
Comprehensive Peak Characterization (CPC) in Untargeted LC–MS Analysis

 ,
, 
 ,
,  and
and
Abstract
:1. Introduction
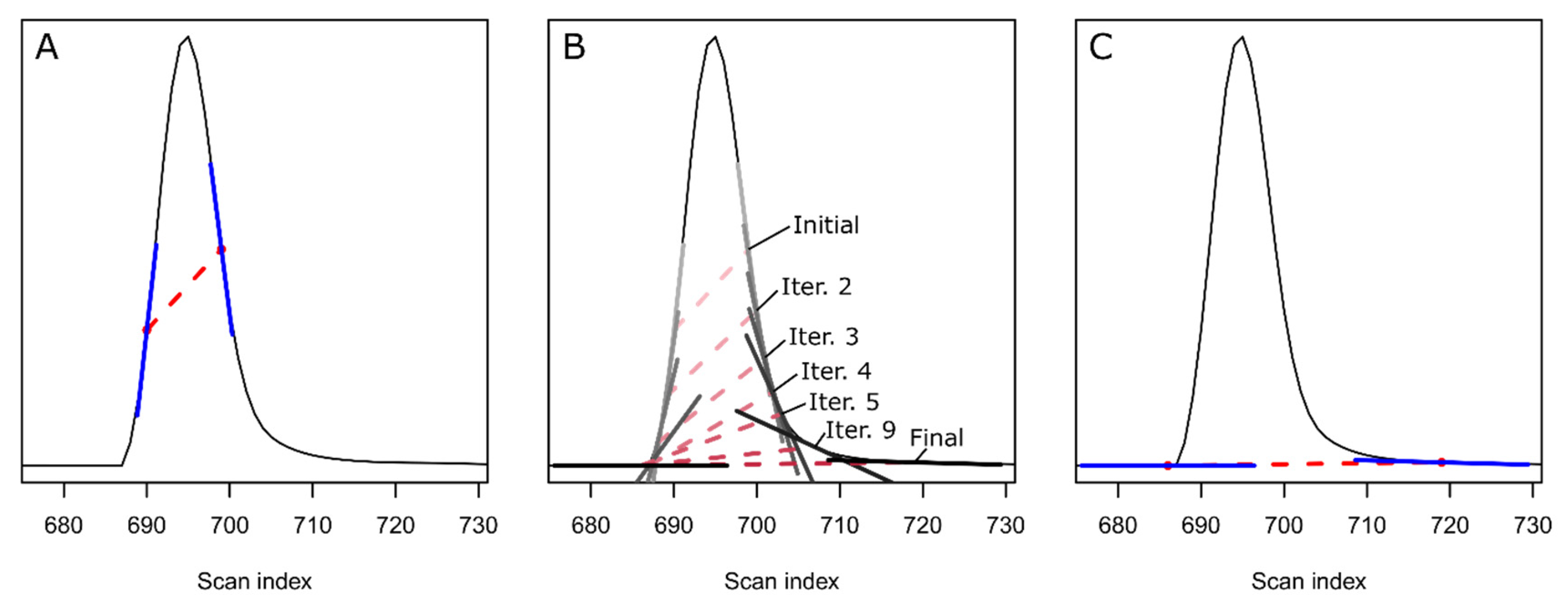
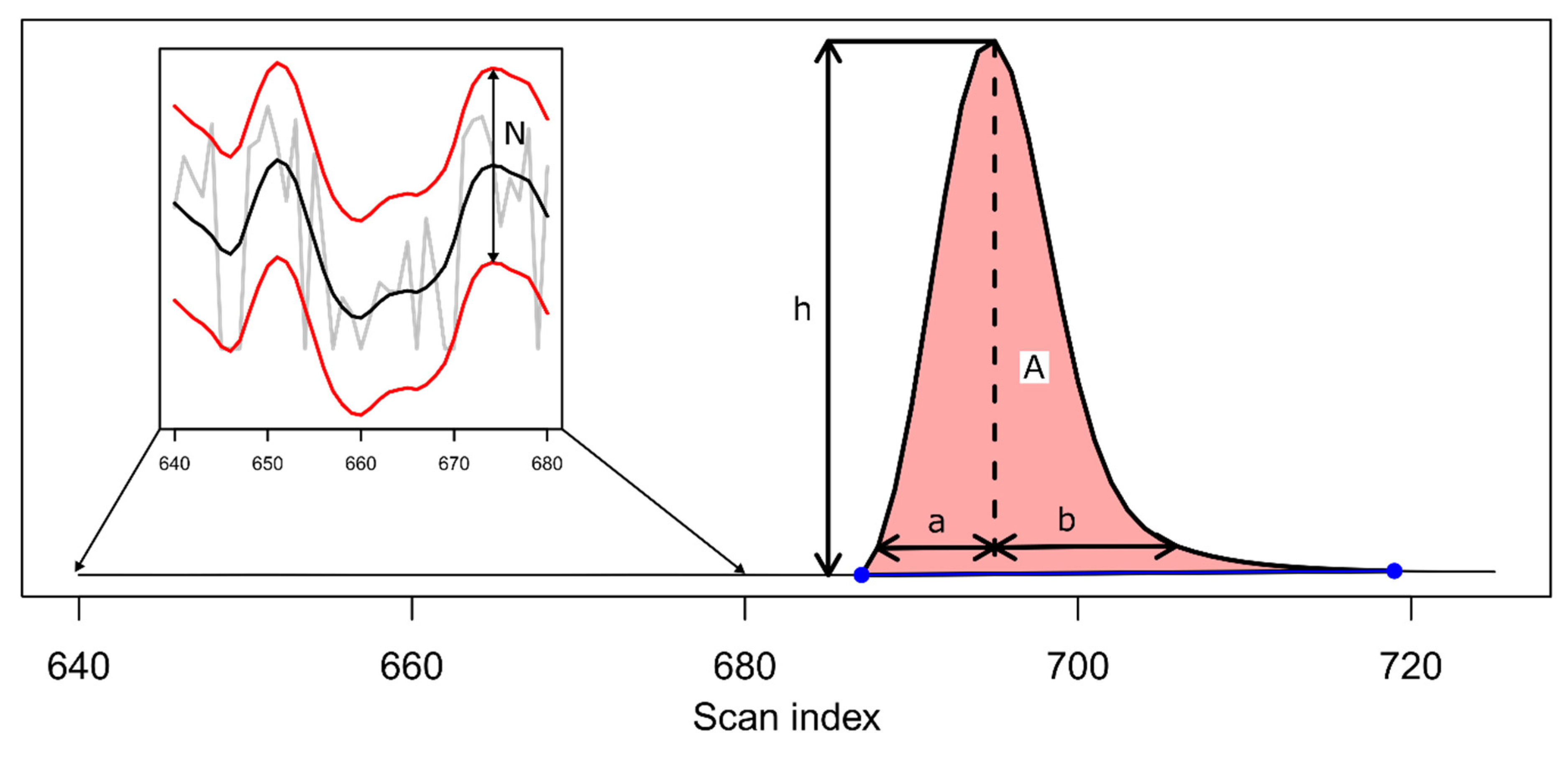
2. Theory and Methodology
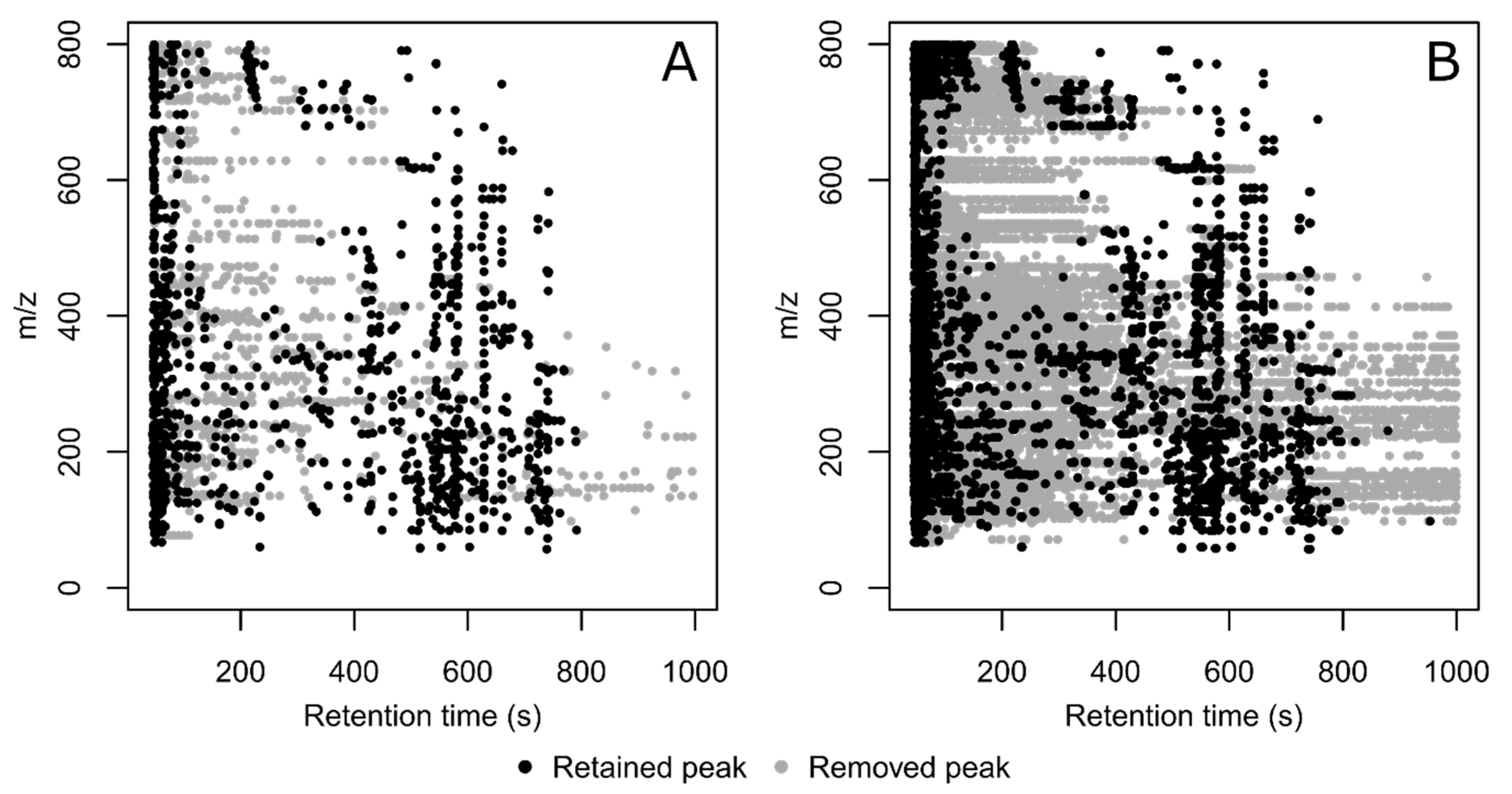
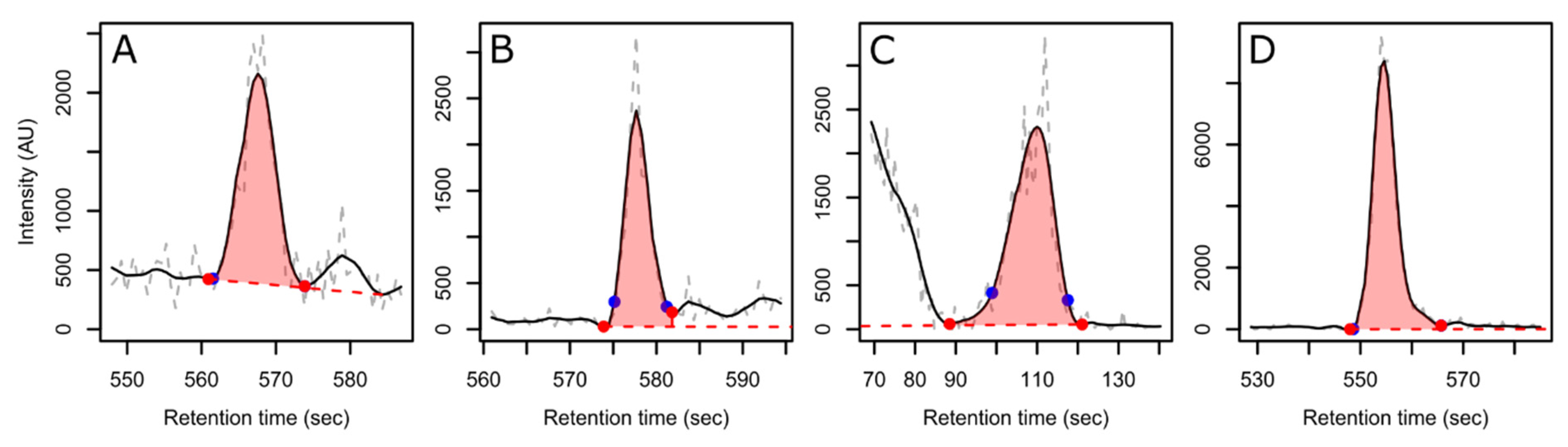
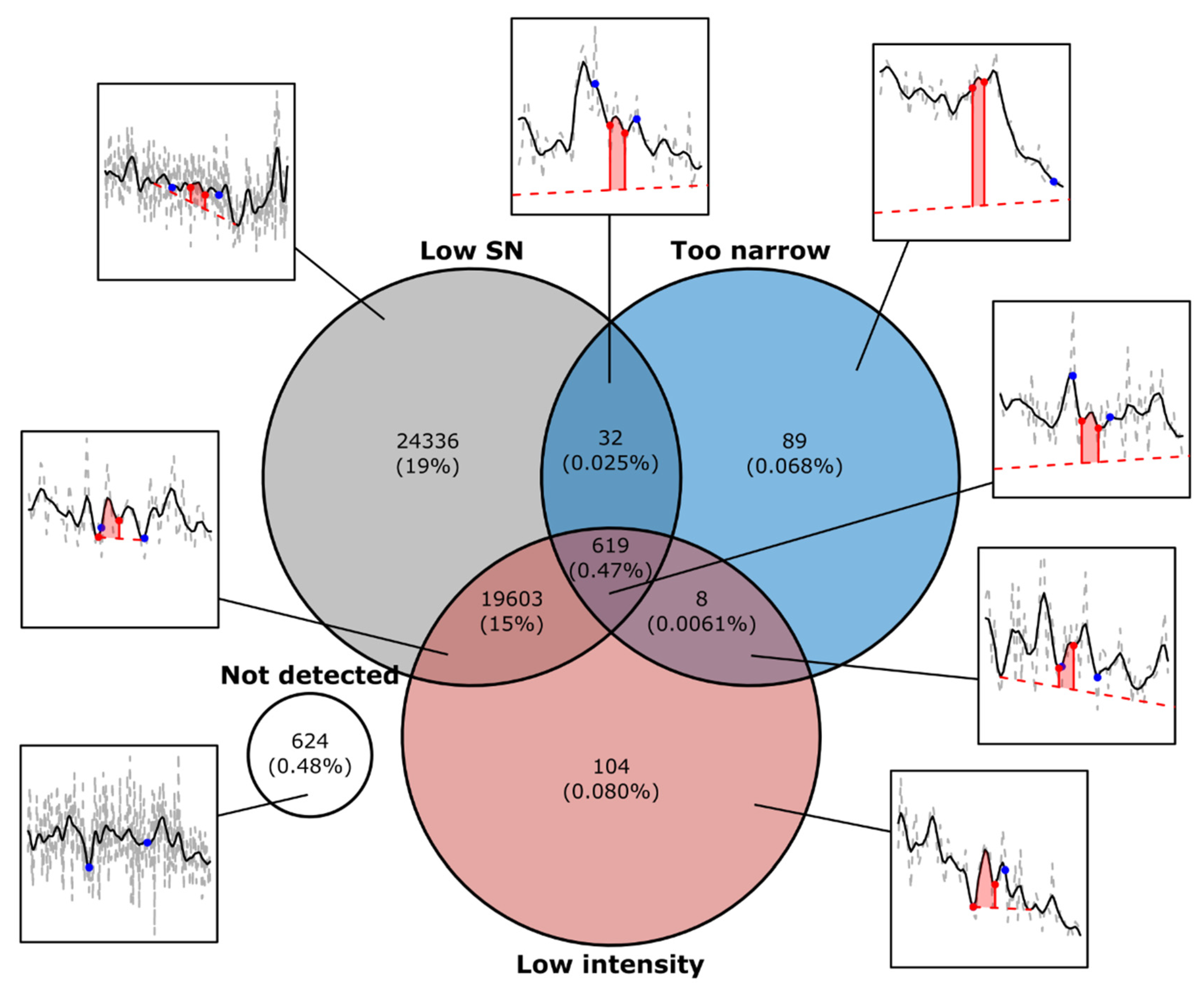
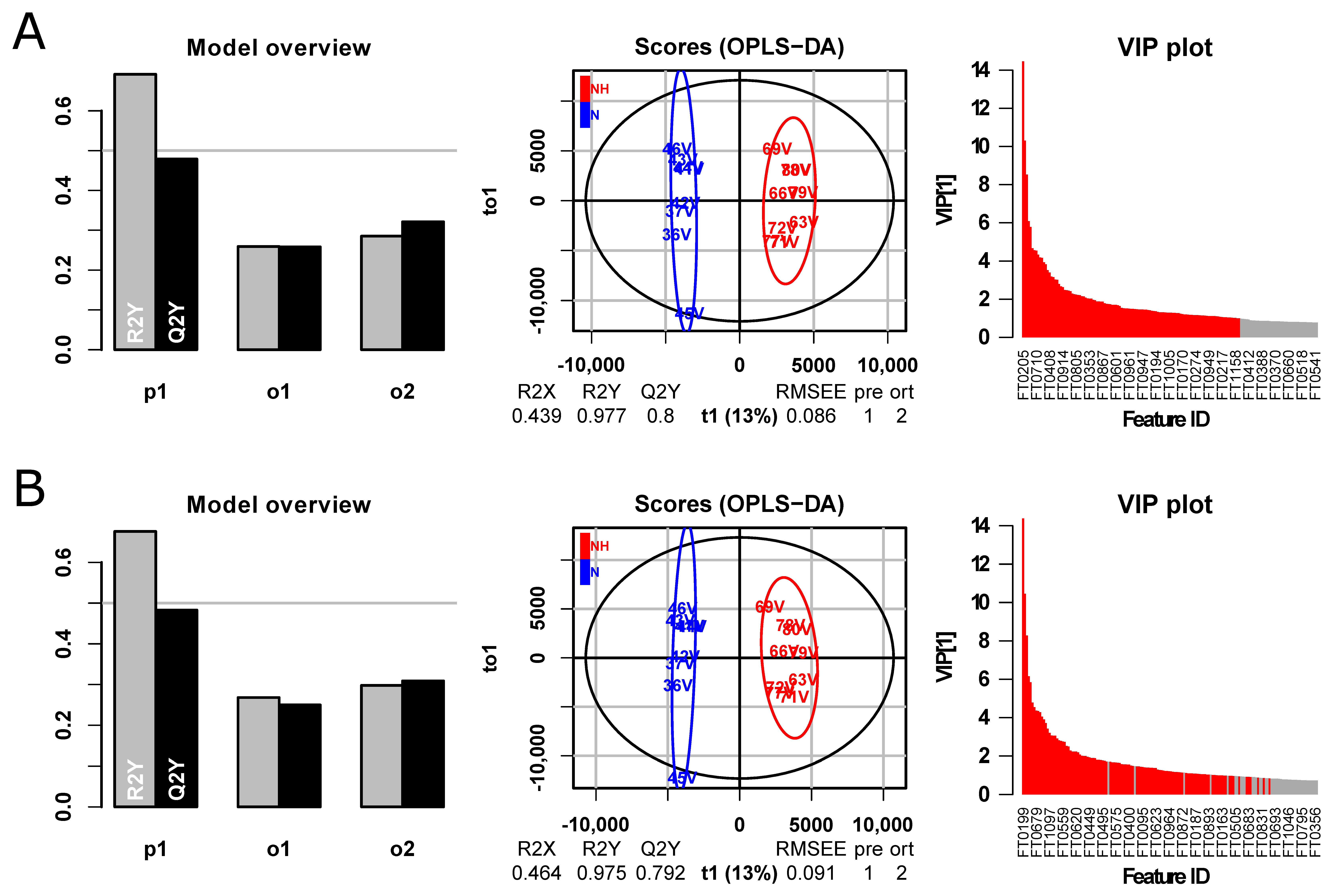
3. Results and Discussion
4. Materials and Methods
4.1. Software
4.2. Untargeted Metabolomics Analysis of Guinea Pig Perilymph Samples
4.3. Data Processing with XCMS and CPC
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wishart, D.S.; Jewison, T.; Guo, A.C.; Wilson, M.; Knox, C.; Liu, Y.; Djoumbou, Y.; Mandal, R.; Aziat, F.; Dong, E.; et al. HMDB 3.0—The Human Metabolome Database in 2013. Nucleic Acids Res. 2012, 41, D801–D807. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.P. Complementing the Genome with an “Exposome”: The Outstanding Challenge of Environmental Exposure Measurement in Molecular Epidemiology. Cancer Epidemiol. Prev. Biomark. 2005, 14, 1847–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scalbert, A.; Brennan, L.; Manach, C.; Andres-Lacueva, C.; Dragsted, L.O.; Draper, J.; Rappaport, S.M.; van der Hooft, J.J.; Wishart, D.S. The Food Metabolome: A Window over Dietary Exposure. Am. J. Clin. Nutr. 2014, 99, 1286–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.H.; Ivanisevic, J.; Siuzdak, G. Metabolomics: Beyond Biomarkers and towards Mechanisms. Nat. Rev. Mol. Cell Biol. 2016, 17, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Bletsou, A.A.; Jeon, J.; Hollender, J.; Archontaki, E.; Thomaidis, N.S. Targeted and Non-Targeted Liquid Chromatography-Mass Spectrometric Workflows for Identification of Transformation Products of Emerging Pollutants in the Aquatic Environment. Trends Anal. Chem. 2015, 66, 32–44. [Google Scholar] [CrossRef] [Green Version]
- Hogenboom, A.C.; van Leerdam, J.A.; de Voogt, P. Accurate Mass Screening and Identification of Emerging Contaminants in Environmental Samples by Liquid Chromatography–Hybrid Linear Ion Trap Orbitrap Mass Spectrometry. J. Chromatogr. A 2009, 1216, 510–519. [Google Scholar] [CrossRef] [Green Version]
- Al-Khelaifi, F.; Diboun, I.; Donati, F.; Botrè, F.; Alsayrafi, M.; Georgakopoulos, C.; Suhre, K.; Yousri, N.A.; Elrayess, M.A. A Pilot Study Comparing the Metabolic Profiles of Elite-Level Athletes from Different Sporting Disciplines. Sports Med. 2018, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narduzzi, L.; Dervilly, G.; Marchand, A.; Audran, M.; Le Bizec, B.; Buisson, C. Applying Metabolomics to Detect Growth Hormone Administration in Athletes: Proof of Concept. Drug Test. Anal. 2020, 12, 887–899. [Google Scholar] [CrossRef]
- Jamin, E.L.; Bonvallot, N.; Tremblay-Franco, M.; Cravedi, J.-P.; Chevrier, C.; Cordier, S.; Debrauwer, L. Untargeted Profiling of Pesticide Metabolites by LC–HRMS: An Exposomics Tool for Human Exposure Evaluation. Anal. Bioanal. Chem. 2014, 406, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The Human Serum Metabolome. PLoS ONE 2011, 6, e16957. [Google Scholar] [CrossRef] [Green Version]
- Dunn, W.B.; Bailey, N.J.C.; Johnson, H.E. Measuring the Metabolome: Current Analytical Technologies. Analyst 2005, 130, 606. [Google Scholar] [CrossRef] [PubMed]
- Schug, K.; McNair, H.M. Adduct Formation in Electrospray Ionization. Part 1: Common Acidic Pharmaceuticals. J. Sep. Sci. 2002, 25, 759–766. [Google Scholar] [CrossRef]
- Katajamaa, M.; Orešič, M. Data Processing for Mass Spectrometry-Based Metabolomics. J. Chromatogr. A 2007, 1158, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Orešič, M. MZmine 2: Modular Framework for Processing, Visualizing, and Analyzing Mass Spectrometry-Based Molecular Profile Data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, O.D.; Sumner, S.J.; Li, S.; Barnes, S.; Du, X. Detailed Investigation and Comparison of the XCMS and MZmine 2 Chromatogram Construction and Chromatographic Peak Detection Methods for Preprocessing Mass Spectrometry Metabolomics Data. Anal. Chem. 2017, 89, 8689–8695. [Google Scholar] [CrossRef] [PubMed]
- Tautenhahn, R.; Böttcher, C.; Neumann, S. Highly Sensitive Feature Detection for High Resolution LC/MS. BMC Bioinform. 2008, 9, 504. [Google Scholar] [CrossRef] [Green Version]
- Coble, J.B.; Fraga, C.G. Comparative Evaluation of Preprocessing Freeware on Chromatography/Mass Spectrometry Data for Signature Discovery. J. Chromatogr. A 2014, 1358, 155–164. [Google Scholar] [CrossRef]
- Rafiei, A.; Sleno, L. Comparison of Peak-Picking Workflows for Untargeted Liquid Chromatography/High-Resolution Mass Spectrometry Metabolomics Data Analysis: Comparing Peak Picking of LC/HRMS Data. Rapid Commun. Mass Spectrom. 2015, 29, 119–127. [Google Scholar] [CrossRef]
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and Considerations for the Use of System Suitability and Quality Control Samples in Mass Spectrometry Assays Applied in Untargeted Clinical Metabolomic Studies. Metabolomics 2018, 14, 72. [Google Scholar] [CrossRef] [Green Version]
- Want, E.J.; Wilson, I.D.; Gika, H.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Holmes, E.; Nicholson, J.K. Global Metabolic Profiling Procedures for Urine Using UPLC–MS. Nat. Protoc. 2010, 5, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Myers, O.D.; Sumner, S.J.; Li, S.; Barnes, S.; Du, X. One Step Forward for Reducing False Positive and False Negative Compound Identifications from Mass Spectrometry Metabolomics Data: New Algorithms for Constructing Extracted Ion Chromatograms and Detecting Chromatographic Peaks. Anal. Chem. 2017, 89, 8696–8703. [Google Scholar] [CrossRef] [PubMed]
- Borgsmüller, N.; Gloaguen, Y.; Opialla, T.; Blanc, E.; Sicard, E.; Royer, A.L.; Le Bizec, B.; Durand, S.; Migné, C.; Pétéra, M.; et al. WiPP: Workflow for Improved Peak Picking for Gas Chromatography-Mass Spectrometry (GC-MS) Data. Metabolites 2019, 9, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chetnik, K.; Petrick, L.; Pandey, G. MetaClean: A Machine Learning-Based Classifier for Reduced False Positive Peak Detection in Untargeted LC–MS Metabolomics Data. Metabolomics 2020, 16, 117. [Google Scholar] [CrossRef]
- Kantz, E.D.; Tiwari, S.; Watrous, J.D.; Cheng, S.; Jain, M. Deep Neural Networks for Classification of LC-MS Spectral Peaks. Anal. Chem. 2019, 91, 12407–12413. [Google Scholar] [CrossRef]
- Melnikov, A.D.; Tsentalovich, Y.P.; Yanshole, V.V. Deep Learning for the Precise Peak Detection in High-Resolution LC–MS Data. Anal. Chem. 2020, 92, 588–592. [Google Scholar] [CrossRef] [Green Version]
- Gloaguen, Y.; Kirwan, J.; Beule, D. Deep Learning Assisted Peak Curation for Large Scale LC-MS Metabolomics. bioRxiv 2020. [CrossRef]
- Jirayupat, C.; Nagashima, K.; Hosomi, T.; Takahashi, T.; Tanaka, W.; Samransuksamer, B.; Zhang, G.; Liu, J.; Kanai, M.; Yanagida, T. Image Processing and Machine Learning for Automated Identification of Chemo-/Biomarkers in Chromatography–Mass Spectrometry. Anal. Chem. 2021, 93, 14708–14715. [Google Scholar] [CrossRef]
- ApexTrack Integration: Theory and Application. In Empower 3 Software; Waters Corp.: Milford, MA, USA, 2016.
- Council of Europe. European Pharmacopoeia, 10th ed.; Council of Europe: Strasbourg, France, 2019; ISBN 92-871-8921-8. [Google Scholar]
- Miller, J.M. Chromatography: Concepts and Contrasts, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; ISBN 978-0-471-98058-2. [Google Scholar]
- Pirttilä, K.; Videhult Pierre, P.; Haglöf, J.; Engskog, M.; Hedeland, M.; Laurell, G.; Arvidsson, T.; Pettersson, C. An LCMS-Based Untargeted Metabolomics Protocol for Cochlear Perilymph: Highlighting Metabolic Effects of Hydrogen Gas on the Inner Ear of Noise Exposed Guinea Pigs. Metabolomics 2019, 15, 138. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | No. Detected Peaks in All Injections | % Filled Peaks in the Dataset | % of Peaks Associated to a Feature | No. Features | % of Features with RSDQC ≤ 30% |
|---|---|---|---|---|---|
| Without CPC filtering | 130,351 | 13.7% | 49.3% | 1270 | 85.7% |
| With CPC filtering | 84,936 | 18.9% | 69.1% | 1213 | 87.0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pirttilä, K.; Balgoma, D.; Rainer, J.; Pettersson, C.; Hedeland, M.; Brunius, C. Comprehensive Peak Characterization (CPC) in Untargeted LC–MS Analysis. Metabolites 2022, 12, 137. https://doi.org/10.3390/metabo12020137
Pirttilä K, Balgoma D, Rainer J, Pettersson C, Hedeland M, Brunius C. Comprehensive Peak Characterization (CPC) in Untargeted LC–MS Analysis. Metabolites. 2022; 12(2):137. https://doi.org/10.3390/metabo12020137
Chicago/Turabian StylePirttilä, Kristian, David Balgoma, Johannes Rainer, Curt Pettersson, Mikael Hedeland, and Carl Brunius. 2022. "Comprehensive Peak Characterization (CPC) in Untargeted LC–MS Analysis" Metabolites 12, no. 2: 137. https://doi.org/10.3390/metabo12020137