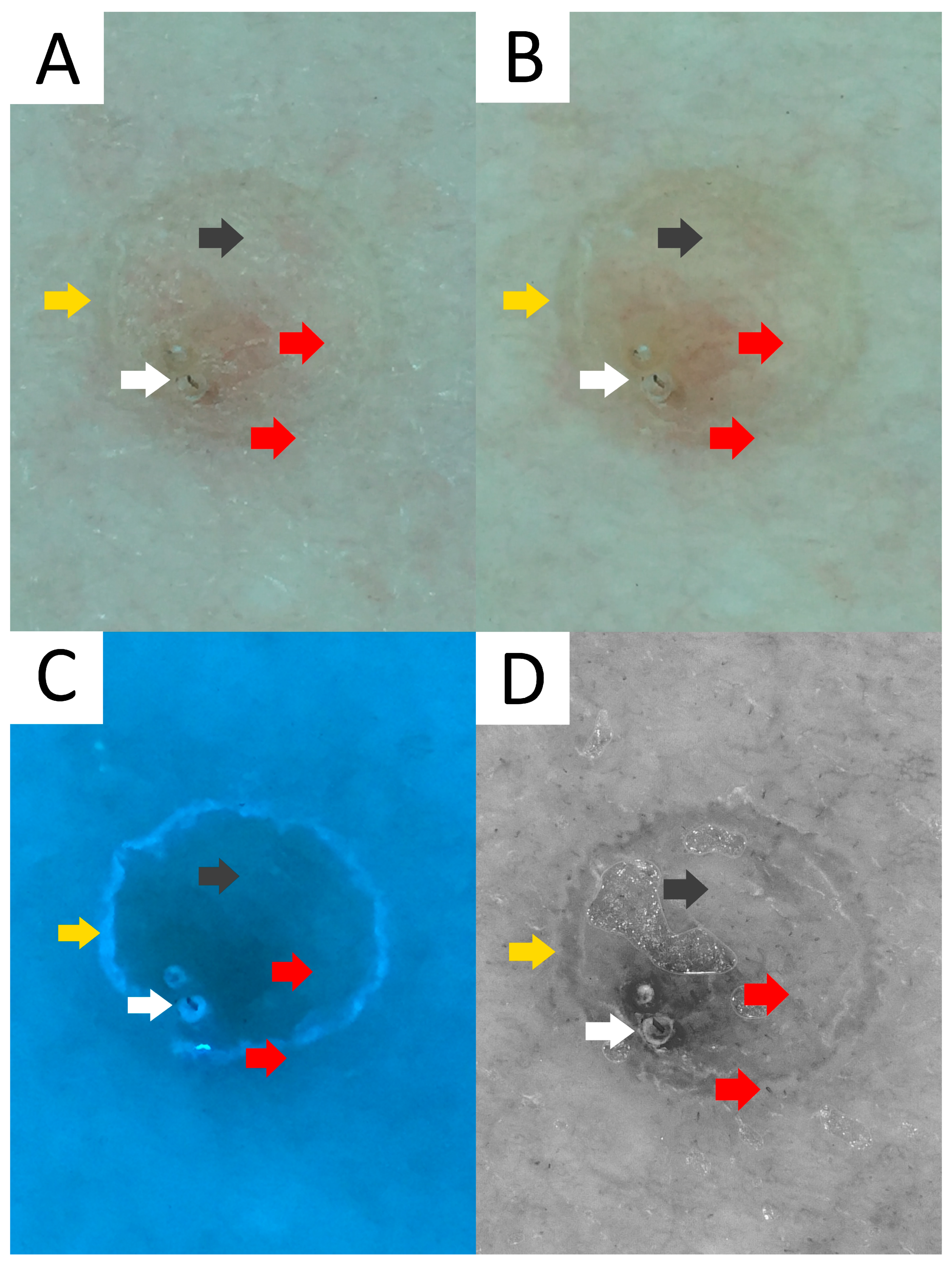{kind=link}
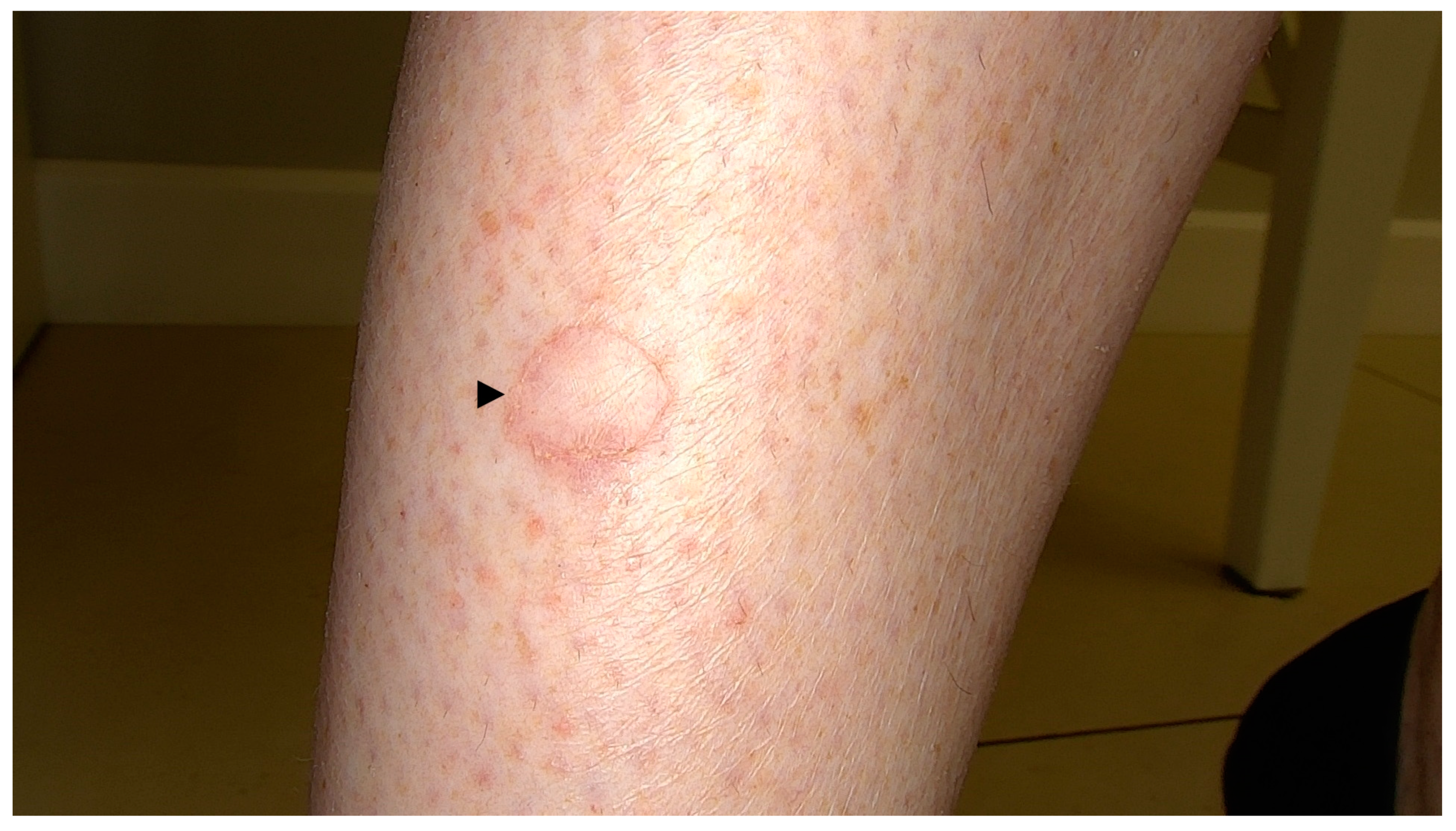{kind=link}
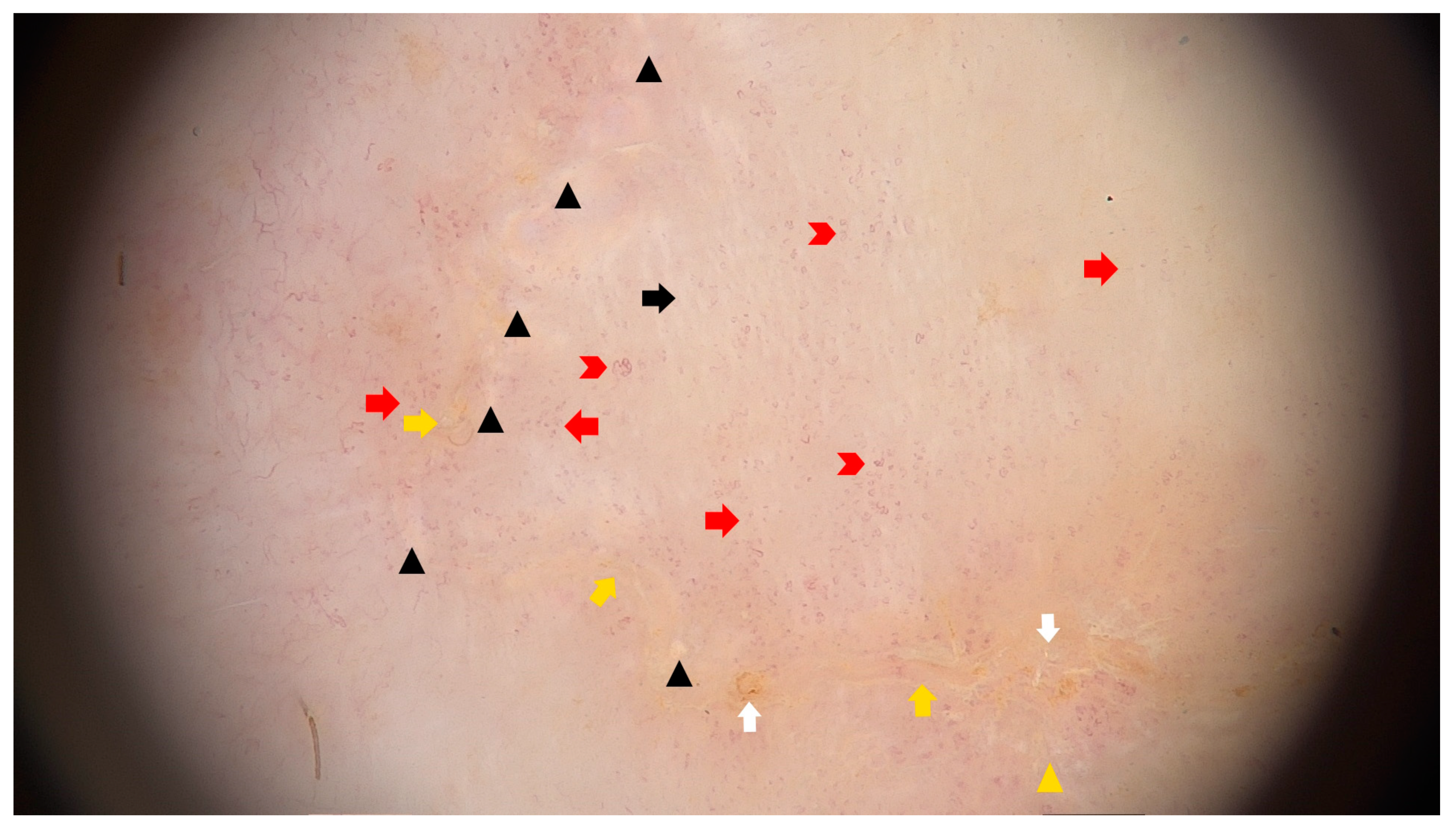{kind=link}
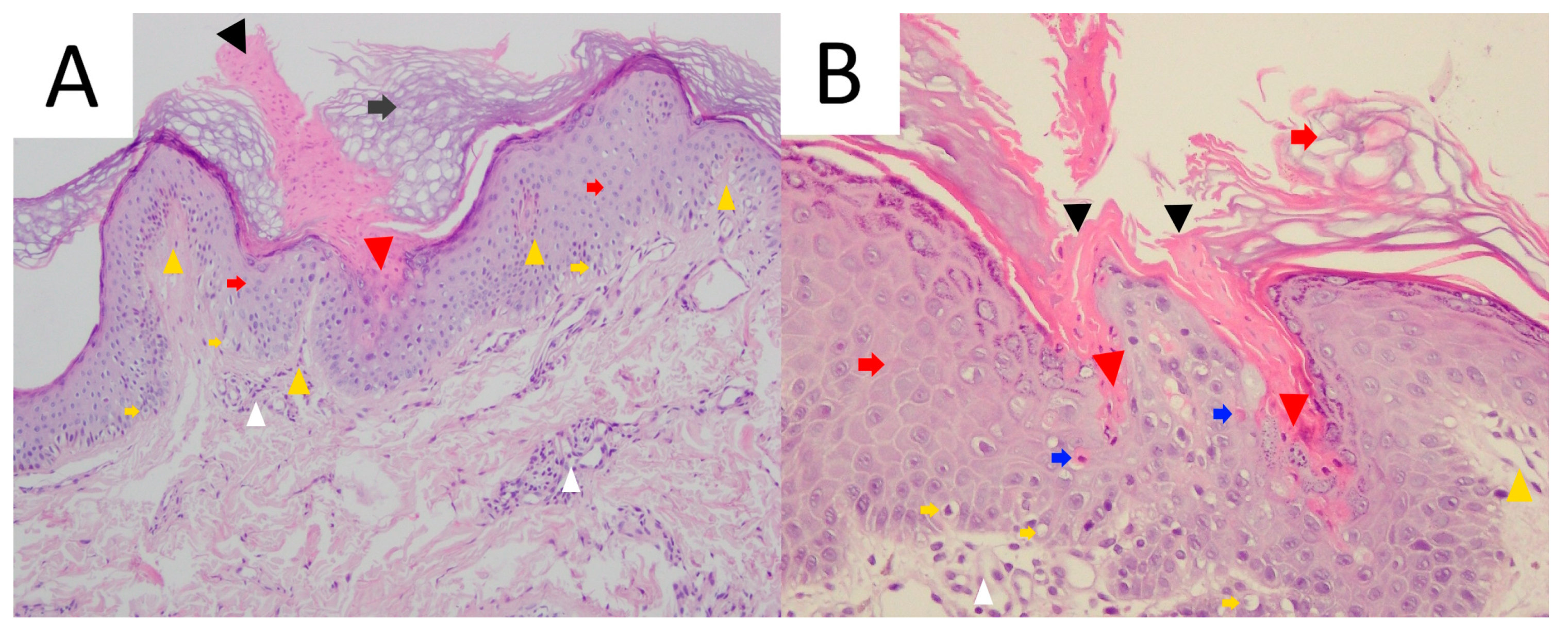{kind=link}
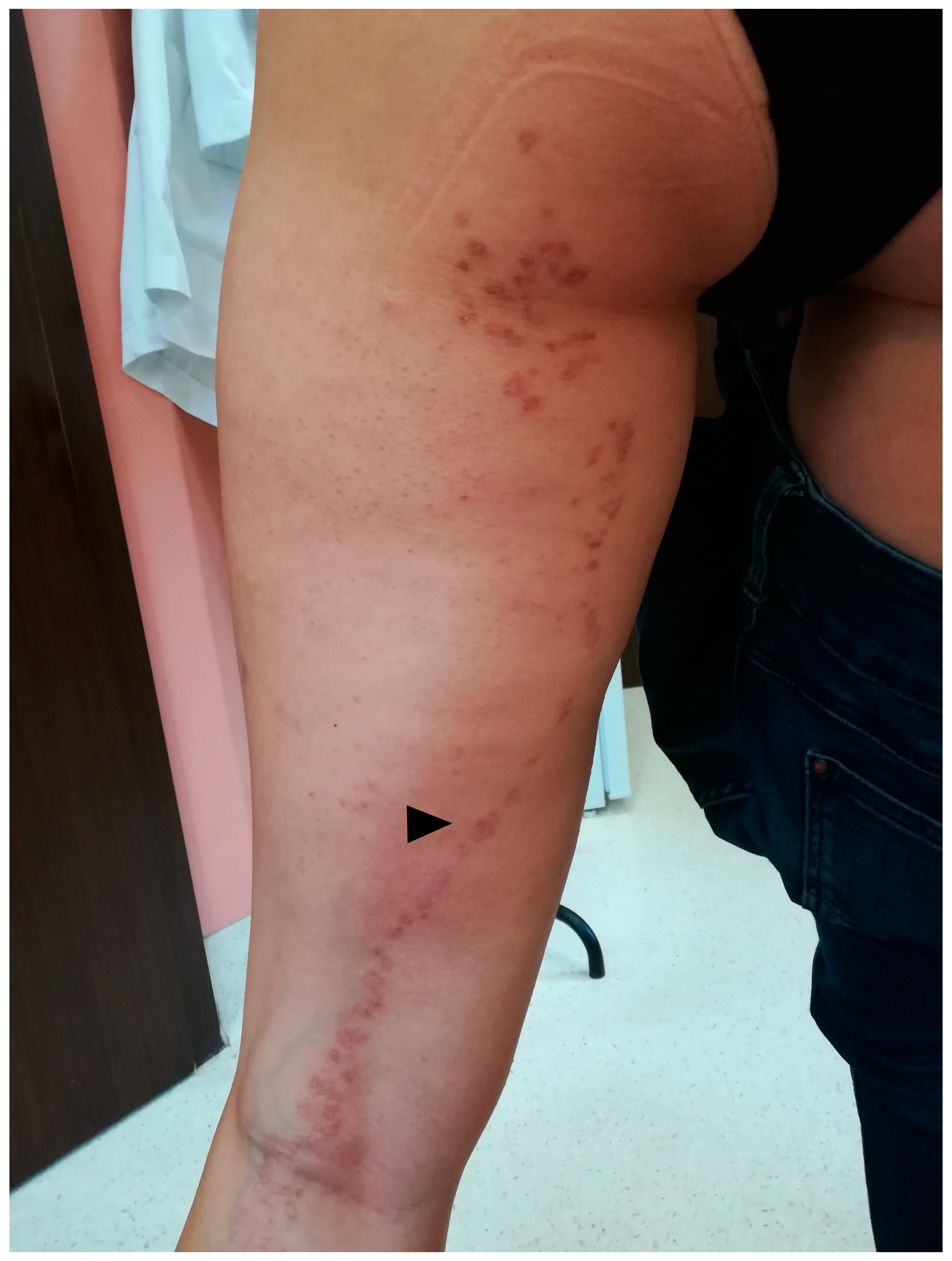{kind=link}
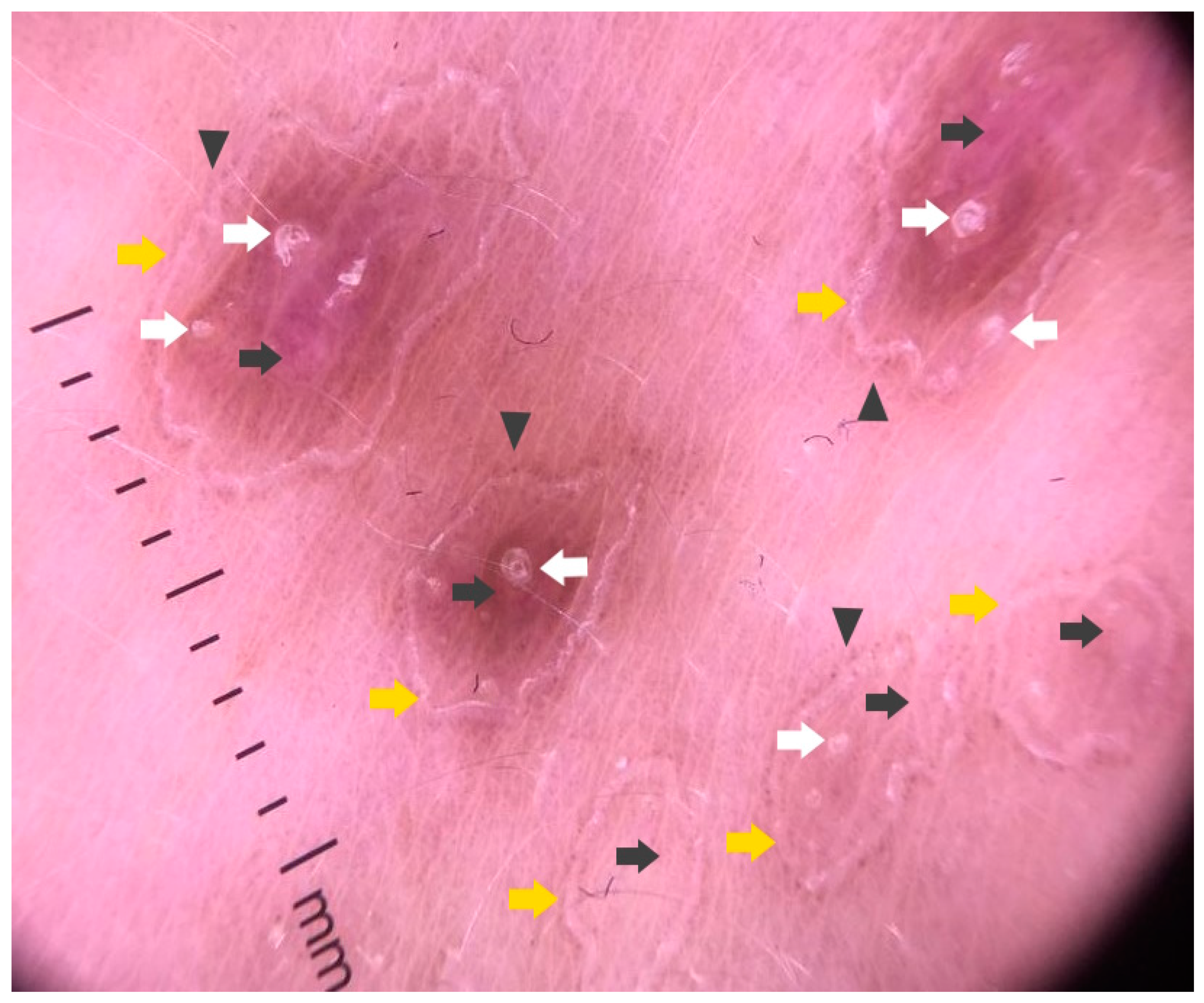{kind=link}
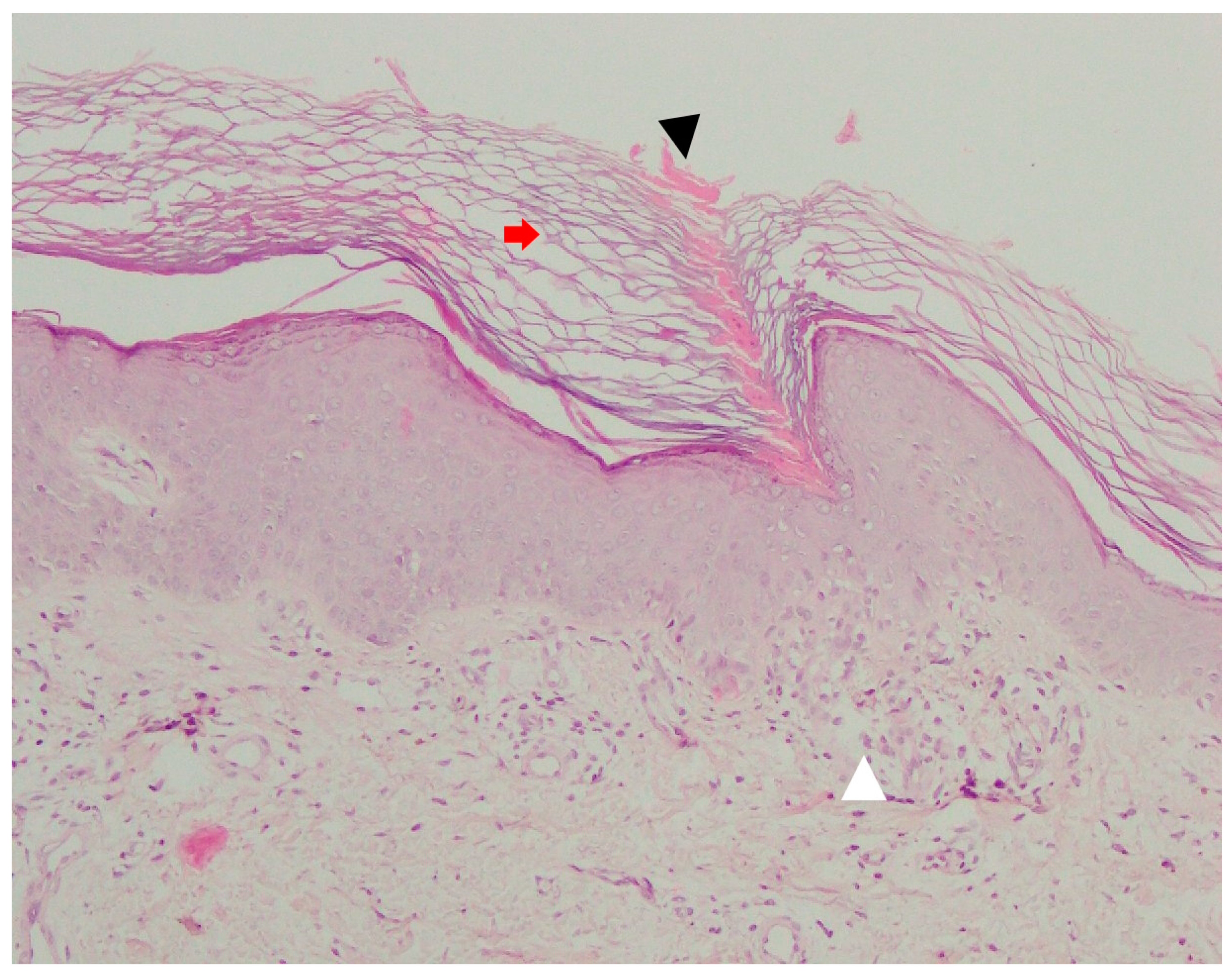{kind=link}
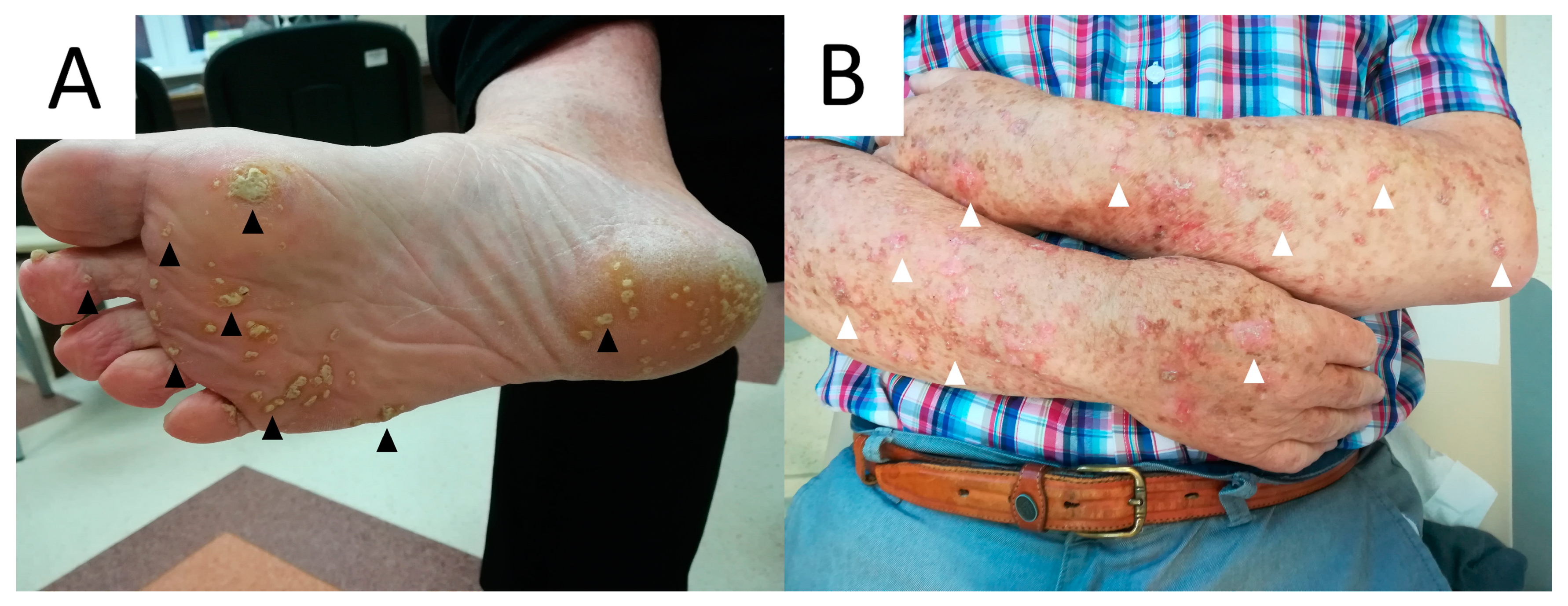{kind=link}
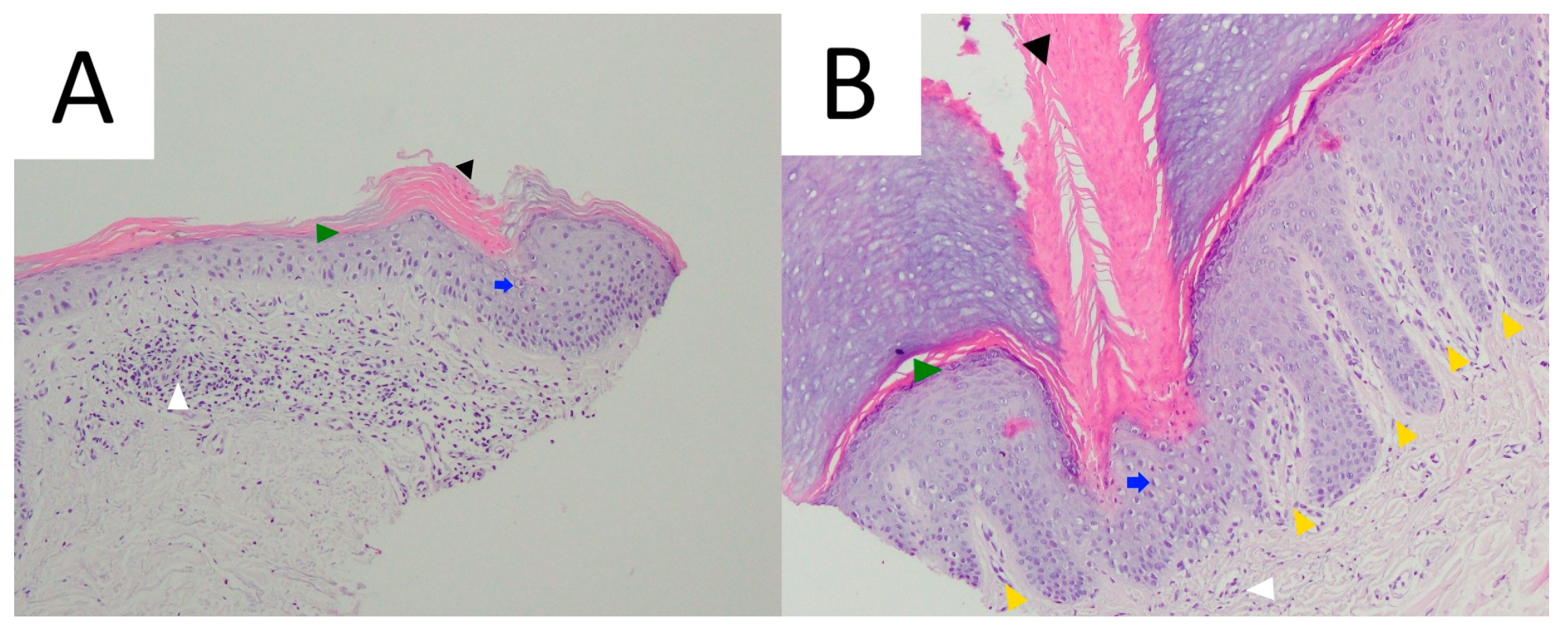{kind=link}
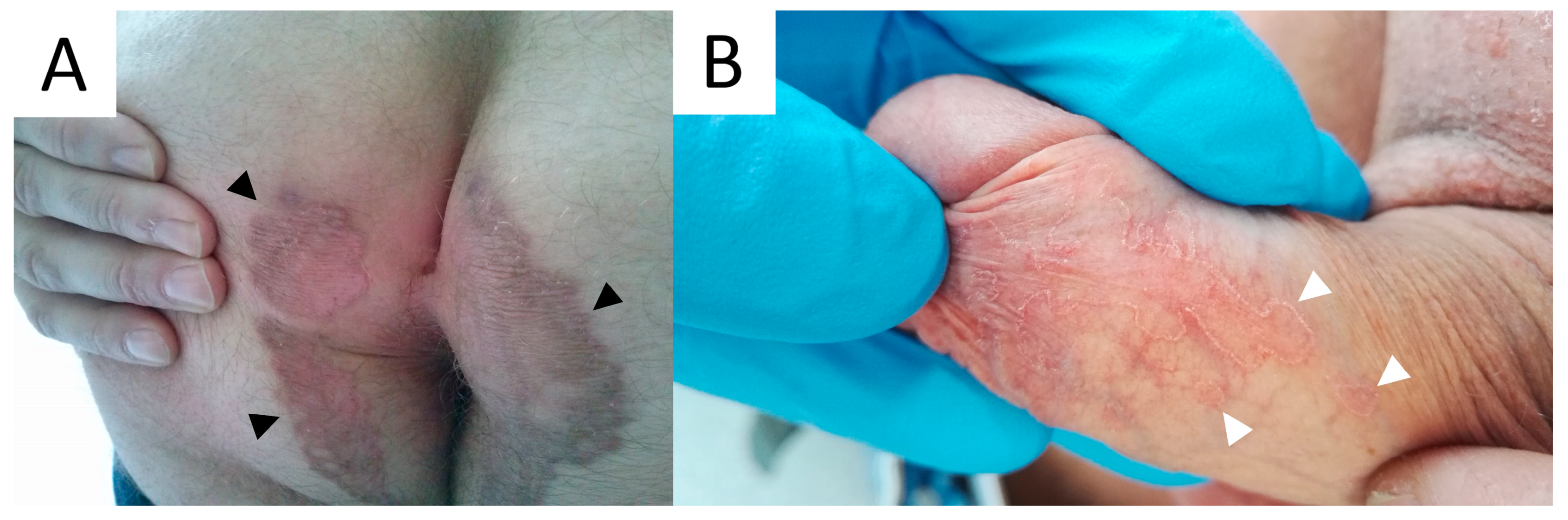{kind=link}
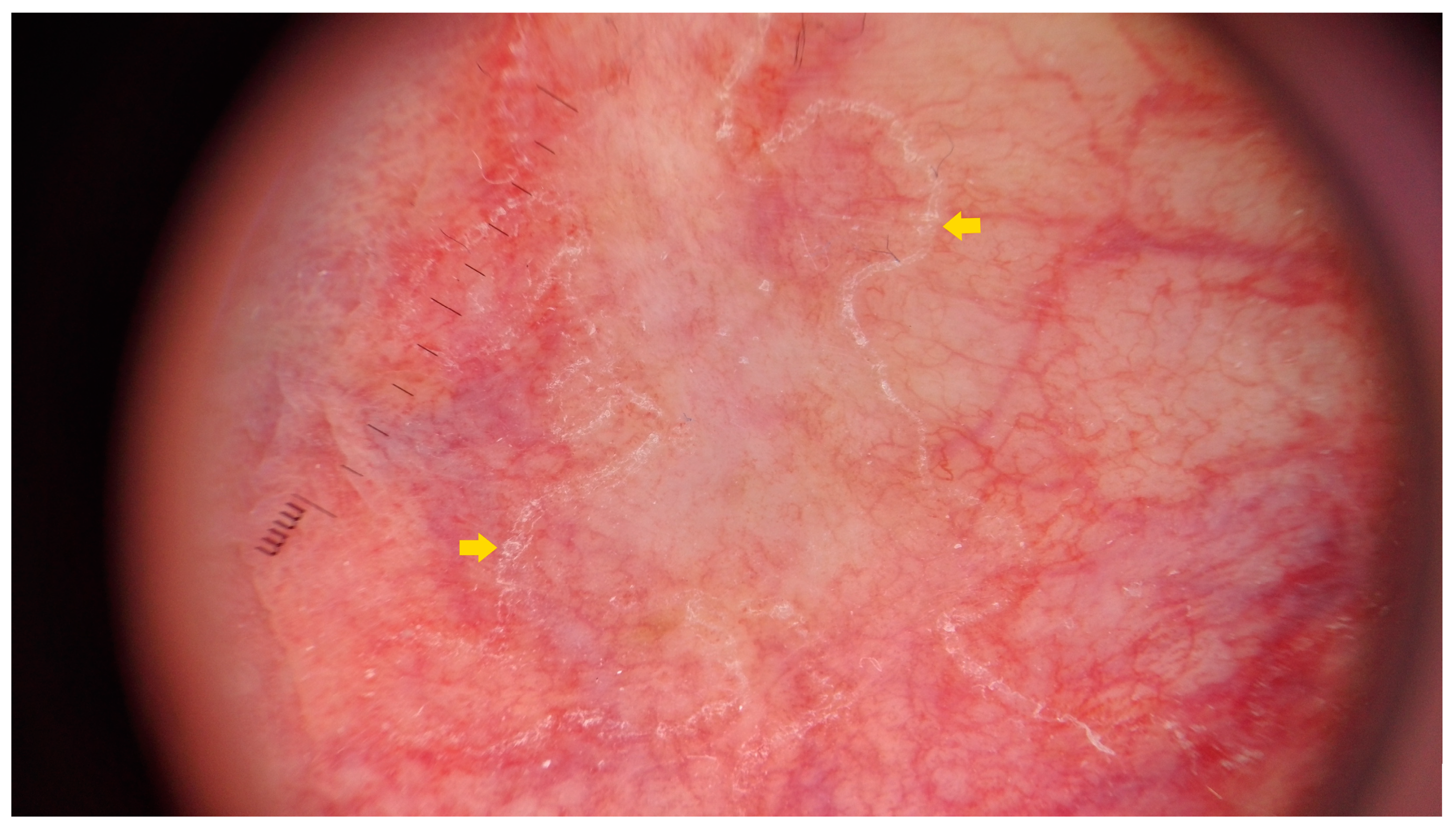{kind=link}
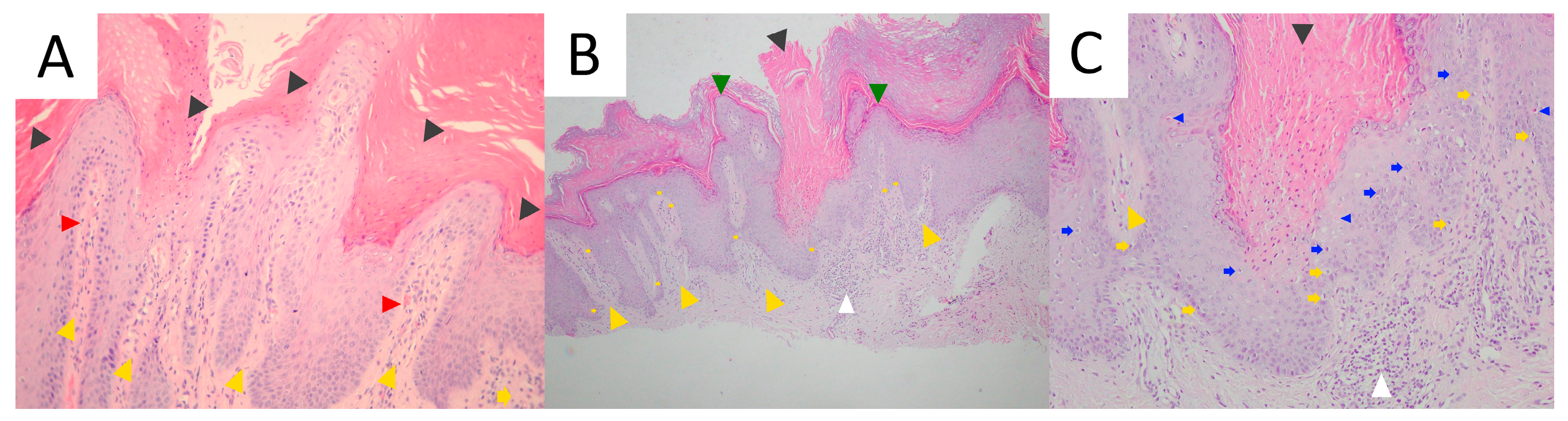{kind=link}
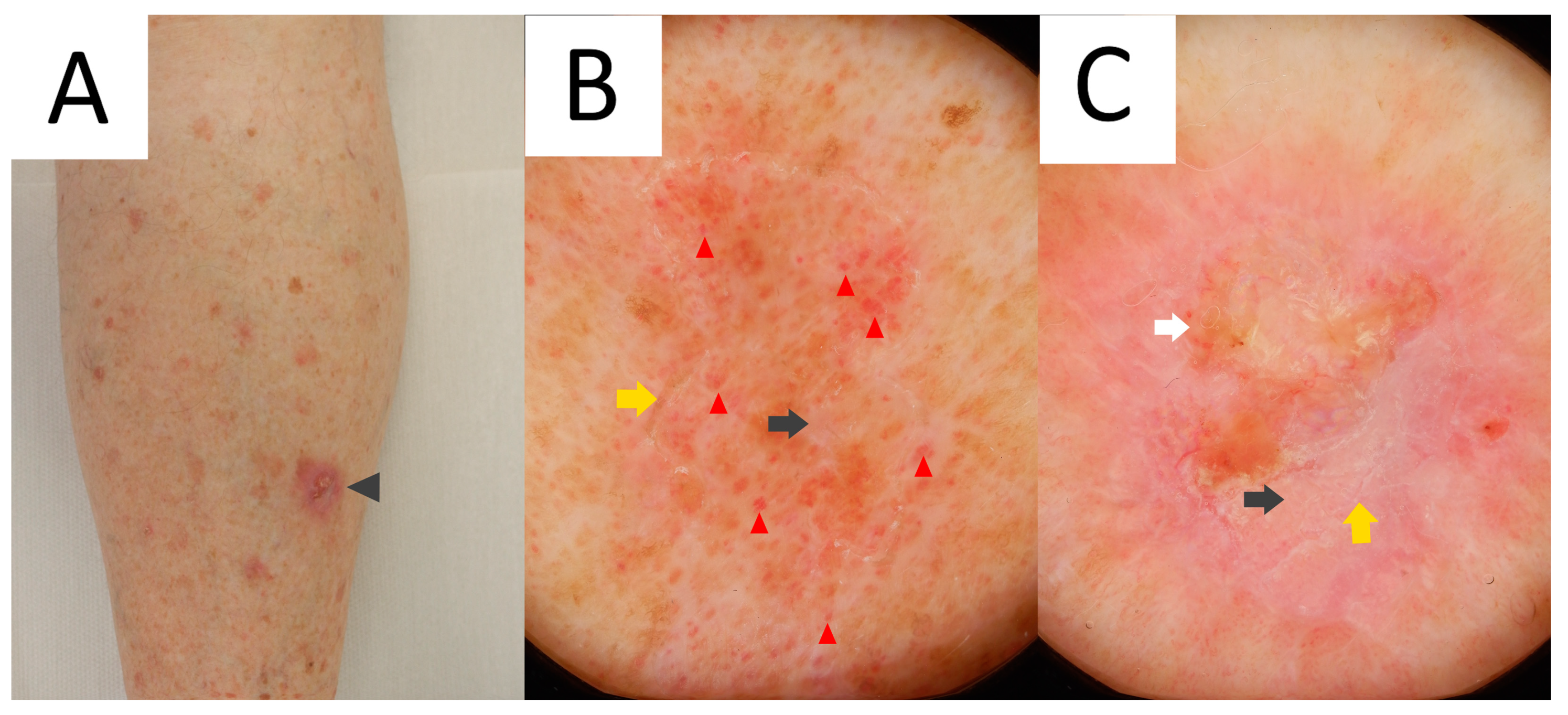{kind=link}
| Clinical Variant | Onset | Gender | Key Clinical Features | Distribution | Key Pathological Features | Estimated Risk for Malignant Transformation |
|---|---|---|---|---|---|---|
| DSAP | 3rd to 4th (familial cases) or 3rd to 5th decade (sporadic cases) | F > M | Multiple atrophic/keratotic macules and papules < 1 cm in size | Sun-exposed areas | Single >> doubled CL | 3.4% |
| DSP | 4th to 5th decade | F > M | Same as DSAP | Generalised | Same as DSAP | Low |
| EDP | 6th to 7th decade | M > F | Self-limiting, extremely pruritic lesions; possible paraneoplastic character | Generalised | Same as DSAP, eosinophilic infiltrate | None (no data) |
| PM | Childhood (familial cases) or at any age (sporadic cases) | M > F | Solitary plaque with prominent border | Limbs | Single or multiple CLs; epidermal invagination and papillomatosis | 7.6% |
| LP | Childhood | F ≥ M | Linear, verrucous plaques | Limbs (unilateral and blaschkoid) | Multiple CLs | 11–19% |
| PPPD | Adolescence and 2nd decade | M > F | Multiple keratotic papules | Palms and soles (initially), limbs and trunk (later) | CL | Low |
| PuP | 3rd to 5th decade | M > F | Multiple tender pits filled with keratotic plugs | Palms and soles (bilateral and linear or diffuse) | CL; keratin plugs | None(no data) |
| VP | 3rd to 5th decade | M > F | Multiple, itching or burning, verrucous or psoriasiform patches | Anogenital region and skin folds (butterfly-shape appearance) | Multiple large CLs; papillomatosis and/or psoriasiform epidermal hyperplasia and papillary dermis telangiectasia | Low |
| FP | 2nd to 6th decade | M > F | Folliculocentric papules and/or nodules < 1 cm in size | Any site (except palms and soles) | Infundibulocentric CL | None (no data) |
| Porokeratoma | 5th decade | M > F | Isolated verrucous plaque of nodule | Limbs | Multiple discrete or broad confluent CLs; hypertrophic centre | None (no data) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietkiewicz, P.; Korecka, K.; Salwowska, N.; Kohut, I.; Adhikari, A.; Bowszyc-Dmochowska, M.; Pogorzelska-Antkowiak, A.; Navarrete-Dechent, C. Porokeratoses—A Comprehensive Review on the Genetics and Metabolomics, Imaging Methods and Management of Common Clinical Variants. Metabolites 2023, 13, 1176. https://doi.org/10.3390/metabo13121176
Pietkiewicz P, Korecka K, Salwowska N, Kohut I, Adhikari A, Bowszyc-Dmochowska M, Pogorzelska-Antkowiak A, Navarrete-Dechent C. Porokeratoses—A Comprehensive Review on the Genetics and Metabolomics, Imaging Methods and Management of Common Clinical Variants. Metabolites. 2023; 13(12):1176. https://doi.org/10.3390/metabo13121176
Chicago/Turabian StylePietkiewicz, Paweł, Katarzyna Korecka, Natalia Salwowska, Ihor Kohut, Adarsha Adhikari, Monika Bowszyc-Dmochowska, Anna Pogorzelska-Antkowiak, and Cristian Navarrete-Dechent. 2023. "Porokeratoses—A Comprehensive Review on the Genetics and Metabolomics, Imaging Methods and Management of Common Clinical Variants" Metabolites 13, no. 12: 1176. https://doi.org/10.3390/metabo13121176