
Mechanistic Insights of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis: An Update on a Lasting Relationship

 , and
, and
Abstract
:1. Introduction
2. Search Methods, Eligibility Criteria, and Screening
3. Involvement of fALS-Associated Proteins in Mitochondrial Dysfunction: An Update
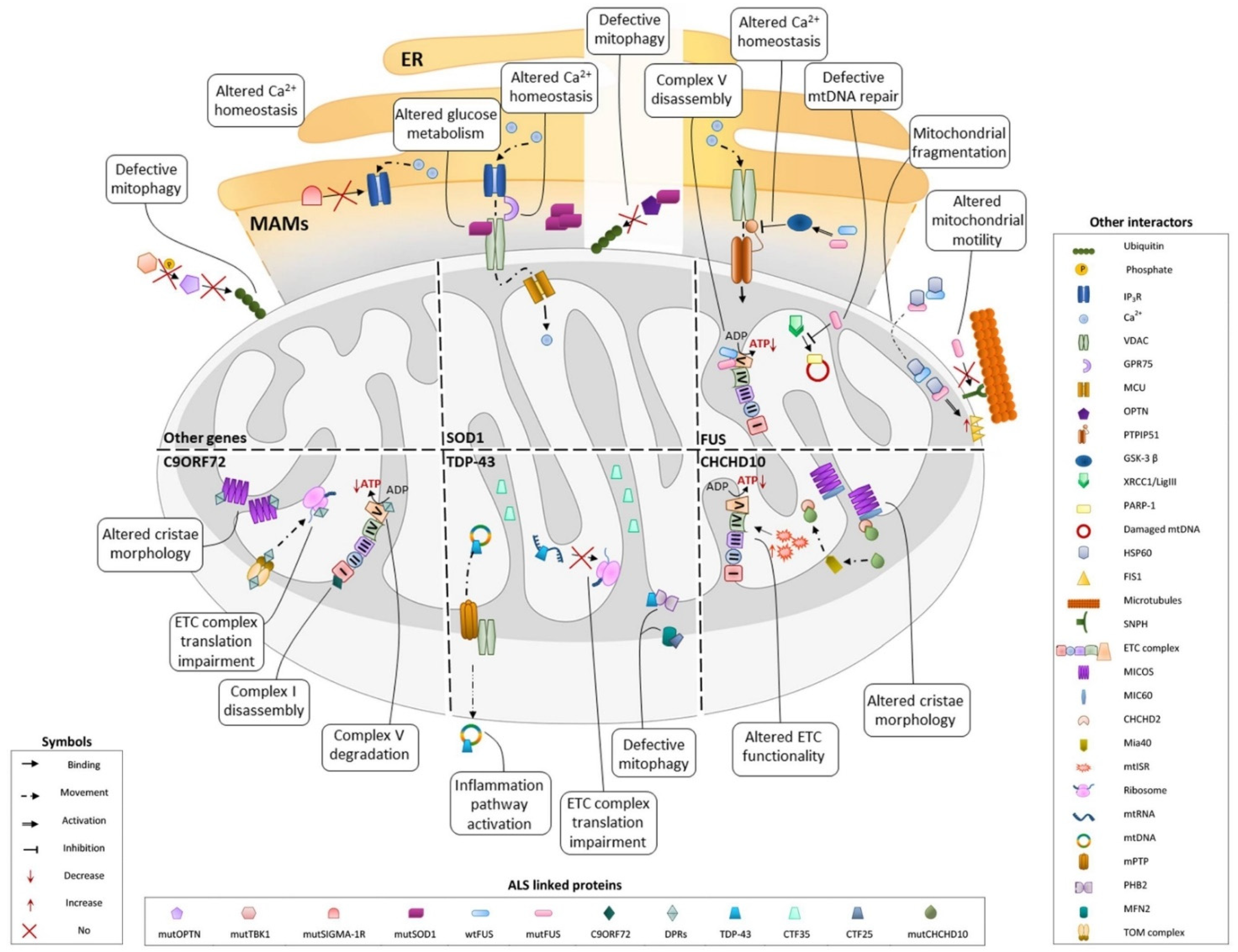
3.1. SOD1
3.2. TDP-43
3.3. C9ORF72
3.4. FUS
3.5. CHCHD10
3.6. Other Genes
3.6.1. Mitophagy
3.6.2. Ca2+ Homeostasis
4. Mitochondrial Alterations in Sporadic ALS Cases
5. The Threshold Effect of Mitochondrial Dysfunction
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| MNs | Motor neurons |
| sALS | Sporadic ALS |
| fALS | Familial ALS |
| SOD1 | Cu–Zn superoxide dismutase 1 |
| ROS | Reactive oxygen species |
| OXPHOS | Oxidative phosphorylation |
| C9ORF72 | Chromosome 9 open reading frame 72 |
| TDP-43 | TAR DNA-binding protein 43 |
| FUS | Fused in sarcoma |
| CHCHD10 | Coiled-coil-helix domain-containing protein 10 |
| MAMs | Mitochondria-associated membrane |
| mutSOD1 | Mutant SOD1 |
| IMS | Mitochondrial intermembrane space |
| VDAC1 | Voltage-dependent anion channel 1 |
| OMM | Outer mitochondrial membrane |
| ER | Endoplasmic reticulum |
| Sigma-1R | Sigma-1 receptor |
| OPTN | Optineurin |
| CTF-25 | TDP-43 C-terminal fragment of 25 kDa |
| CTF-35 | TDP-43 C-terminal fragment of 35 kDa |
| UPRmt | Mitochondrial unfolded protein response |
| m∆Φ | Mitochondrial inner membrane potential |
| IMM | Inner mitochondrial membrane |
| mtRNA | Mitochondrial RNA |
| mtDNA | Mitochondrial DNA |
| MICU | Mitochondrial Ca2+ uptake regulator |
| PHB | Prohibitin |
| G4C2 | Noncoding hexanucleotide GGGGCC of c9orf72 intron 1 |
| DPR | Dipeptide repeats proteins |
| ETC | Electron transport chain |
| MICOS | Mitochondrial contact site and cristae organizing system |
| TIMMDC1 | Translocase of the mitochondrial membrane domain internal containing 1 |
| mtISR | Mitochondrial integrated stress response |
| SNP | Syntaphilin |
| OPA1 | Optic atrophy 1 |
| TBK1 | TANK-binding kinase 1 |
| DPR1 | Dynamin-related protein 1 |
| PP1 | Protein phosphatases 1 |
References
- van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17085. [Google Scholar] [CrossRef] [PubMed]
- Orrell, R.W. Motor neuron disease: Systematic reviews of treatment for ALS and SMA. Br. Med. Bull. 2010, 93, 145–159. [Google Scholar] [CrossRef] [Green Version]
- Mathis, S.; Goizet, C.; Soulages, A.; Vallat, J.M.; Masson, G. Le Genetics of amyotrophic lateral sclerosis: A review. J. Neurol. Sci. 2019, 399, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Alsultan, A.A.; Waller, R.; Heath, P.R.; Kirby, J. The genetics of amyotrophic lateral sclerosis: Current insights. Degener. Neurol. Neuromuscul. Dis. 2016, 6, 49. [Google Scholar] [CrossRef] [Green Version]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.C.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Kwiatkowski, T.J.; Bosco, D.A.; LeClerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Cozzolino, M.; Ferri, A.; Valle, C.; Carrì, M.T. Mitochondria and ALS: Implications from novel genes and pathways. Mol. Cell. Neurosci. 2013, 55, 44–49. [Google Scholar] [CrossRef]
- Bannwarth, S.; Ait-El-Mkadem, S.; Chaussenot, A.; Genin, E.C.; Lacas-Gervais, S.; Fragaki, K.; Berg-Alonso, L.; Kageyama, Y.; Serre, V.; Moore, D.G.; et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 2014, 137, 2329–2345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, S.; Watanabe, S.; Komine, O.; Sobue, A.; Yamanaka, K. Novel reporters of mitochondria-associated membranes (MAM), MAMtrackers, demonstrate MAM disruption as a common pathological feature in amyotrophic lateral sclerosis. FASEB J. 2021, 35, e21688. [Google Scholar] [CrossRef] [PubMed]
- Cacabelos, D.; Ramírez-Núñez, O.; Granado-Serrano, A.B.; Torres, P.; Ayala, V.; Moiseeva, V.; Povedano, M.; Ferrer, I.; Pamplona, R.; Portero-Otin, M.; et al. Early and gender-specific differences in spinal cord mitochondrial function and oxidative stress markers in a mouse model of ALS. Acta Neuropathol. Commun. 2016, 4, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, P.; Gal, J.; Kwinter, D.M.; Liu, X.; Zhu, H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2010, 1802, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PLoS ONE 2013, 8, e0082112. [Google Scholar] [CrossRef]
- Pickles, S.; Semmler, S.; Broom, H.R.; Destroismaisons, L.; Legroux, L.; Arbour, N.; Meiering, E.; Cashman, N.R.; Vande Velde, C. ALS-linked misfolded SOD1 species have divergent impacts on mitochondria. Acta Neuropathol. Commun. 2016, 4, 43. [Google Scholar] [CrossRef] [Green Version]
- Shteinfer-Kuzmine, A.; Argueti, S.; Gupta, R.; Shvil, N.; Abu-Hamad, S.; Gropper, Y.; Hoeber, J.; Magrì, A.; Messina, A.; Kozlova, E.N.; et al. A VDAC1-Derived N-Terminal Peptide Inhibits Mutant SOD1-VDAC1 Interactions and Toxicity in the SOD1 Model of ALS. Front. Cell. Neurosci. 2019, 13, 346. [Google Scholar] [CrossRef] [Green Version]
- Magrì, A.; Risiglione, P.; Caccamo, A.; Formicola, B.; Tomasello, M.F.; Arrigoni, C.; Zimbone, S.; Guarino, F.; Re, F.; Messina, A. Small Hexokinase 1 Peptide against Toxic SOD1 G93A Mitochondrial Accumulation in ALS Rescues the ATP-Related Respiration. Biomedicines 2021, 9, 948. [Google Scholar] [CrossRef]
- D’Eletto, M.; Rossin, F.; Occhigrossi, L.; Farrace, M.G.; Faccenda, D.; Desai, R.; Marchi, S.; Refolo, G.; Falasca, L.; Antonioli, M.; et al. Transglutaminase Type 2 Regulates ER-Mitochondria Contact Sites by Interacting with GRP75. Cell Rep. 2018, 25, 3573–3581.e4. [Google Scholar] [CrossRef] [Green Version]
- Damiano, M.; Starkov, A.A.; Petri, S.; Kipiani, K.; Kiaei, M.; Mattiazzi, M.; Beal, M.F.; Manfredi, G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J. Neurochem. 2006, 96, 1349–1361. [Google Scholar] [CrossRef]
- Semmler, S.; Gagné, M.; Garg, P.; Pickles, S.R.; Baudouin, C.; Hamon-Keromen, E.; Destroismaisons, L.; Khalfallah, Y.; Chaineau, M.; Caron, E.; et al. TNF receptor-associated factor 6 interacts with ALS-linked misfolded superoxide dismutase 1 and promotes aggregation. J. Biol. Chem. 2020, 295, 3808–3825. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.; Lu, J.; Siedlak, S.L.; Fujioka, H.; Liang, J.; Jiang, S.; Ma, X.; Jiang, Z.; Da Rocha, E.L.; et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016, 22, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Izumikawa, K.; Nobe, Y.; Yoshikawa, H.; Ishikawa, H.; Miura, Y.; Nakayama, H.; Nonaka, T.; Hasegawa, M.; Egawa, N.; Inoue, H.; et al. TDP-43 stabilises the processing intermediates of mitochondrial transcripts. Sci. Rep. 2017, 7, 7709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef] [PubMed]
- Altman, T.; Ionescu, A.; Ibraheem, A.; Priesmann, D.; Gradus-Pery, T.; Farberov, L.; Alexandra, G.; Shelestovich, N.; Dafinca, R.; Shomron, N.; et al. Axonal TDP-43 condensates drive neuromuscular junction disruption through inhibition of local synthesis of nuclear encoded mitochondrial proteins. Nat. Commun. 2021, 12, 6914. [Google Scholar] [CrossRef]
- Dafinca, R.; Barbagallo, P.; Farrimond, L.; Candalija, A.; Scaber, J.; Ababneh, N.A.; Sathyaprakash, C.; Vowles, J.; Cowley, S.A.; Talbot, K. Impairment of Mitochondrial Calcium Buffering Links Mutations in C9ORF72 and TARDBP in iPS-Derived Motor Neurons from Patients with ALS/FTD. Stem Cell Rep. 2020, 14, 892–908. [Google Scholar] [CrossRef]
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15. [Google Scholar] [CrossRef]
- Jun, M.H.; Jang, J.W.; Jeon, P.; Lee, S.K.; Lee, S.H.; Choi, H.E.; Lee, Y.K.; Choi, H.; Park, S.W.; Kim, J.; et al. Nonmuscle myosin IIB regulates Parkin-mediated mitophagy associated with amyotrophic lateral sclerosis-linked TDP-43. Cell Death Dis. 2020, 11, 952. [Google Scholar] [CrossRef]
- Gautam, M.; Xie, E.F.; Kocak, N.; Ozdinler, P.H. Mitoautophagy: A Unique Self-Destructive Path Mitochondria of Upper Motor Neurons With TDP-43 Pathology Take, Very Early in ALS. Front. Cell. Neurosci. 2019, 13, 489. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.L.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.L.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.R.; Gregory, J.M.; Dando, O.; Carter, R.N.; Burr, K.; Nanda, J.; Story, D.; McDade, K.; Smith, C.; Morton, N.M.; et al. Mitochondrial bioenergetic deficits in C9orf72 amyotrophic lateral sclerosis motor neurons cause dysfunctional axonal homeostasis. Acta Neuropathol. 2021, 141, 257–279. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wu, Z.; Tantray, I.; Li, Y.; Chen, S.; Dong, J.; Glynn, S.; Vogel, H.; Snyder, M.; Lu, B. Quality-control mechanisms targeting translationally stalled and C-terminally extended poly(GR) associated with ALS/FTD. Proc. Natl. Acad. Sci. USA 2020, 117, 25104–25115. [Google Scholar] [CrossRef]
- Choi, S.Y.; Lopez-Gonzalez, R.; Krishnan, G.; Phillips, H.L.; Li, A.N.; Seeley, W.W.; Yao, W.D.; Almeida, S.; Gao, F.B. C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat. Neurosci. 2019, 22, 851–862. [Google Scholar] [CrossRef]
- Lynch, E.; Semrad, T.; Belsito, V.S.; FitzGibbons, C.; Reilly, M.; Hayakawa, K.; Suzuki, M. C9ORF72-related cellular pathology in skeletal myocytes derived from ALS-patient induced pluripotent stem cells. Dis. Model. Mech. 2019, 12, dmm039552. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Wu, Z.; Li, Y.; Tantray, I.; De Stefani, D.; Mattarei, A.; Krishnan, G.; Gao, F.B.; Vogel, H.; Lu, B. Altered MICOS Morphology and Mitochondrial Ion Homeostasis Contribute to Poly(GR) Toxicity Associated with C9-ALS/FTD. Cell Rep. 2020, 32, 107989. [Google Scholar] [CrossRef]
- Wang, T.; Liu, H.; Itoh, K.; Oh, S.; Zhao, L.; Murata, D.; Sesaki, H.; Hartung, T.; Na, C.H.; Wang, J. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 2021, 33, 531–546.e9. [Google Scholar] [CrossRef]
- Ishigaki, S.; Sobue, G. Importance of Functional Loss of FUS in FTLD/ALS. Front. Mol. Biosci. 2018, 5, 44. [Google Scholar] [CrossRef]
- Wang, H.; Guo, W.; Mitra, J.; Hegde, P.M.; Vandoorne, T.; Eckelmann, B.J.; Mitra, S.; Tomkinson, A.E.; Van Den Bosch, L.; Hegde, M.L. Mutant FUS causes DNA ligation defects to inhibit oxidative damage repair in Amyotrophic Lateral Sclerosis. Nat. Commun. 2018, 9, 3683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Yang, M.; Chen, Y.; Chen, X.; Liu, J.; Sun, S.; Cheng, H.; Li, Y.; Bigio, E.H.; Mesulam, M.; et al. FUS Interacts with HSP60 to Promote Mitochondrial Damage. PLoS Genet. 2015, 11, e1005357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salam, S.; Tacconelli, S.; Smith, B.N.; Mitchell, J.C.; Glennon, E.; Nikolaou, N.; Houart, C.; Vance, C. Identification of a novel interaction of FUS and syntaphilin may explain synaptic and mitochondrial abnormalities caused by ALS mutations. Sci. Rep. 2021, 11, 13613. [Google Scholar] [CrossRef]
- Deng, J.; Wang, P.; Chen, X.; Cheng, H.; Liu, J.; Fushimi, K.; Zhu, L.; Wu, J.Y. FUS interacts with ATP synthase beta subunit and induces mitochondrial unfolded protein response in cellular and animal models. Proc. Natl. Acad. Sci. USA 2018, 115, E9678–E9686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandoorne, T.; Veys, K.; Guo, W.; Sicart, A.; Vints, K.; Swijsen, A.; Moisse, M.; Eelen, G.; Gounko, N.V.; Fumagalli, L.; et al. Differentiation but not ALS mutations in FUS rewires motor neuron metabolism. Nat. Commun. 2019, 10, 4147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, S.J.; Choi, H.-J.; Kim, H.-J.; Choi, E.J.; Song, K.-H.; Im, D.S.; Kim, K. Parkin expression reverses mitochondrial dysfunction in fused in sarcoma-induced amyotrophic lateral sclerosis. Insect Mol. Biol. 2020, 29, 56–65. [Google Scholar] [CrossRef]
- Chen, Y.; Deng, J.; Wang, P.; Yang, M.; Chen, X.; Zhu, L.; Liu, J.; Lu, B.; Shen, Y.; Fushimi, K.; et al. PINK1 and Parkin are genetic modifiers for FUS-induced neurodegeneration. Hum. Mol. Genet. 2016, 25, 5059–5068. [Google Scholar] [CrossRef] [Green Version]
- Stoica, R.; Paillusson, S.; Gomez-Suaga, P.; Mitchell, J.C.; Lau, D.H.; Gray, E.H.; Sancho, R.M.; Vizcay-Barrena, G.; De Vos, K.J.; Shaw, C.E.; et al. ALS/FTD-associated FUS activates GSK-3β to disrupt the VAPB–PTPIP 51 interaction and ER –mitochondria associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef]
- Wollweber, F.; von der Malsburg, K.; van der Laan, M. Mitochondrial contact site and cristae organizing system: A central player in membrane shaping and crosstalk. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1481–1489. [Google Scholar] [CrossRef]
- Zhou, W.; Ma, D.; Tan, E.K. Mitochondrial CHCHD2 and CHCHD10: Roles in Neurological Diseases and Therapeutic Implications. Neuroscientist 2020, 26, 170–184. [Google Scholar] [CrossRef]
- Eramo, M.J.; Lisnyak, V.; Formosa, L.E.; Ryan, M.T. The “mitochondrial contact site and cristae organising system” (MICOS) in health and human disease. J. Biochem. 2020, 167, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Mourier, A.; Yamada, J.; Michael McCaffery, J.; Nunnari, J. MICOS coordinates with respiratory complexes and lipids to establish mitochondrial inner membrane architecture. eLife 2015, 2015, e07739. [Google Scholar] [CrossRef] [PubMed]
- Rieger, B.; Junge, W.; Busch, K.B. Lateral pH gradient between OXPHOS complex IV and F0F1 ATP-synthase in folded mitochondrial membranes. Nat. Commun. 2014, 5, 3103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavallaro, G. Genome-wide analysis of eukaryotic twin CX9C proteins. Mol. Biosyst. 2010, 6, 2459–2470. [Google Scholar] [CrossRef]
- Khan, N.A.; Nikkanen, J.; Yatsuga, S.; Jackson, C.; Wang, L.; Pradhan, S.; Kivelä, R.; Pessia, A.; Velagapudi, V.; Suomalainen, A. mTORC1 Regulates Mitochondrial Integrated Stress Response and Mitochondrial Myopathy Progression. Cell Metab. 2017, 26, 419–428.e5. [Google Scholar] [CrossRef]
- Anderson, C.J.; Bredvik, K.; Burstein, S.R.; Davis, C.; Meadows, S.M.; Dash, J.; Case, L.; Milner, T.A.; Kawamata, H.; Zuberi, A.; et al. ALS/FTD mutant CHCHD10 mice reveal a tissue-specific toxic gain-of-function and mitochondrial stress response. Acta Neuropathol. 2019, 138, 103. [Google Scholar] [CrossRef]
- Liu, T.; Woo, J.A.A.; Bukhari, M.Z.; LePochat, P.; Chacko, A.; Selenica, M.L.B.; Yan, Y.; Kotsiviras, P.; Buosi, S.C.; Zhao, X.; et al. CHCHD10-regulated OPA1-mitofilin complex mediates TDP-43-induced mitochondrial phenotypes associated with frontotemporal dementia. FASEB J. 2020, 34, 8493–8509. [Google Scholar] [CrossRef]
- Glytsou, C.; Calvo, E.; Cogliati, S.; Mehrotra, A.; Anastasia, I.; Rigoni, G.; Raimondi, A.; Shintani, N.; Loureiro, M.; Vazquez, J.; et al. Optic Atrophy 1 Is Epistatic to the Core MICOS Component MIC60 in Mitochondrial Cristae Shape Control. Cell Rep. 2016, 17, 3024–3034. [Google Scholar] [CrossRef] [Green Version]
- Cho, B.; Cho, H.M.; Jo, Y.; Kim, H.D.; Song, M.; Moon, C.; Kim, H.; Kim, K.; Sesaki, H.; Rhyu, I.J.; et al. Constriction of the mitochondrial inner compartment is a priming event for mitochondrial division. Nat. Commun. 2017, 8, 15754. [Google Scholar] [CrossRef] [Green Version]
- Harding, O.; Evans, C.S.; Ye, J.; Cheung, J.; Maniatis, T.; Holzbaur, E.L.F. ALS- And FTD-associated missense mutations in TBK1 differentially disrupt mitophagy. Proc. Natl. Acad. Sci. USA 2021, 118, e2025053118. [Google Scholar] [CrossRef]
- Li, F.; Xie, X.; Wang, Y.; Liu, J.; Cheng, X.; Guo, Y.; Gong, Y.; Hu, S.; Pan, L. Structural insights into the interaction and disease mechanism of neurodegenerative disease-associated optineurin and TBK1 proteins. Nat. Commun. 2016, 7, 12708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Cheung, J.; Gerbino, V.; Ahlsén, G.; Zimanyi, C.; Hirsh, D.; Maniatis, T. Effects of ALS-associated TANK binding kinase 1 mutations on protein–protein interactions and kinase activity. Proc. Natl. Acad. Sci. USA 2019, 116, 24517–24526. [Google Scholar] [CrossRef] [PubMed]
- de Majo, M.; Topp, S.D.; Smith, B.N.; Nishimura, A.L.; Chen, H.J.; Gkazi, A.S.; Miller, J.; Wong, C.H.; Vance, C.; Baas, F.; et al. ALS-associated missense and nonsense TBK1 mutations can both cause loss of kinase function. Neurobiol. Aging 2018, 71, 266.e1–266.e10. [Google Scholar] [CrossRef]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, C.S.; Holzbaur, E.L.F. Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. eLife 2020, 9, e50260. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Xu, D.; Wang, Y.; Zhou, Z.; Liu, J.; Hu, S.; Gong, Y.; Yuan, J.; Pan, L. Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1-mediated phosphorylation. Autophagy 2018, 14, 66–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, A.S.; Holzbaur, E.L.F. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Manfredi, G.; Kawamata, H. Mitochondria and endoplasmic reticulum crosstalk in amyotrophic lateral sclerosis. Neurobiol. Dis. 2016, 90, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with High Ca2+ Close to IP3-Sensitive Channels that Are Sensed by Neighboring Mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef]
- Rizzuto, R.; Duchen, M.R.; Pozzan, T. Flirting in little space: The ER/mitochondria Ca2+ liaison. Sci. STKE 2004, 2004, ref1. [Google Scholar] [CrossRef] [PubMed]
- Bannister, W.H. From Haemocuprein to Copper-Zinc Superoxide Dismutase: A History on the Fiftieth Anniversary of the Discovery of Haemocuprein and the Twentieth Anniversary of the Discovery of Superoxide Dismutase. Free Radic. Res. Commun. 2009, 5, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Redler, R.L.; Dokholyan, N.V. The Complex Molecular Biology of Amyotrophic Lateral Sclerosis (ALS). Prog. Mol. Biol. Transl. Sci. 2012, 107, 215–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guissart, C.; Mouzat, K.; Kantar, J.; Louveau, B.; Vilquin, P.; Polge, A.; Raoul, C.; Lumbroso, S. Premature termination codons in SOD1 causing Amyotrophic Lateral Sclerosis are predicted to escape the nonsense-mediated mRNA decay. Sci. Rep. 2020, 10, 20738. [Google Scholar] [CrossRef] [PubMed]
- Israelson, A.; Arbel, N.; Da Cruz, S.; Ilieva, H.; Yamanaka, K.; Shoshan-Barmatz, V.; Cleveland, D.W. Misfolded Mutant SOD1 Directly Inhibits VDAC1 Conductance in a Mouse Model of Inherited ALS. Neuron 2010, 67, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoshan-Barmatz, V.; Shteinfer-Kuzmine, A.; Verma, A. VDAC1 at the Intersection of Cell Metabolism, Apoptosis, and Diseases. Biomolecules 2020, 10, 1485. [Google Scholar] [CrossRef]
- César Rosa, J.; de Cerqueira César, M. Role of Hexokinase and VDAC in Neurological Disorders. Curr. Mol. Pharmacol. 2016, 9, 320–331. [Google Scholar] [CrossRef]
- Araujo, B.G.; Souza e Silva, L.F.; de Barros Torresi, J.L.; Siena, A.; Valerio, B.C.O.; Brito, M.D.; Rosenstock, T.R. Decreased Mitochondrial Function, Biogenesis, and Degradation in Peripheral Blood Mononuclear Cells from Amyotrophic Lateral Sclerosis Patients as a Potential Tool for Biomarker Research. Mol. Neurobiol. 2020, 57, 5084–5102. [Google Scholar] [CrossRef]
- Yang, M.; Li, C.; Yang, S.; Xiao, Y.; Xiong, X.; Chen, W.; Zhao, H.; Zhang, Q.; Han, Y.; Sun, L. Mitochondria-Associated ER Membranes—The Origin Site of Autophagy. Front. Cell Dev. Biol. 2020, 8, 595. [Google Scholar] [CrossRef]
- Johri, A.; Chandra, A. Connection Lost, MAM: Errors in ER-Mitochondria Connections in Neurodegenerative Diseases. Brain Sci. 2021, 11, 1437. [Google Scholar] [CrossRef]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef] [PubMed]
- van Rheenen, W.; van der Spek, R.A.A.; Bakker, M.K.; van Vugt, J.J.F.A.; Hop, P.J.; Zwamborn, R.A.J.; de Klein, N.; Westra, H.-J.; Bakker, O.B.; Deelen, P.; et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 2021, 53, 1636–1648. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Zhou, X.; Lu, J.-H.; Yue, Z. Autophagy deficiency in neurodevelopmental disorders. Cell Biosci. 2021, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Buratti, E.; Baralle, F.E. The multiple roles of TDP-43 in pre-mRNA processing and gene expression regulation. RNA Biol. 2010, 7, 420–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, I.R.A.; Bigio, E.H.; Ince, P.G.; Geser, F.; Neumann, M.; Cairns, N.J.; Kwong, L.K.; Forman, M.S.; Ravits, J.; Stewart, H.; et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007, 61, 427–434. [Google Scholar] [CrossRef]
- Bozzo, F.; Salvatori, I.; Iacovelli, F.; Mirra, A.; Rossi, S.; Cozzolino, M.; Falconi, M.; Valle, C.; Carrì, M.T. Structural insights into the multi-determinant aggregation of TDP-43 in motor neuron-like cells. Neurobiol. Dis. 2016, 94, 63–72. [Google Scholar] [CrossRef]
- Gasset-Rosa, F.; Lu, S.; Yu, H.; Chen, C.; Melamed, Z.; Guo, L.; Shorter, J.; Da Cruz, S.; Cleveland, D.W. Cytoplasmic TDP-43 De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron 2019, 102, 339–357.e7. [Google Scholar] [CrossRef] [Green Version]
- Clippinger, A.K.; D’Alton, S.; Lin, W.L.; Gendron, T.F.; Howard, J.; Borchelt, D.R.; Cannon, A.; Carlomagno, Y.; Chakrabarty, P.; Cook, C.; et al. Robust cytoplasmic accumulation of phosphorylated TDP-43 in transgenic models of tauopathy. Acta Neuropathol. 2013, 126, 39–50. [Google Scholar] [CrossRef] [Green Version]
- Mori, F.; Tanji, K.; Zhang, H.X.; Nishihira, Y.; Tan, C.F.; Takahashi, H.; Wakabayashi, K. Maturation process of TDP-43-positive neuronal cytoplasmic inclusions in amyotrophic lateral sclerosis with and without dementia. Acta Neuropathol. 2008, 116, 193–203. [Google Scholar] [CrossRef]
- Xu, Y.-F.; Gendron, T.F.; Zhang, Y.-J.; Lin, W.-L.; Alton, S.D.; Sheng, H.; Casey, M.C.; Tong, J.; Knight, J.; Yu, X.; et al. Neurobiology of Disease Wild-Type Human TDP-43 Expression Causes TDP-43 Phosphorylation, Mitochondrial Aggregation, Motor Deficits, and Early Mortality in Transgenic Mice. J. Neurosci. 2010, 30, 10851–10859. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Arakawa, H.; Wang, L.; Okolo, O.; Siedlak, S.L.; Jiang, Y.; Gao, J.; Xie, F.; Petersen, R.B.; Wang, X. Motor-Coordinative and Cognitive Dysfunction Caused by Mutant TDP-43 Could Be Reversed by Inhibiting Its Mitochondrial Localization. Mol. Ther. 2017, 25, 127–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvatori, I.; Ferri, A.; Scaricamazza, S.; Giovannelli, I.; Serrano, A.; Rossi, S.; D’Ambrosi, N.; Cozzolino, M.; Di Giulio, A.; Moreno, S.; et al. Differential toxicity of TAR DNA-binding protein 43 isoforms depends on their submitochondrial localization in neuronal cells. J. Neurochem. 2018, 146, 585–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simón-Sánchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, T.P.; Daniels, R.D.; Gatta, A.T.; Wong, L.H.; Hayes, M.J. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 2013, 29, 499–503. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.A.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, L.; Swaminathan, K.; Herrlinger, S.; Lai, F.; Shiekhattar, R.; Chen, J.F. A C9ORF72/SMCR8-containing complex regulates ULK1 and plays a dual role in autophagy. Sci. Adv. 2016, 2, 1601167. [Google Scholar] [CrossRef] [Green Version]
- Smeyers, J.; Banchi, E.G.; Latouche, M. C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell. Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef]
- Lee, Y.B.; Chen, H.J.; Peres, J.N.; Gomez-Deza, J.; Attig, J.; Štalekar, M.; Troakes, C.; Nishimura, A.L.; Scotter, E.L.; Vance, C.; et al. Hexanucleotide repeats in ALS/FTD form length-dependent RNA Foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013, 5, 1178–1186. [Google Scholar] [CrossRef] [Green Version]
- Rossi, S.; Serrano, A.; Gerbino, V.; Giorgi, A.; Di Francesco, L.; Nencini, M.; Bozzo, F.; Schininà, M.E.; Bagni, C.; Cestra, G.; et al. Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. J. Cell Sci. 2015, 128, 1787–1799. [Google Scholar] [CrossRef] [Green Version]
- Koppers, M.; Blokhuis, A.M.; Westeneng, H.J.; Terpstra, M.L.; Zundel, C.A.C.; Vieira De Sá, R.; Schellevis, R.D.; Waite, A.J.; Blake, D.J.; Veldink, J.H.; et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 2015, 78, 426–438. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.B.; Baskaran, P.; Gomez-Deza, J.; Chen, H.J.; Nishimura, A.L.; Smith, B.N.; Troakes, C.; Adachi, Y.; Stepto, A.; Petrucelli, L.; et al. C9orf72 poly GA RAN-translated protein plays a key role in amyotrophic lateral sclerosis via aggregation and toxicity. Hum. Mol. Genet. 2017, 26, 4765–4777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Z.; Wang, H.; Xia, Q.; Li, K.; Li, K.; Jiang, X.; Xu, G.; Wang, G.; Ying, Z. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Hum. Mol. Genet. 2015, 24, 2426–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, X.; Tan, W.; Westergard, T.; Krishnamurthy, K.; Markandaiah, S.S.; Shi, Y.; Lin, S.; Shneider, N.A.; Monaghan, J.; Pandey, U.B.; et al. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate invitro and invivo neuronal death. Neuron 2014, 84, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Shi, K.Y.; Mori, E.; Nizami, Z.F.; Lin, Y.; Kato, M.; Xiang, S.; Wu, L.C.; Ding, M.; Yu, Y.; Gall, J.G.; et al. Toxic PRn poly-dipeptides encoded by the C9orf72 repeat expansion block nuclear import and export. Proc. Natl. Acad. Sci. USA 2017, 114, E1111–E1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farg, M.A.; Konopka, A.; Soo, K.Y.; Ito, D.; Atkin, J.D. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2017, 26, 2882–2896. [Google Scholar] [CrossRef] [PubMed]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debska-Vielhaber, G.; Miller, I.; Peeva, V.; Zuschratter, W.; Walczak, J.; Schreiber, S.; Petri, S.; Machts, J.; Vogt, S.; Szczepanowska, J.; et al. Impairment of mitochondrial oxidative phosphorylation in skin fibroblasts of SALS and FALS patients is rescued by in vitro treatment with ROS scavengers. Exp. Neurol. 2021, 339, 113620. [Google Scholar] [CrossRef]
- Masuda, A.; Takeda, J.; Ohno, K. FUS-mediated regulation of alternative RNA processing in neurons: Insights from global transcriptome analysis. Wiley Interdiscip. Rev. RNA 2016, 7, 2–5. [Google Scholar] [CrossRef]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009, 323, 1208–1211. [Google Scholar] [CrossRef] [Green Version]
- Dormann, D.; Haass, C. Fused in sarcoma (FUS): An oncogene goes awry in neurodegeneration. Mol. Cell. Neurosci. 2013, 56, 475–486. [Google Scholar] [CrossRef]
- Gerbino, V.; Carrì, M.T.; Cozzolino, M.; Achsel, T. Mislocalised FUS mutants stall spliceosomal snRNPs in the cytoplasm. Neurobiol. Dis. 2013, 55, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Mirra, A.; Rossi, S.; Scaricamazza, S.; DI Salvio, M.; Salvatori, I.; Valle, C.; Rusmini, P.; Poletti, A.; Cestra, G.; Carrì, M.T.; et al. Functional interaction between FUS and SMN underlies SMA-like splicing changes in wild-type hFUS mice. Sci. Rep. 2017, 7, 2033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, T.; Chen, S.; Yu, Y.; Yan, B.; Haertlein, T.C.; Carrasco, M.A.; Tapia, J.C.; Zhai, B.; Das, R.; Lalancette-Hebert, M.; et al. FUS-SMN Protein Interactions Link the Motor Neuron Diseases ALS and SMA. Cell Rep. 2012, 2, 799–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dini Modigliani, S.; Morlando, M.; Errichelli, L.; Sabatelli, M.; Bozzoni, I. An ALS-associated mutation in the FUS 3′2-UTR disrupts a microRNA-FUS regulatory circuitry. Nat. Commun. 2014, 5, 4335. [Google Scholar] [CrossRef] [PubMed]
- Sabatelli, M.; Moncada, A.; Conte, A.; Lattante, S.; Marangi, G.; Luigetti, M.; Lucchini, M.; Mirabella, M.; Romano, A.; Del Grande, A.; et al. Mutations in the 3′ untranslated region of FUS causing FUS overexpression are associated with amyotrophic lateral sclerosis. Hum. Mol. Genet. 2013, 22, 4748–4755. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.A.; Ansorge, O.; Strong, M.; Bilbao, J.; Zinman, L.; Ang, L.C.; Baker, M.; Stewart, H.; Eisen, A.; Rademakers, R.; et al. Pathological heterogeneity in amyotrophic lateral sclerosis with FUS mutations: Two distinct patterns correlating with disease severity and mutation. Acta Neuropathol. 2011, 122, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.W.; Brent, J.R.; Tomlinson, A.; Shneider, N.A.; McCabe, B.D. The ALS-associated proteins FUS and TDP-43 function together to affect Drosophila locomotion and life span. J. Clin. Investig. 2011, 121, 4118–4126. [Google Scholar] [CrossRef] [Green Version]
- Kodavati, M.; Wang, H.; Hegde, M.L. Altered Mitochondrial Dynamics in Motor Neuron Disease: An Emerging Perspective. Cells 2020, 9, 1065. [Google Scholar] [CrossRef]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef]
- Zhou, Q.Q.; Chen, Y.P.; Wei, Q.Q.; Cao, B.; Wu, Y.; Zhao, B.; Ou, R.W.; Yang, J.; Chen, X.P.; Hadano, S.; et al. Mutation Screening of the CHCHD10 Gene in Chinese Patients with Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2017, 54, 3189–3194. [Google Scholar] [CrossRef]
- Perrone, F.; Nguyen, H.P.; Van Mossevelde, S.; Moisse, M.; Sieben, A.; Santens, P.; De Bleecker, J.; Vandenbulcke, M.; Engelborghs, S.; Baets, J.; et al. Investigating the role of ALS genes CHCHD10 and TUBA4A in Belgian FTD-ALS spectrum patients. Neurobiol. Aging 2017, 51, 177.e9–177.e16. [Google Scholar] [CrossRef] [PubMed]
- Lehmer, C.; Schludi, M.H.; Ransom, L.; Greiling, J.; Junghänel, M.; Exner, N.; Riemenschneider, H.; Zee, J.; Van Broeckhoven, C.; Weydt, P.; et al. A novel CHCHD10 mutation implicates a Mia40-dependent mitochondrial import deficit in ALS. EMBO Mol. Med. 2018, 10, e8558. [Google Scholar] [CrossRef] [PubMed]
- Keith, J.L.; Swinkin, E.; Gao, A.; Alminawi, S.; Zhang, M.; Mcgoldrick, P.; Mckeever, P.; Robertson, J.; Rogaeva, E.; Zinman, L. Neuropathologic description of CHCHD10 mutated amyotrophic lateral sclerosis. Neurol. Genet. 2020, 6, e394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Wu, B.P.; Nguyen, D.; Liu, Y.T.; Marani, M.; Hench, J.; Bénit, P.; Kozjak-Pavlovic, V.; Rustin, P.; Frank, S.; et al. CHCHD2 accumulates in distressed mitochondria and facilitates oligomerization of CHCHD10. Hum. Mol. Genet. 2018, 27, 3881–3900, Erratum in Hum. Mol. Genet. 2019, 28, 349. [Google Scholar]
- Woo, J.A.A.; Liu, T.; Trotter, C.; Fang, C.C.; De Narvaez, E.; Lepochat, P.; Maslar, D.; Bukhari, A.; Zhao, X.; Deonarine, A.; et al. Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat. Commun. 2017, 8, 15558. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Saw, W.T.; Tan, E.K. Mitochondrial CHCHD-Containing Proteins: Physiologic Functions and Link with Neurodegenerative Diseases. Mol. Neurobiol. 2017, 54, 5534–5546. [Google Scholar] [CrossRef]
- Ronchi, D.; Riboldi, G.; Del Bo, R.; Ticozzi, N.; Scarlato, M.; Galimberti, D.; Corti, S.; Silani, V.; Bresolin, N.; Comi, G. Pietro CHCHD10 mutations in Italian patients with sporadic amyotrophic lateral sclerosis. Brain 2015, 138, e372. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.S.; Holzbaur, E.L.F. Autophagy and mitophagy in ALS. Neurobiol. Dis. 2019, 122, 35–40. [Google Scholar] [CrossRef]
- Fivenson, E.M.; Lautrup, S.; Sun, N.; Scheibye-Knudsen, M.; Stevnsner, T.; Nilsen, H.; Bohr, V.A.; Fang, E.F. Mitophagy in neurodegeneration and aging. Neurochem. Int. 2017, 109, 202–209. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirulli, E.T.; Lasseigne, B.N.; Petrovski, S.; Sapp, P.C.; Dion, P.A.; Leblond, C.S.; Couthouis, J.; Lu, Y.F.; Wang, Q.; Krueger, B.J.; et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015, 347, 1436–1441. [Google Scholar] [CrossRef] [Green Version]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Kawakami, H. Optineurin and amyotrophic lateral sclerosis. Geriatr. Gerontol. Int. 2013, 13, 528–532. [Google Scholar] [CrossRef]
- Gerbino, V.; Kaunga, E.; Ye, J.; Canzio, D.; O’Keeffe, S.; Rudnick, N.D.; Guarnieri, P.; Lutz, C.M.; Maniatis, T. The Loss of TBK1 Kinase Activity in Motor Neurons or in All Cell Types Differentially Impacts ALS Disease Progression in SOD1 Mice. Neuron 2020, 106, 789–805.e5. [Google Scholar] [CrossRef]
- Oakes, J.A.; Davies, M.C.; Collins, M.O. TBK1: A new player in ALS linking autophagy and neuroinflammation. Mol. Brain 2017, 10, 5. [Google Scholar] [CrossRef] [Green Version]
- Brenner, D.; Yilmaz, R.; Müller, K.; Grehl, T.; Petri, S.; Meyer, T.; Grosskreutz, J.; Weydt, P.; Ruf, W.; Neuwirth, C.; et al. Hot-spot KIF5A mutations cause familial ALS. Brain 2018, 141, 688–697. [Google Scholar] [CrossRef] [Green Version]
- Le Ber, I.; De Septenville, A.; Millecamps, S.; Camuzat, A.; Caroppo, P.; Couratier, P.; Blanc, F.; Lacomblez, L.; Sellal, F.; Fleury, M.C.; et al. TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol. Aging 2015, 36, 3116.e5–3116.e8. [Google Scholar] [CrossRef]
- Freischmidt, A.; Müller, K.; Ludolph, A.C.; Weishaupt, J.H.; Andersen, P.M. Association of mutations in TBK1 with sporadic and familial amyotrophic lateral sclerosis and frontotemporal dementia. JAMA Neurol. 2017, 74, 110–113. [Google Scholar] [CrossRef]
- Shen, W.C.; Li, H.Y.; Chen, G.C.; Chern, Y.; Tu, P.H. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy- mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy 2015, 11, 685–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korac, J.; Schaeffer, V.; Kovacevic, I.; Clement, A.M.; Jungblut, B.; Behl, C.; Terzic, J.; Dikic, I. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J. Cell Sci. 2013, 126, 580–592. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef] [PubMed]
- Zachari, M.; Gudmundsson, S.R.; Li, Z.; Manifava, M.; Shah, R.; Smith, M.; Stronge, J.; Karanasios, E.; Piunti, C.; Kishi-Itakura, C.; et al. Selective Autophagy of Mitochondria on a Ubiquitin-Endoplasmic-Reticulum Platform. Dev. Cell 2019, 50, 627–643.e5. [Google Scholar] [CrossRef] [Green Version]
- Grosskreutz, J.; Van Den Bosch, L.; Keller, B.U. Calcium dysregulation in amyotrophic lateral sclerosis. Cell Calcium 2010, 47, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, F.R.; Winkler, K.; Kuznetsov, A.V.; Bartels, C.; Vielhaber, S.; Feistner, H.; Kunz, W.S. Impairment of mitochondrial function in skeletal muscle of patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 1998, 156, 65–72. [Google Scholar] [CrossRef]
- Shaw, P.; Eggett, C.J. Molecular factors underlying selective vulnerability of motor neurons to neurodegeneration in amyotrophic lateral sclerosis. J. Neurol. 2000, 247, I17–I27. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bosch, L.; Vandenberghe, W.; Klaassen, H.; Van Houtte, E.; Robberecht, W. Ca(2+)-permeable AMPA receptors and selective vulnerability of motor neurons. J. Neurol. Sci. 2000, 180, 29–34. [Google Scholar] [CrossRef]
- Engelen-Lee, J.; Blokhuis, A.M.; Spliet, W.G.M.; Pasterkamp, R.J.; Aronica, E.; Demmers, J.A.A.; Broekhuizen, R.; Nardo, G.; Bovenschen, N.; Van Den Berg, L.H. Proteomic profiling of the spinal cord in ALS: Decreased ATP5D levels suggest synaptic dysfunction in ALS pathogenesis. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Benson, B.C.; Shaw, P.J.; Azzouz, M.; Highley, J.R.T.; Hautbergue, G.M. Proteinopathies as Hallmarks of Impaired Gene Expression, Proteostasis and Mitochondrial Function in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2021, 15, 783624. [Google Scholar] [CrossRef]
- Walczak, J.; Debska-Vielhaber, G.; Vielhaber, S.; Szymański, J.; Charzyńska, A.; Duszyński, J.; Szczepanowska, J. Distinction of sporadic and familial forms of ALS based on mitochondrial characteristics. FASEB J. 2019, 33, 4388–4403. [Google Scholar] [CrossRef] [PubMed]
- Ladd, A.C.; Brohawn, D.G.; Thomas, R.R.; Keeney, P.M.; Berr, S.S.; Khan, S.M.; Portell, F.R.; Shakenov, M.Z.; Antkowiak, P.F.; Kundu, B.; et al. RNA-seq analyses reveal that cervical spinal cords and anterior motor neurons from amyotrophic lateral sclerosis subjects show reduced expression of mitochondrial DNA-encoded respiratory genes, and rhTFAM may correct this respiratory deficiency. Brain Res. 2017, 1667, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Veyrat-Durebex, C.; Bris, C.; Codron, P.; Bocca, C.; Chupin, S.; Corcia, P.; Vourc’h, P.; Hergesheimer, R.; Cassereau, J.; Funalot, B.; et al. Metabo-lipidomics of Fibroblasts and Mitochondrial-Endoplasmic Reticulum Extracts from ALS Patients Shows Alterations in Purine, Pyrimidine, Energetic, and Phospholipid Metabolisms. Mol. Neurobiol. 2019, 56, 5780–5791. [Google Scholar] [CrossRef] [PubMed]
- Delic, V.; Kurien, C.; Cruz, J.; Zivkovic, S.; Barretta, J.; Thomson, A.; Hennessey, D.; Joseph, J.; Ehrhart, J.; Willing, A.E.; et al. Discrete mitochondrial aberrations in the spinal cord of sporadic ALS patients. J. Neurosci. Res. 2018, 96, 1353–1366. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Jiao, Y.; Ferrando, L.M.; Yablonska, S.; Li, F.; Horoszko, E.C.; Lacomis, D.; Friedlander, R.M.; Carlisle, D.L. Neuronal mitochondrial dysfunction in sporadic amyotrophic lateral sclerosis is developmentally regulated. Sci. Rep. 2021, 11, 18916. [Google Scholar] [CrossRef]
- Rahman, S. Mitochondrial disease in children. J. Intern. Med. 2020, 287, 609–633. [Google Scholar] [CrossRef]
- Son, J.M.; Lee, C. Mitochondria: Multifaceted regulators of aging. BMB Rep. 2019, 52, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Webb, M.; Sideris, D.P. Intimate Relations-Mitochondria and Ageing. Int. J. Mol. Sci. 2020, 21, 7580. [Google Scholar] [CrossRef]
- Akbari, M.; Kirkwood, T.B.L.; Bohr, V.A. Mitochondria in the signaling pathways that control longevity and health span. Ageing Res. Rev. 2019, 54, 100940. [Google Scholar] [CrossRef]
- Rosenbohm, A.; Peter, R.; Dorst, J.; Kassubek, J.; Rothenbacher, D.; Nagel, G.; Ludolph, A.C. Life Course of Physical Activity and Risk and Prognosis of Amyotrophic Lateral Sclerosis in a German ALS Registry. Neurology 2021, 97, e1955–e1963. [Google Scholar] [CrossRef]
- Julian, T.H.; Glascow, N.; Barry, A.D.F.; Moll, T.; Harvey, C.; Klimentidis, Y.C.; Newell, M.; Zhang, S.; Snyder, M.P.; Cooper-Knock, J.; et al. Physical exercise is a risk factor for amyotrophic lateral sclerosis: Convergent evidence from Mendelian randomisation, transcriptomics and risk genotypes. EBioMedicine 2021, 68, 103397. [Google Scholar] [CrossRef] [PubMed]
- Raymond, J.; Mehta, P.; Larson, T.; Factor-Litvak, P.; Davis, B.; Horton, K. History of vigorous leisure-time physical activity and early onset amyotrophic lateral sclerosis (ALS), data from the national ALS registry: 2010–2018. Amyotroph. Lateral Scler. Front. Degener. 2021, 22, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.; Lanfranconi, F.; Corna, G.; Bonazzi, R.; Marchese, S.; Magnoni, A.; Tremolizzo, L. Tailored Exercise Training Counteracts Muscle Disuse and Attenuates Reductions in Physical Function in Individuals With Amyotrophic Lateral Sclerosis. Front. Physiol. 2019, 10, 1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaricamazza, S.; Salvatori, I.; Ferri, A.; Valle, C. Skeletal Muscle in ALS: An Unappreciated Therapeutic Opportunity? Cells 2021, 10, 525. [Google Scholar] [CrossRef]
- Sassani, M.; Alix, J.J.; McDermott, C.J.; Baster, K.; Hoggard, N.; Wild, J.M.; Mortiboys, H.J.; Shaw, P.J.; Wilkinson, I.D.; Jenkins, T.M. Magnetic resonance spectroscopy reveals mitochondrial dysfunction in amyotrophic lateral sclerosis. Brain 2021, 143, 3603–3618. [Google Scholar] [CrossRef]
- Park, S.K.; Park, S.; Liebman, S.W. Respiration Enhances TDP-43 Toxicity, but TDP-43 Retains Some Toxicity in the Absence of Respiration. J. Mol. Biol. 2019, 431, 2050–2059. [Google Scholar] [CrossRef]
- Giachin, G.; Bouverot, R.; Acajjaoui, S.; Pantalone, S.; Soler-López, M. Dynamics of Human Mitochondrial Complex I Assembly: Implications for Neurodegenerative Diseases. Front. Mol. Biosci. 2016, 3, 43. [Google Scholar] [CrossRef] [Green Version]
- Joshi, A.U.; Saw, N.L.; Vogel, H.; Cunnigham, A.D.; Shamloo, M.; Mochly-Rosen, D. Inhibition of Drp1/Fis1 interaction slows progression of amyotrophic lateral sclerosis. EMBO Mol. Med. 2018, 10, e8166. [Google Scholar] [CrossRef]
- Magrané, J.; Hervias, I.; Henning, M.S.; Damiano, M.; Kawamata, H.; Manfredi, G. Mutant SOD1 in neuronal mitochondria causes toxicity and mitochondrial dynamics abnormalities. Hum. Mol. Genet. 2009, 18, 4552–4564. [Google Scholar] [CrossRef] [Green Version]
- Scaricamazza, S.; Salvatori, I.; Giacovazzo, G.; Loeffler, J.P.; Renè, F.; Rosina, M.; Quessada, C.; Proietti, D.; Heil, C.; Rossi, S.; et al. Skeletal-Muscle Metabolic Reprogramming in ALS-SOD1 G93A Mice Predates Disease Onset and Is a Promising Therapeutic Target. iScience 2020, 23, 101087. [Google Scholar] [CrossRef]
- Scaricamazza, S.; Salvatori, I.; Amadio, S.; Nesci, V.; Torcinaro, A.; Giacovazzo, G.; Primiano, A.; Gloriani, M.; Candelise, N.; Pieroni, L.; et al. Repurposing of Trimetazidine for amyotrophic lateral sclerosis: A study in SOD1 G93A mice. Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Gene Symbol | Protein Symbol | Physiological Functions | Mitochondrial Related Dysfunction | Mitochondrial Localization | References |
|---|---|---|---|---|---|
| sod1 | SOD1 | Cytosolic antioxidant | ER-Ca2+ homeostasis Mitophagy Apoptosis | IMS OMM MAMs | [16,17,18,19,20,21] |
| tardbp | TDP-43 | Splicing regulation RNA transport miRNA biogenesis Autophagy Stress response | ETC impairment Ca2+ homeostasis and excitotoxicity Mitophagy Inflammation mt genes regulation | IMS IMM cristae Matrix | [22,23,24,25,26,27,28,29,30,31] |
| c9orf72 | C9ORF72 | Transcription Splicing regulation Ribosome-associated quality control Endosomal trafficking Autophagy Axonal maintenance | ETC impairment Bioenergetic deficit Oxidative stress ER-Ca2+ homeostasis Morphology Quality control | IMM IMS cristae OMM proximity | [32,33,34,35,36,37,38,39] |
| fus | FUS | Splicing regulation RNA transport Maintenance of genomic integrity miRNA processing ER–mitochondria trafficking | ETC/oxidative stress ER-Ca2+ homeostasis Dynamics (axonal transport) mtDNA repair | IMM MAMs Matrix | [40,41,42,43,44,45,46,47,48] |
| chchd10 | CHCHD10 | MICOS integrity Oxidative phosphorylation | ETC impairment Structural integrity Dynamics (fusion/fission) Stress response | IMS IMM cristae | [49,50,51,52,53,54,55,56,57,58,59] |
| tbk1 | TBK1 | Autophagy Innate immunity signaling | Mitophagy | OMM proximity | [60,61,62,63] |
| optn | OPTN | Golgi maintenance and membrane trafficking Autophagy | Mitophagy | OMM proximity | [60,64,65,66,67] |
| sigma1r | SIGMA1R | ER–mitochondria trafficking Antioxidant response | ER-Ca2+ homeostasis | MAMs | [68,69,70,71] |
| Altered Mitochondrial Function(s) | Experimental Model(s) | References |
|---|---|---|
| Respiratory chain and mitochondrial bioenergetic | Spinal cord sections | [149] |
| iPSCs-derived MNs | [107,150] | |
| Fibroblasts | [107] | |
| PBMCs | [20] | |
| Oxidative stress | iPSCs-derived MNs | [151] |
| Fibroblasts | [107,150] | |
| PBMCs | [20] | |
| Ca2+ homeostasis | Fibroblasts | [107,150] |
| PBMCs | [20] | |
| Mitochondrial distribution | Spinal cord sections | [107,150] |
| Mitochondrial biogenesis | PBMCs | [20] |
| mtDNA expression and protein involved in mitochondrial function | Spinal cord sections and laser-captured motor neurons | [152] |
| Fibroblasts | [153] | |
| iPSCs-derived MNs | [151] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Candelise, N.; Salvatori, I.; Scaricamazza, S.; Nesci, V.; Zenuni, H.; Ferri, A.; Valle, C. Mechanistic Insights of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis: An Update on a Lasting Relationship. Metabolites 2022, 12, 233. https://doi.org/10.3390/metabo12030233
Candelise N, Salvatori I, Scaricamazza S, Nesci V, Zenuni H, Ferri A, Valle C. Mechanistic Insights of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis: An Update on a Lasting Relationship. Metabolites. 2022; 12(3):233. https://doi.org/10.3390/metabo12030233
Chicago/Turabian StyleCandelise, Niccolò, Illari Salvatori, Silvia Scaricamazza, Valentina Nesci, Henri Zenuni, Alberto Ferri, and Cristiana Valle. 2022. "Mechanistic Insights of Mitochondrial Dysfunction in Amyotrophic Lateral Sclerosis: An Update on a Lasting Relationship" Metabolites 12, no. 3: 233. https://doi.org/10.3390/metabo12030233