
Metabolic Role of Autophagy in the Pathogenesis and Development of NAFLD
 , , , , ,
, , , , , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Forms and Working Mechanisms of Autophagy
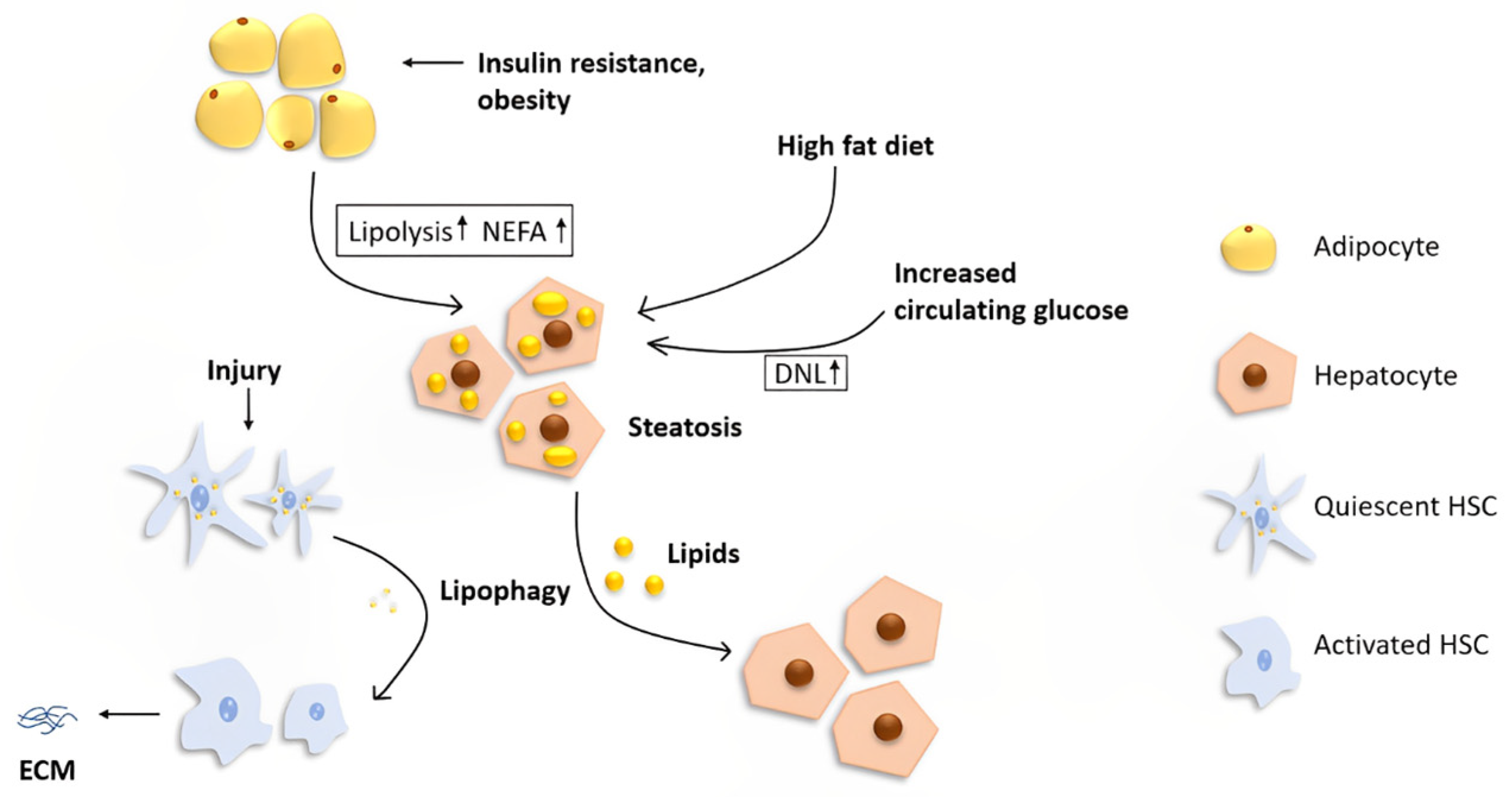
3. Lipid Metabolism in the Liver
4. The Role of Autophagy and Its Regulation in Lipid Metabolism within the Liver
5. Autophagy and Hepatic Stellate Cell Activation
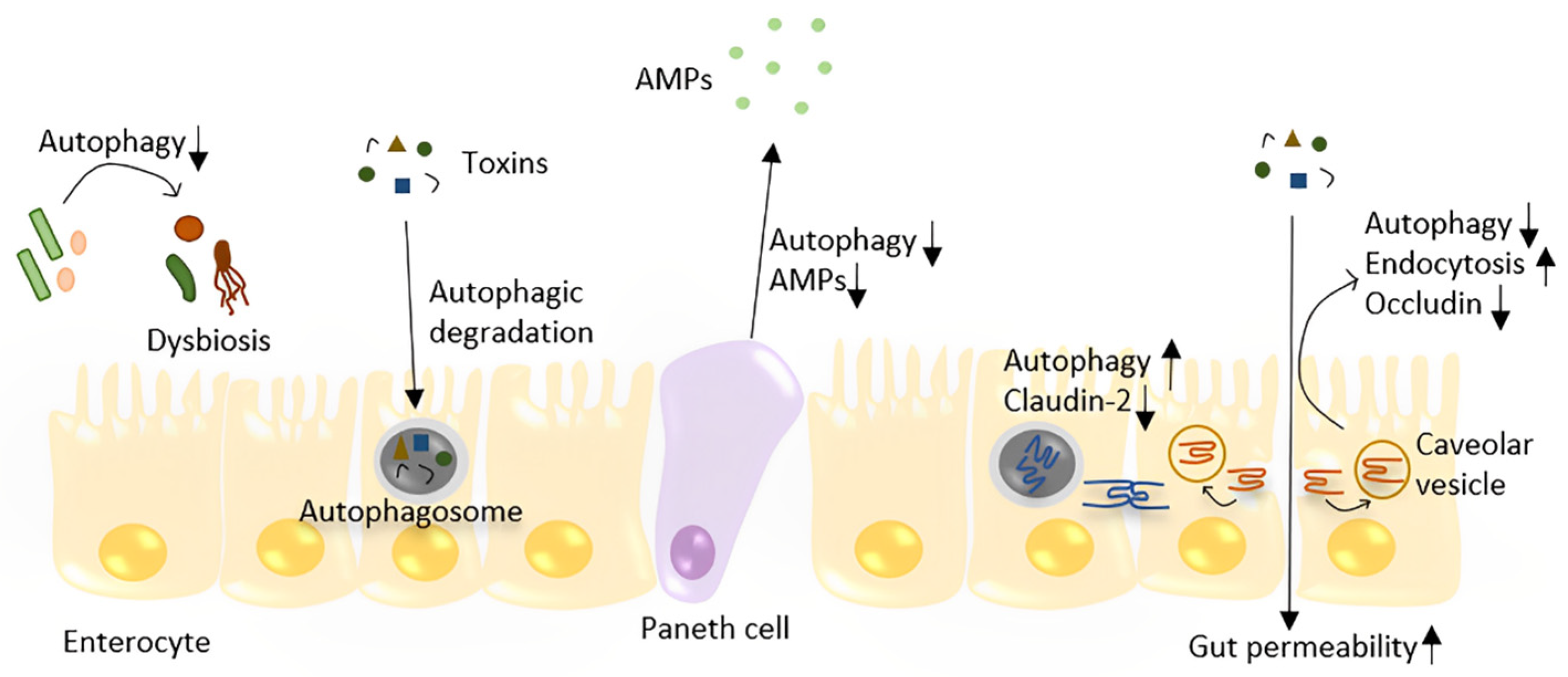
6. Autophagy and Gut Barrier Function
7. Potential Therapeutics and Future Directions
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hardy, T.; Oakley, F.; Anstee, Q.M.; Day, C.P. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 451–496. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Kader, S.M.; El-Den Ashmawy, E.M. Non-alcoholic fatty liver disease: The diagnosis and management. World J. Hepatol. 2015, 7, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Cotter, T.G.; Charlton, M. Nonalcoholic Steatohepatitis After Liver Transplantation. Liver Transpl. 2020, 26, 141–159. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarus, J.V.; Mark, H.E.; Anstee, Q.M.; Arab, J.P.; Batterham, R.L.; Castera, L.; Cortez-Pinto, H.; Crespo, J.; Cusi, K.; Dirac, M.A.; et al. Advancing the global public health agenda for NAFLD: A consensus statement. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 60–78. [Google Scholar] [CrossRef] [PubMed]
- Attia, S.L.; Softic, S.; Mouzaki, M. Evolving Role for Pharmacotherapy in NAFLD/NASH. Clin. Transl. Sci. 2021, 14, 11–19. [Google Scholar] [CrossRef]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, 1–8. [Google Scholar]
- Czaja, M.J.; Ding, W.X.; Donohue, T.M., Jr.; Friedman, S.L.; Kim, J.S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.H.; et al. Functions of autophagy in normal and diseased liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef] [Green Version]
- Deter, R.L.; Baudhuin, P.; De Duve, C. Participation of lysosomes in cellular autophagy induced in rat liver by glucagon. J. Cell Biol. 1967, 35, C11–C16. [Google Scholar] [CrossRef] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Marzella, L.; Ahlberg, J.; Glaumann, H. Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Archiv B Cell Pathol. 1981, 36, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Schuck, S. Microautophagy—distinct molecular mechanisms handle cargoes of many sizes. J. Cell Sci. 2020, 133, jcs246322. [Google Scholar] [CrossRef] [PubMed]
- Schulze, R.J.; Krueger, E.W.; Weller, S.G.; Johnson, K.M.; Casey, C.A.; Schott, M.B.; McNiven, M.A. Direct lysosome-based autophagy of lipid droplets in hepatocytes. Proc. Natl. Acad. Sci. USA 2020, 117, 32443–32452. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Cregg, J.M.; Dunn, W.A., Jr.; Emr, S.D.; Sakai, Y.; Sandoval, I.V.; Sibirny, A.; Subramani, S.; Thumm, M.; Veenhuis, M.; et al. A unified nomenclature for yeast autophagy-related genes. Dev. Cell 2003, 5, 539–545. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Kirisako, T.; Kamada, Y.; Mizushima, N.; Noda, T.; Ohsumi, Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001, 20, 5971–5981. [Google Scholar] [CrossRef]
- Morel, E. Endoplasmic Reticulum Membrane and Contact Site Dynamics in Autophagy Regulation and Stress Response. Front. Cell Dev. Biol. 2020, 8, 343. [Google Scholar] [CrossRef]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.Y.; Li, L.; Zhu, X.M.; Lu, J.P.; Liu, X.H.; Lin, F.C. The crucial role of the regulatory mechanism of the Atg1/ULK1 complex in fungi. Front. Microbiol. 2022, 13, 1019543. [Google Scholar] [CrossRef]
- Wong, S.Q.; Kumar, A.V.; Mills, J.; Lapierre, L.R. Autophagy in aging and longevity. Hum. Genet. 2020, 139, 277–290. [Google Scholar] [CrossRef]
- Ohsumi, Y. Molecular dissection of autophagy: Two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Mancias, J.D.; Kimmelman, A.C. Mechanisms of Selective Autophagy in Normal Physiology and Cancer. J. Mol. Biol. 2016, 428, 1659–1680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef]
- Jing, K.; Lim, K. Why is autophagy important in human diseases? Exp. Mol. Med. 2012, 44, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Mathew, R.; White, E. Autophagy in tumorigenesis and energy metabolism: Friend by day, foe by night. Curr. Opin. Genet. Dev. 2011, 21, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H. Regulation of mammalian acetyl-coenzyme A carboxylase. Annu. Rev. Nutr. 1997, 17, 77–99. [Google Scholar] [CrossRef]
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; Le Bloc’h, J.; Siliart, B.; Dumon, H. Liver lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283. [Google Scholar] [CrossRef] [PubMed]
- Geisler, C.E.; Renquist, B.J. Hepatic lipid accumulation: Cause and consequence of dysregulated glucoregulatory hormones. J. Endocrinol. 2017, 234, R1–R21. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.E.; Ahmadian, M.; Jaworski, K.; Sarkadi-Nagy, E.; Sul, H.S. Regulation of lipolysis in adipocytes. Annu. Rev. Nutr. 2007, 27, 79–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sears, B.; Perry, M. The role of fatty acids in insulin resistance. Lipids Health Dis. 2015, 14, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zambo, V.; Simon-Szabo, L.; Szelenyi, P.; Kereszturi, E.; Banhegyi, G.; Csala, M. Lipotoxicity in the liver. World J. Hepatol. 2013, 5, 550–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Czaja, M.J. Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 2013, 20, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Han, J.; Wang, L.; Yang, X.; Yan, Z.; Qu, M.; Zhou, H.; Bilal, H.; Wang, F.; Ge, H.; et al. The vesicular transporter STX11 governs ATGL-mediated hepatic lipolysis and lipophagy. iScience 2022, 25, 104085. [Google Scholar] [CrossRef]
- Edwards, M.; Mohiuddin, S.S. Biochemistry, Lipolysis; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Shelness, G.S.; Sellers, J.A. Very-low-density lipoprotein assembly and secretion. Curr. Opin. Lipidol. 2001, 12, 151–157. [Google Scholar] [CrossRef]
- Linton, M.R.F.; Yancey, P.G.; Davies, S.S.; Jerome, W.G.; Linton, E.F.; Song, W.L.; Doran, A.C.; Vickers, K.C. The Role of Lipids and Lipoproteins in Atherosclerosis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-alcoholic fatty liver disease and risk of incident cardiovascular disease: A meta-analysis. J. Hepatol. 2016, 65, 589–600. [Google Scholar] [CrossRef] [Green Version]
- Kloska, A.; Wesierska, M.; Malinowska, M.; Gabig-Ciminska, M.; Jakobkiewicz-Banecka, J. Lipophagy and Lipolysis Status in Lipid Storage and Lipid Metabolism Diseases. Int. J. Mol. Sci. 2020, 21, 6113. [Google Scholar] [CrossRef]
- Zechner, R.; Kienesberger, P.C.; Haemmerle, G.; Zimmermann, R.; Lass, A. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J. Lipid Res. 2009, 50, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Schott, M.B.; Weller, S.G.; Schulze, R.J.; Krueger, E.W.; Drizyte-Miller, K.; Casey, C.A.; McNiven, M.A. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J. Cell Biol. 2019, 218, 3320–3335. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Cuervo, A.M. Liver autophagy: Much more than just taking out the trash. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 187–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.Y.; Han, J.; Cao, S.Y.; Hong, T.; Zhuo, D.; Shi, J.; Liu, Z.; Cao, W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: Inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J. Biol. Chem. 2009, 284, 31484–31492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Li, P.; Fu, S.; Calay, E.S.; Hotamisligil, G.S. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010, 11, 467–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizaso, A.; Tan, K.T.; Lee, Y.H. beta-adrenergic receptor-stimulated lipolysis requires the RAB7-mediated autolysosomal lipid degradation. Autophagy 2013, 9, 1228–1243. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, B.; Schulze, R.J.; Weller, S.G.; Sletten, A.C.; Casey, C.A.; McNiven, M.A. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology 2015, 61, 1896–1907. [Google Scholar] [CrossRef] [Green Version]
- Sinha, R.A.; You, S.H.; Zhou, J.; Siddique, M.M.; Bay, B.H.; Zhu, X.; Privalsky, M.L.; Cheng, S.Y.; Stevens, R.D.; Summers, S.A.; et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J. Clin. Investig. 2012, 122, 2428–2438. [Google Scholar] [CrossRef] [Green Version]
- Byun, S.; Seok, S.; Kim, Y.C.; Zhang, Y.; Yau, P.; Iwamori, N.; Xu, H.E.; Ma, J.; Kemper, B.; Kemper, J.K. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat. Commun. 2020, 11, 807. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Wang, K.; Long, A.; Jia, L.; Zhang, Y.; Deng, H.; Li, Y.; Han, J.; Wang, Y. Fasting-induced hormonal regulation of lysosomal function. Cell Res. 2017, 27, 748–763. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Wang, K.; He, J.; Zhang, G.; Zhang, D.; Chen, F. TFE3 Alleviates Hepatic Steatosis through Autophagy-Induced Lipophagy and PGC1alpha-Mediated Fatty Acid beta-Oxidation. Int. J. Mol. Sci. 2016, 17, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr J. 2014, 13, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef] [Green Version]
- Yamagata, K.; Daitoku, H.; Shimamoto, Y.; Matsuzaki, H.; Hirota, K.; Ishida, J.; Fukamizu, A. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. J. Biol. Chem. 2004, 279, 23158–23165. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ding, W.X.; Li, T. Cholesterol and bile acid-mediated regulation of autophagy in fatty liver diseases and atherosclerosis. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 726–733. [Google Scholar] [CrossRef]
- Seok, S.; Fu, T.; Choi, S.E.; Li, Y.; Zhu, R.; Kumar, S.; Sun, X.; Yoon, G.; Kang, Y.; Zhong, W.; et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature 2014, 516, 108–111. [Google Scholar] [CrossRef] [Green Version]
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Gea, V.; Friedman, S.L. Autophagy fuels tissue fibrogenesis. Autophagy 2012, 8, 849–850. [Google Scholar] [CrossRef] [Green Version]
- Bobowski-Gerard, M.; Zummo, F.P.; Staels, B.; Lefebvre, P.; Eeckhoute, J. Retinoids Issued from Hepatic Stellate Cell Lipid Droplet Loss as Potential Signaling Molecules Orchestrating a Multicellular Liver Injury Response. Cells 2018, 7, 137. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2009, 1791, 467–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, E.; De Minicis, S.; Osterreicher, C.H.; Kluwe, J.; Osawa, Y.; Brenner, D.A.; Schwabe, R.F. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat. Med. 2007, 13, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, J.; Yang, W.; Ling, W. Lipopolysaccharide mediates hepatic stellate cell activation by regulating autophagy and retinoic acid signaling. Autophagy 2017, 13, 1813–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Wang, B. Role of gut barrier function in the pathogenesis of nonalcoholic Fatty liver disease. Gastroenterol. Res. Pract. 2015, 2015, 287348. [Google Scholar] [CrossRef] [Green Version]
- Harte, A.L.; da Silva, N.F.; Creely, S.J.; McGee, K.C.; Billyard, T.; Youssef-Elabd, E.M.; Tripathi, G.; Ashour, E.; Abdalla, M.S.; Sharada, H.M.; et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J. Inflamm. 2010, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Niu, X.; Wang, Y.; Kong, L.; Wang, R.; Zhang, Y.; Zhao, S.; Nan, Y. MiR-146a-5p suppresses activation and proliferation of hepatic stellate cells in nonalcoholic fibrosing steatohepatitis through directly targeting Wnt1 and Wnt5a. Sci. Rep. 2015, 5, 16163. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yu, Y.; Li, S.; Liu, Y.; Zhou, S.; Cao, S.; Yin, J.; Li, G. MicroRNA-30a ameliorates hepatic fibrosis by inhibiting Beclin1-mediated autophagy. J. Cell Mol. Med. 2017, 21, 3679–3692. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhao, S.; Yao, Z.; Wang, L.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Autophagy regulates turnover of lipid droplets via ROS-dependent Rab25 activation in hepatic stellate cell. Redox Biol. 2017, 11, 322–334. [Google Scholar] [CrossRef]
- Meng, D.; Li, Z.; Wang, G.; Ling, L.; Wu, Y.; Zhang, C. Carvedilol attenuates liver fibrosis by suppressing autophagy and promoting apoptosis in hepatic stellate cells. Biomed. Pharmacother. 2018, 108, 1617–1627. [Google Scholar] [CrossRef]
- Zhang, X.L.; Chen, Z.N.; Huang, Q.F.; Bai, F.C.; Nie, J.L.; Lu, S.J.; Wei, J.B.; Lin, X. Methyl Helicterate Inhibits Hepatic Stellate Cell Activation Through Modulation of Apoptosis and Autophagy. Cell Physiol. Biochem. 2018, 51, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Y.; Huang, H.; Shi, M.; Yang, W.; Kuang, J.; Yan, J. Autophagy mediated by endoplasmic reticulum stress enhances the caffeine-induced apoptosis of hepatic stellate cells. Int. J. Mol. Med. 2017, 40, 1405–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtani, N.; Kawada, N. Role of the Gut-Liver Axis in Liver Inflammation, Fibrosis, and Cancer: A Special Focus on the Gut Microbiota Relationship. Hepatol. Commun. 2019, 3, 456–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roderburg, C.; Luedde, T. The role of the gut microbiome in the development and progression of liver cirrhosis and hepatocellular carcinoma. Gut Microbes 2014, 5, 441–445. [Google Scholar] [CrossRef]
- Konturek, P.C.; Harsch, I.A.; Konturek, K.; Schink, M.; Konturek, T.; Neurath, M.F.; Zopf, Y. Gut—Liver Axis: How Do Gut Bacteria Influence the Liver? Med. Sci. 2018, 6, 79. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.S.; Wang, J.; Yannie, P.J.; Ghosh, S. Intestinal barrier function and metabolic/liver diseases. Liver Res. 2020, 4, 81–87. [Google Scholar] [CrossRef]
- An, L.; Wirth, U.; Koch, D.; Schirren, M.; Drefs, M.; Koliogiannis, D.; Niess, H.; Andrassy, J.; Guba, M.; Bazhin, A.V.; et al. The Role of Gut-Derived Lipopolysaccharides and the Intestinal Barrier in Fatty Liver Diseases. J. Gastrointest. Surg. 2022, 26, 671–683. [Google Scholar] [CrossRef]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef]
- Benjamin, J.L.; Sumpter, R., Jr.; Levine, B.; Hooper, L.V. Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe 2013, 13, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Conway, K.L.; Kuballa, P.; Song, J.H.; Patel, K.K.; Castoreno, A.B.; Yilmaz, O.H.; Jijon, H.B.; Zhang, M.; Aldrich, L.N.; Villablanca, E.J.; et al. Atg16l1 is Required for Autophagy in Intestinal Epithelial Cells and Protection of Mice From Salmonella Infection. Gastroenterology 2013, 145, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa-Ishimoto, Y.; Shono, Y.; Gomez, L.E.; Hubbard-Lucey, V.M.; Cammer, M.; Neil, J.; Dewan, M.Z.; Lieberman, S.R.; Lazrak, A.; Marinis, J.M.; et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J. Exp. Med. 2017, 214, 3687–3705. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Leal-Ekman, J.S.; Lu, Q.; Stappenbeck, T.S. Atg14 protects the intestinal epithelium from TNF-triggered villus atrophy. Autophagy 2019, 15, 1990–2001. [Google Scholar] [CrossRef] [PubMed]
- Trentesaux, C.; Fraudeau, M.; Pitasi, C.L.; Lemarchand, J.; Jacques, S.; Duche, A.; Letourneur, F.; Naser, E.; Bailly, K.; Schmitt, A.; et al. Essential role for autophagy protein ATG7 in the maintenance of intestinal stem cell integrity. Proc. Natl. Acad. Sci. USA 2020, 117, 11136–11146. [Google Scholar] [CrossRef]
- Asano, J.; Sato, T.; Ichinose, S.; Kajita, M.; Onai, N.; Shimizu, S.; Ohteki, T. Intrinsic Autophagy Is Required for the Maintenance of Intestinal Stem Cells and for Irradiation-Induced Intestinal Regeneration. Cell Rep. 2017, 20, 1050–1060. [Google Scholar] [CrossRef] [Green Version]
- Zeisel, M.B.; Dhawan, P.; Baumert, T.F. Tight junction proteins in gastrointestinal and liver disease. Gut 2019, 68, 547–561. [Google Scholar] [CrossRef]
- Nighot, P.K.; Hu, C.A.; Ma, T.Y. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. J. Biol. Chem. 2015, 290, 7234–7246. [Google Scholar] [CrossRef] [Green Version]
- Saha, K.; Ganapathy, A.S.; Wang, A.; Morris, N.M.; Suchanec, E.; Ding, W.; Yochum, G.; Koltun, W.; Nighot, M.; Ma, T.; et al. Autophagy Reduces the Degradation and Promotes Membrane Localization of Occludin to Enhance the Intestinal Epithelial Tight Junction Barrier against Paracellular Macromolecule Flux. J. Crohns Colitis 2022, jjac148. [Google Scholar] [CrossRef]
- Schwenger, K.J.; Clermont-Dejean, N.; Allard, J.P. The role of the gut microbiome in chronic liver disease: The clinical evidence revised. JHEP Rep. 2019, 1, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Lapaquette, P.; Bizeau, J.B.; Acar, N.; Bringer, M.A. Reciprocal interactions between gut microbiota and autophagy. World J. Gastroenterol. 2021, 27, 8283–8301. [Google Scholar] [CrossRef]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef]
- Yang, L.; Liu, C.; Zhao, W.; He, C.; Ding, J.; Dai, R.; Xu, K.; Xiao, L.; Luo, L.; Liu, S.; et al. Impaired Autophagy in Intestinal Epithelial Cells Alters Gut Microbiota and Host Immune Responses. Appl. Environ. Microbiol. 2018, 84, e00880-18. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Lee, J.Y.; Shin, S.G.; Kim, J.K.; Silwal, P.; Kim, Y.J.; Shin, N.R.; Kim, P.S.; Won, M.; Lee, S.H.; et al. ESRRA (estrogen related receptor alpha) is a critical regulator of intestinal homeostasis through activation of autophagic flux via gut microbiota. Autophagy 2021, 17, 2856–2875. [Google Scholar] [CrossRef]
- Patel, N.S.; Doycheva, I.; Peterson, M.R.; Hooker, J.; Kisselva, T.; Schnabl, B.; Seki, E.; Sirlin, C.B.; Loomba, R. Effect of weight loss on magnetic resonance imaging estimation of liver fat and volume in patients with nonalcoholic steatohepatitis. Clin. Gastroenterol. Hepatol. 2015, 13, 561–568e1. [Google Scholar] [CrossRef] [Green Version]
- Truong, E.; Noureddin, M. Improvement in nonalcoholic fatty liver disease through bariatric surgery. Clin. Liver Dis. 2022, 20, 13–17. [Google Scholar] [CrossRef]
- Hafeez, S.; Ahmed, M.H. Bariatric surgery as potential treatment for nonalcoholic fatty liver disease: A future treatment by choice or by chance? J. Obes. 2013, 2013, 839275. [Google Scholar] [CrossRef] [Green Version]
- Grefhorst, A.; van de Peppel, I.P.; Larsen, L.E.; Jonker, J.W.; Holleboom, A.G. The Role of Lipophagy in the Development and Treatment of Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2020, 11, 601627. [Google Scholar] [CrossRef]
- Li, Y.; Liu, L.; Wang, B.; Wang, J.; Chen, D. Metformin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Biomed. Rep. 2013, 1, 57–64. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.; Fang, L.; Zhang, H.; Zhang, W.S.; Li, X.O.; Du, S.Y. Metformin Induces Autophagy via the AMPK-mTOR Signaling Pathway in Human Hepatocellular Carcinoma Cells. Cancer Manag. Res. 2020, 12, 5803–5811. [Google Scholar] [CrossRef]
- Song, Y.M.; Lee, Y.H.; Kim, J.W.; Ham, D.S.; Kang, E.S.; Cha, B.S.; Lee, H.C.; Lee, B.W. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy 2015, 11, 46–59. [Google Scholar] [CrossRef] [Green Version]
- Gosis, B.S.; Wada, S.; Thorsheim, C.; Li, K.; Jung, S.; Rhoades, J.H.; Yang, Y.; Brandimarto, J.; Li, L.; Uehara, K.; et al. Inhibition of nonalcoholic fatty liver disease in mice by selective inhibition of mTORC1. Science 2022, 376, eabf8271. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Qiu, S.; Zhou, S.; Tan, Y.; Bai, Y.; Cao, H.; Guo, J.; Su, Z. mTOR: A Potential New Target in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 9196. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Song, J.W.; Park, J.H.; Lim, B.K.; Moon, O.S.; Son, H.Y.; Lee, J.H.; Gao, B.; Won, Y.S.; Kwon, H.J. TXNIP/VDUP1 attenuates steatohepatitis via autophagy and fatty acid oxidation. Autophagy 2021, 17, 2549–2564. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Li, Y.; Li, L. Targeting folliculin to selectively inhibit mTORC1: A promising strategy for treating nonalcoholic fatty liver disease. Signal Transduct. Target. Ther. 2022, 7, 277. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Ma, X.; Geng, S.; Jiang, X.; Li, Y.; Hu, L.; Li, J.; Wang, Y.; Han, X. Fecal Microbiota Transplantation Beneficially Regulates Intestinal Mucosal Autophagy and Alleviates Gut Barrier Injury. mSystems 2018, 3, e00137-18. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cavallaro, P.M.; Kim, B.M.; Liu, T.; Wang, H.; Kuhn, F.; Adiliaghdam, F.; Liu, E.; Vasan, R.; Samarbafzadeh, E.; et al. A role for intestinal alkaline phosphatase in preventing liver fibrosis. Theranostics 2021, 11, 14–26. [Google Scholar] [CrossRef]
- Singh, S.B.; Carroll-Portillo, A.; Coffman, C.; Ritz, N.L.; Lin, H.C. Intestinal Alkaline Phosphatase Exerts Anti-Inflammatory Effects Against Lipopolysaccharide by Inducing Autophagy. Sci. Rep. 2020, 10, 3107. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.W.; Chung, H.Y. The Effects of Calorie Restriction on Autophagy: Role on Aging Intervention. Nutrients 2019, 11, 2923. [Google Scholar] [CrossRef] [Green Version]
- Antunes, F.; Erustes, A.G.; Costa, A.J.; Nascimento, A.C.; Bincoletto, C.; Ureshino, R.P.; Pereira, G.J.S.; Smaili, S.S. Autophagy and intermittent fasting: The connection for cancer therapy? Clinics 2018, 73 (Suppl. 1), e814s. [Google Scholar] [CrossRef]
- Yin, C.; Li, Z.; Xiang, Y.; Peng, H.; Yang, P.; Yuan, S.; Zhang, X.; Wu, Y.; Huang, M.; Li, J. Effect of Intermittent Fasting on Non-Alcoholic Fatty Liver Disease: Systematic Review and Meta-Analysis. Front. Nutr. 2021, 8, 709683. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, L.; Wirth, U.; Koch, D.; Schirren, M.; Drefs, M.; Koliogiannis, D.; Niess, H.; Andrassy, J.; Guba, M.; Bazhin, A.V.; et al. Metabolic Role of Autophagy in the Pathogenesis and Development of NAFLD. Metabolites 2023, 13, 101. https://doi.org/10.3390/metabo13010101
An L, Wirth U, Koch D, Schirren M, Drefs M, Koliogiannis D, Niess H, Andrassy J, Guba M, Bazhin AV, et al. Metabolic Role of Autophagy in the Pathogenesis and Development of NAFLD. Metabolites. 2023; 13(1):101. https://doi.org/10.3390/metabo13010101
Chicago/Turabian StyleAn, Lingxuan, Ulrich Wirth, Dominik Koch, Malte Schirren, Moritz Drefs, Dionysios Koliogiannis, Hanno Niess, Joachim Andrassy, Markus Guba, Alexandr V. Bazhin, and et al. 2023. "Metabolic Role of Autophagy in the Pathogenesis and Development of NAFLD" Metabolites 13, no. 1: 101. https://doi.org/10.3390/metabo13010101