


Comparative Study of the Gut Microbiota Community between the Farmed and Wild Mastacembelus armatus (Zig-Zag Eel)
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. 16 S rRNA Gene Sequencing
2.3. Data Analysis
3. Results
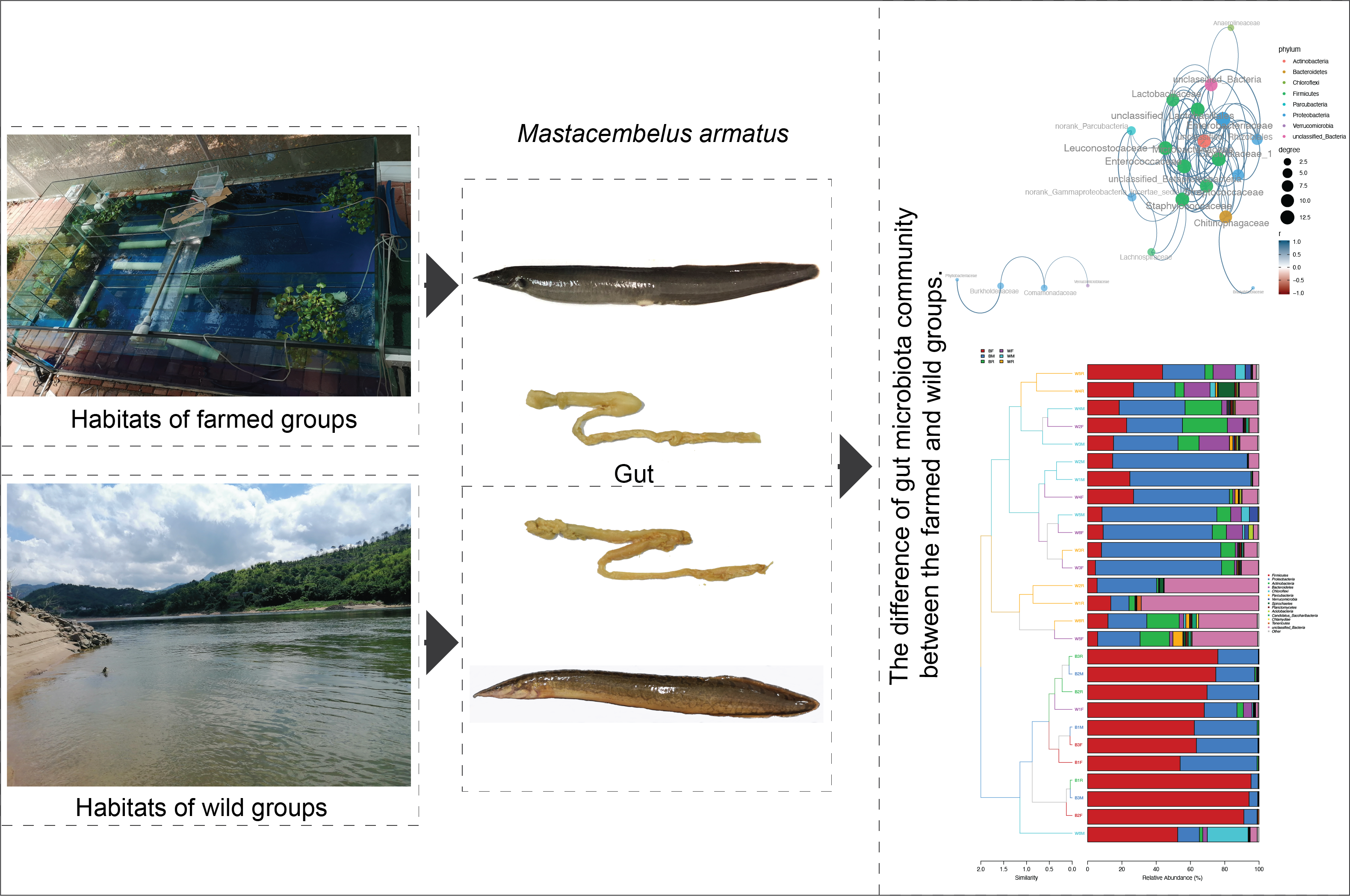
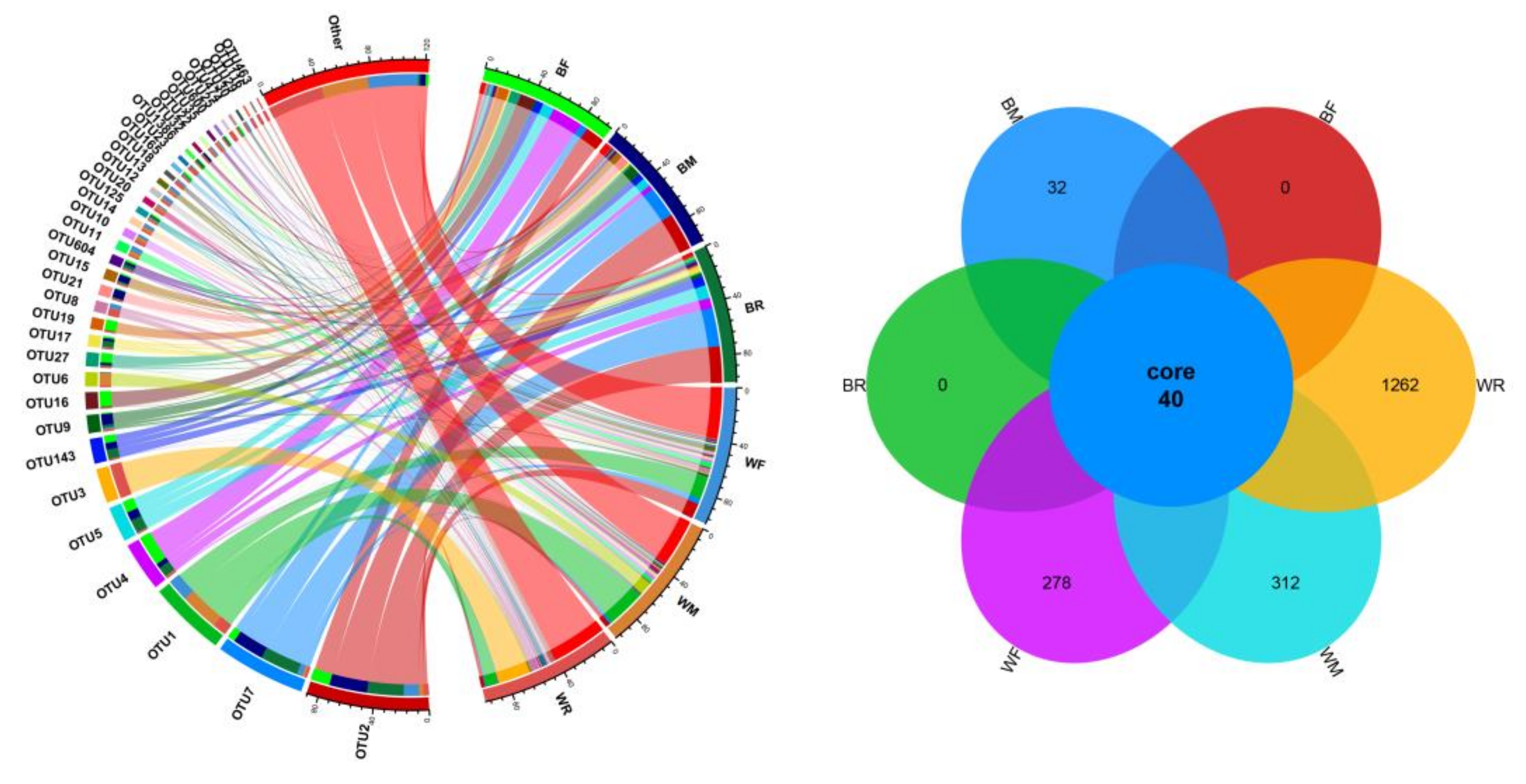
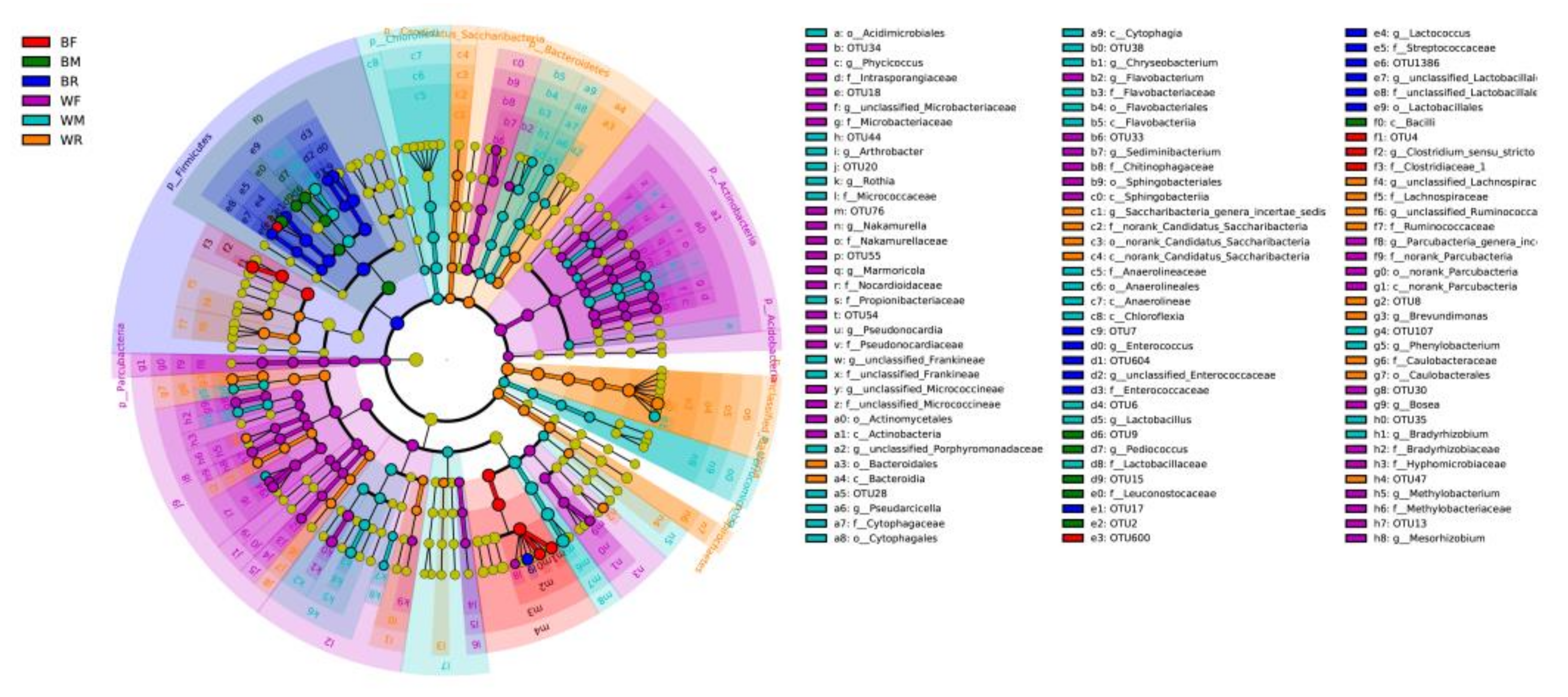
3.1. Composition of the Gut Microbiota in the Cultivated and Wild Zig-Zag Eels
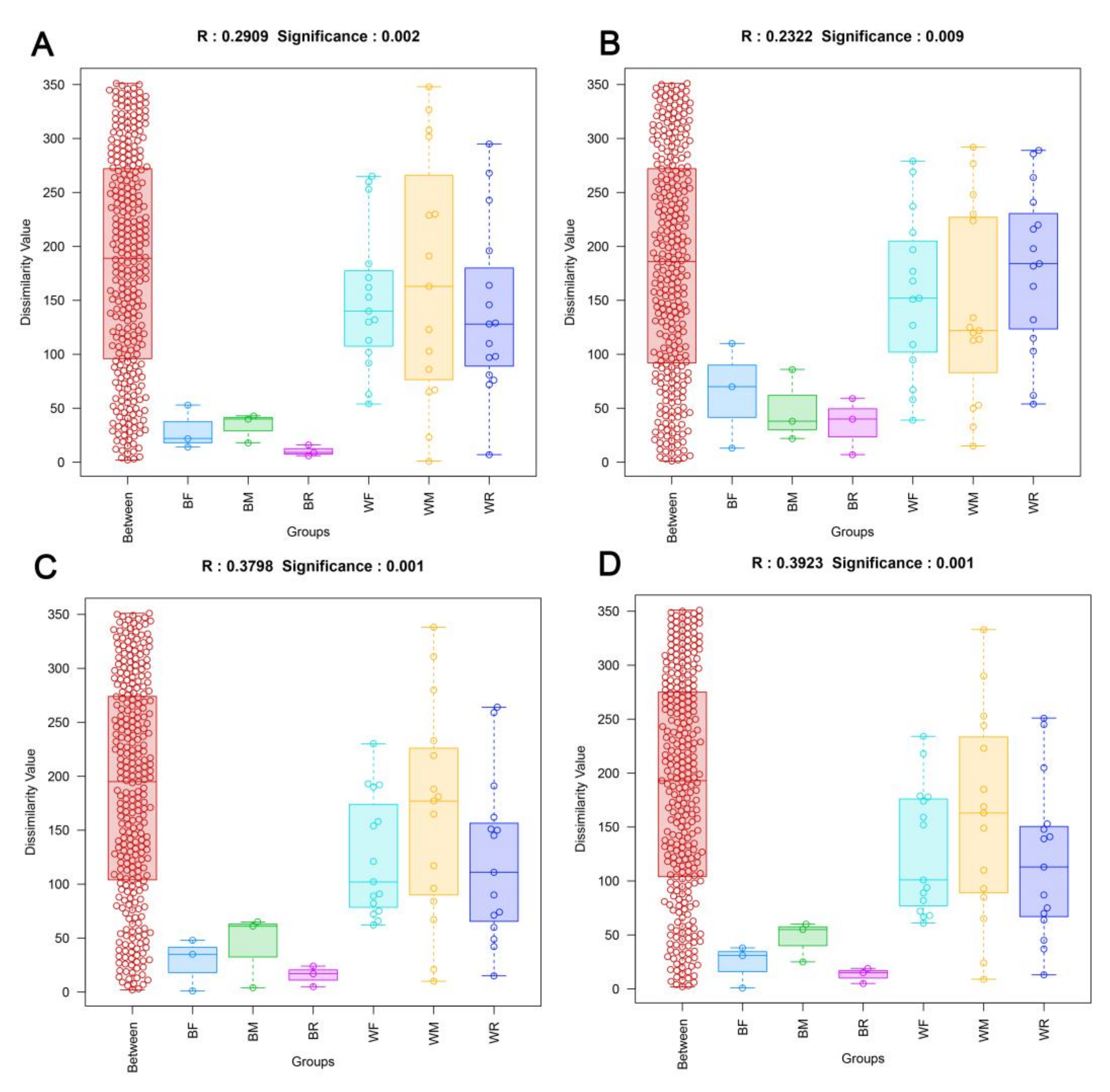
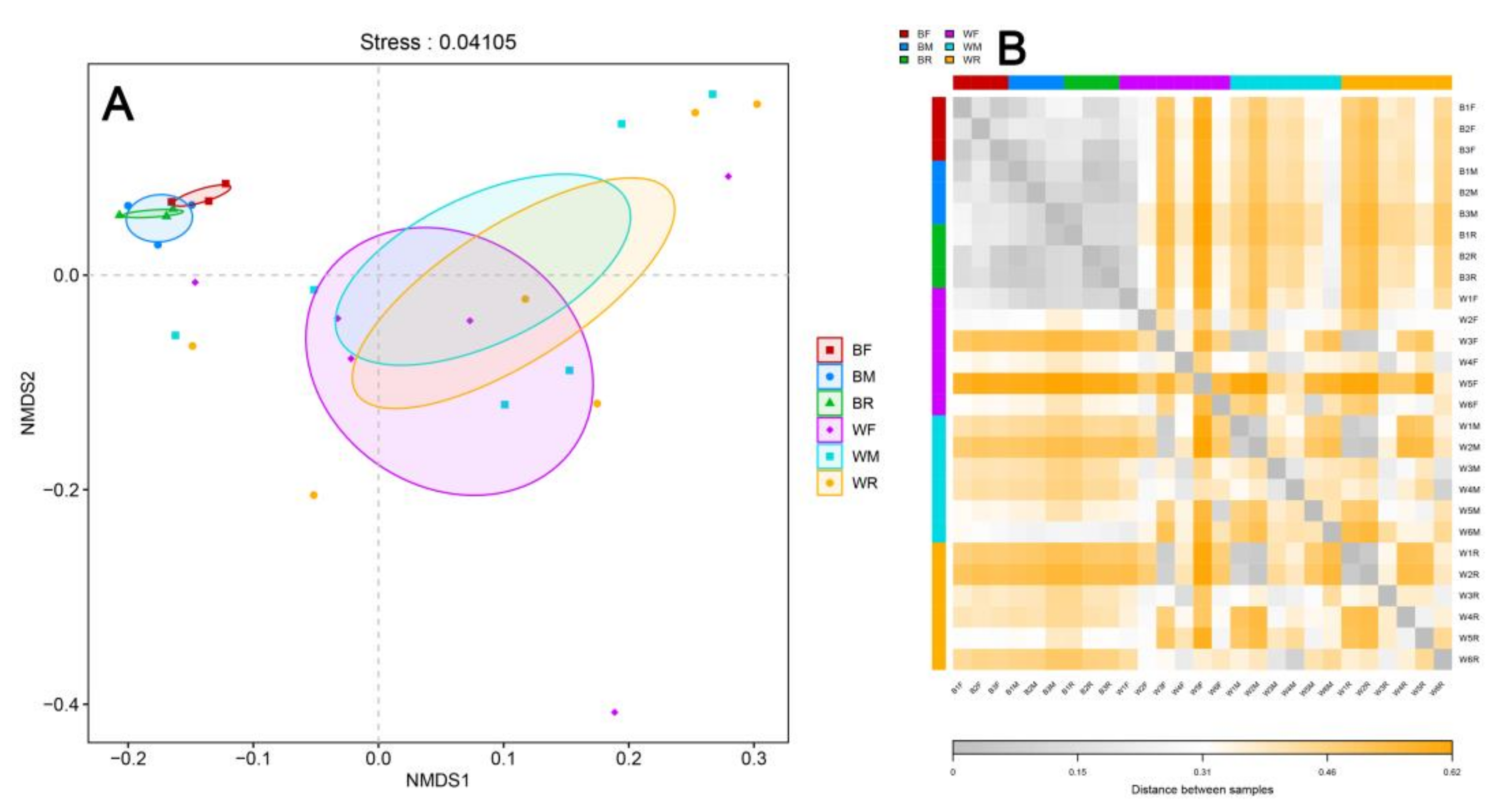
3.2. Diversity of the Gut Microbiota in the Cultivated and Wild Zig-Zag Eels
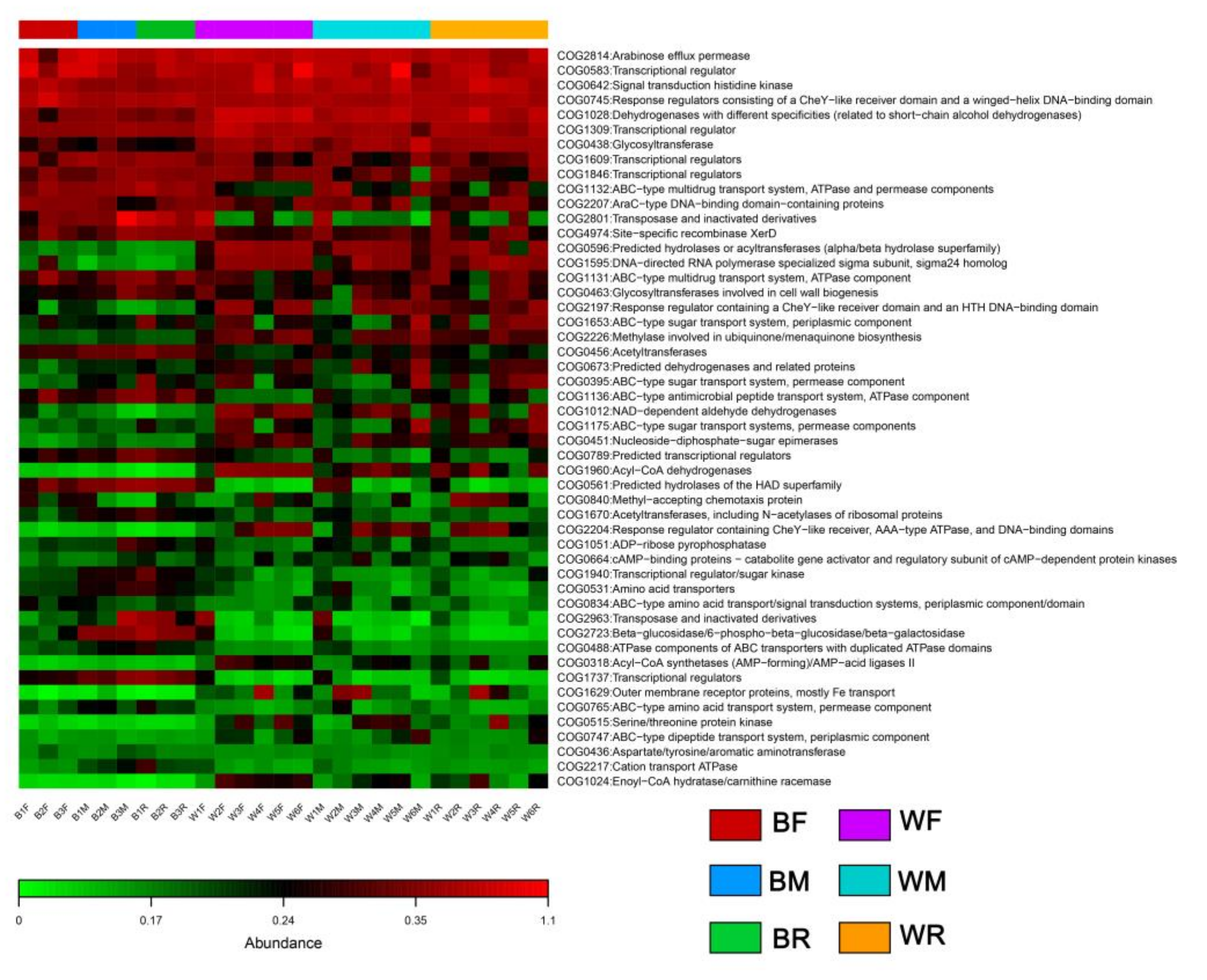
3.3. Gut Microbiota Functional Profile Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stokstad, E. De´tente in the fisheries war. Science 2009, 324, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.; Deng, J.M.; Wang, Z.M.; Zhang, J. Transplantation of Icefish (Salangidae) in China: Glory or Disaster? Rev. Aquacult. 2015, 7, 13–27. [Google Scholar] [CrossRef]
- Reid, A.J.; Carlson, A.K.; Creed, I.F.; Eliason, E.J.; Gell, P.A.; Johnson, P.T.J.; Kidd, K.A.; MacCormack, T.J.; Olden, J.D.; Ormerod, S.J.; et al. Emerging threats and persistent conservation challenges for freshwater biodiversity. Biol. Rev. 2019, 94, 849–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cressey, D. Aquaculture: Future fish. Nature 2009, 458, 398–400. [Google Scholar] [CrossRef] [Green Version]
- Flint, H.J.; Duncan, S.H.; Scott, K.P.; Louis, P. Links between diet, gut microbiota composition and gut metabolism. Proc. Nutr. Soc. 2015, 74, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Wei, J.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- Egert, M.; de Graaf, A.A.; Smidt, H.; de Vos, W.M.; Venema, K. Beyond diversity: Functional microbiomics of the human colon. Trends. Microbiol. 2006, 14, 86–91. [Google Scholar] [CrossRef]
- Sommer, F.; Bäckhed, F. The gut microbiota—Masters of host development and physiology. Nat. Rev. Microbiol. 2013, 11, 227–238. [Google Scholar] [CrossRef]
- Llewellyn, M.S.; McGinnity, P.; Dionne, M.; Letourneau, J.; Thonier, F.; Carvalho, G.R.; Creer, S.; Derome, N. The biogeography of the Atlantic salmon (Salmo salar) gut microbiome. ISME J. 2015, 10, 1280–1284. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.R.; Ran, C.; Ringø, E.; Zhou, Z.G. Progress in fish gastrointestinal microbiota research. Rev. Aquacult. 2018, 10, 626–640. [Google Scholar] [CrossRef]
- Heijtz, R.D.; Wang, S.; Anuar, F.; Qian, Y.; Björkholm, B.; Samuelsson, A.; Hibberd, M.L.; Forssberg, H.; Pettersson, S. Normal gut microbiota modulates brain development and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 3047–3052. [Google Scholar] [CrossRef] [Green Version]
- Semova, I.; Carten, J.D.; Stombaugh, J.; Mackey, L.C.; Knight, B.; Farber, S.A.; Rawls, J.F. Microbiota regulate intestinal absorption and metabolism of fatty acids in the zebrafish. Cell Host Microbe 2012, 12, 277–288. [Google Scholar] [CrossRef] [Green Version]
- LeBlanc, J.G.; Milani, C.; de Giori, G.S.; Sesma, F.; van Sinderen, D.; Ventura, M. Bacteria as vitamin suppliers to their host: A gut microbiota perspective. Curr. Opin. Biotech. 2013, 24, 160–168. [Google Scholar] [CrossRef]
- Fiayyaz, F.; Rehman, K.; Sharif, H.; Irshad, K.; Kamal, S.; Akash, M.S.H. Alteration of Gut Microbiota in EDCs-Induced Metabolic Disorders. In Endocrine Disrupting Chemicals-Induced Metabolic Disorders and Treatment Strategies. Emerging Contaminants and Associated Treatment Technologies; Akash, M.S.H., Rehman, K., Hashmi, M.Z., Eds.; Springer: Cham, Switzerland, 2021. [Google Scholar]
- Akash, M.S.H.; Fiayyaz, F.; Rehman, K.; Sabir, S.; Rasool, M.H. Gut microbiota and metabolic disorders: Advances in therapeutic tnterventions. Crit. Rev. Immunol. 2019, 39, 223–237. [Google Scholar] [CrossRef]
- Muegge, B.D.; Kuczynski, J.; Knights, D.; Clemente, J.C.; González, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon, J.I. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, B.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Moeller, A.H.; Peeters, M.; Ndjango, J.B.; Li, Y.; Hahn, B.H.; Ochman, H. Sympatric chimpanzees and gorillas harbor convergent gut microbial communities. Genome Res. 2013, 23, 1715–1720. [Google Scholar] [CrossRef] [Green Version]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righini, N.; Carbonero, F.; Estrada, A.; Gaskins, H.R.; Stumpf, R.M.; Yildirim, S.; Torralba, M.; et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013, 7, 1344–1353. [Google Scholar] [CrossRef]
- McCord, A.I.; Chapman, C.A.; Weny, G.; Tumukunde, A.; Hyeroba, D.; Klotz, K.; Koblings, A.S.; Mbora, D.N.M.; Cregger, M.A.; White, B.A.; et al. Fecal microbiomes of non-human primates in Western Uganda reveal species-specific communities largely resistant to habitat perturbation. Am. Primatol. 2014, 76, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Bletz, M.C.; Goedbloed, D.J.; Sanchez, E.; Reinhardt, T.; Tebbe, C.C.; Bhuju, S.; Geffers, R.; Jarek, M.; Vences, M.; Steinfartz, S. Amphibian gut microbiota shifts differentially in community structure but converges on habitat-specific predicted functions. Nat. Commun. 2016, 7, 13699. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.I.; Walker, D.W. Role of gut microbiota in aging-related health decline: Insights from invertebrate models. Cell. Mol. Life Sci. 2018, 75, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Grond, K.; Sandercock, B.K.; Jumpponen, A.; Zeglin, L.H. The avian gut microbiota: Community, physiology and function in wild birds. J. Avian Biol. 2018, 49, e01788. [Google Scholar] [CrossRef] [Green Version]
- Trevelline, B.K.; MacLeod, K.J.; Langkilde, T.; Kohl, K.D. Gestation alters the gut microbiota of an oviparous lizard. FEMS Microbiol. Ecol. 2019, 95, fiz086. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.J.; Cao, Y.L.; Ouyang, S.; Wu, X.P. Comparative analysis of gut microbiota diversity in endangered, economical, and common freshwater mussels using 16S rRNA gene sequencing. Ecol. Evol. 2020, 21, 12015–12023. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, T.; Li, Y.; Tang, Y.; Huang, Z. Gut microbiota are associated with sex and age of host: Evidence from semi-provisioned rhesus macaques in southwest Guangxi, China. Ecol. Evol. 2021, 11, 8096–8122. [Google Scholar] [CrossRef]
- Eliades, S.J.; Brown, J.C.; Colston, T.J.; Fisher, R.N.; Niukula, J.B.; Gray, K.; Vadada, J.; Rasalato, S.; Siler, C.D. Gut microbial ecology of the Critically Endangered Fijian crested iguana (Brachylophus vitiensis): Effects of captivity status and host reintroduction on endogenous microbiomes. Ecol. Evol. 2021, 11, 4731–4743. [Google Scholar] [CrossRef]
- Verschuere, L.; Rombaut, G.; Sorgeloos, P.; Verstraete, W. Probiotic bacteria as biological control agents in aquaculture. Microbiol. Mol. Biol. Rev. 2000, 64, 655–671. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, K.; Roy, M.; Kar, N.; Ringo, E. Gastrointestinal bacteria in rohu, Labeo Rohita (Actinopterygii: Cypriniformes: Cyprinidae): Scanning electron microscopy and bacteriological study. Acta Ichthyol. Piscat. 2010, 40, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, E.; Yany, G.; Romero, J. Cultivable intestinal microbiota of yellowtail juveniles (Seriola lalandi) in an aquaculture system. Lat. Am. J. Aquat. Res. 2013, 41, 395–403. [Google Scholar] [CrossRef]
- Al-Hisnawi, A.; Ringø, E.; Davies, S.J.; Waines, P.; Bradley, G.; Merrifield, D.L. First report on the autochthonous gut microbiota of brown trout (Salmo trutta Linnaeus). Aquac. Res. 2015, 46, 2962–2971. [Google Scholar] [CrossRef]
- Clements, K.D.; Angert, E.R.; Montgomery, W.L.; Choat, J.H. Intestinal microbiota in fishes: What’s known and what’s not. Mol. Ecol. 2014, 23, 1891–1898. [Google Scholar] [CrossRef]
- Park, S.H.; Lee, S.I.; Ricke, S.C. Microbial populations in naked neck chicken ceca raised on pasture flock fed with commercial yeast cell wall prebiotics via an Illumina MiSeq platform. PLoS ONE 2016, 11, e0151944. [Google Scholar] [CrossRef]
- Sun, Y.; Zhou, L.P.; Fang, L.D.; Su, Y.; Zhu, W.Y. Responses in colonic microbial community and gene expression of pigs to a longterm high resistant starch diet. Front. Microbiol. 2015, 6, 877. [Google Scholar] [CrossRef] [Green Version]
- Sullam, K.E.; Essinger, S.D.; Lozupone, C.A.; O’Connor, M.P.; Rosen, G.L.; Knight, R.; Kilham, S.S.; Russell, J.A. Environmental and ecological factors that shape the gut bacterial communities of fish: A meta-analysis. Mol. Ecol. 2012, 21, 3363–3378. [Google Scholar] [CrossRef] [Green Version]
- Bolnick, D.I.; Snowberg, L.K.; Hirsch, P.E.; Lauber, C.L.; Org, E.; Parks, B.; Lusis, A.J.; Knight, R.; Caporaso, J.G.; Svanbäck, R. Individual diet has sex-dependent effects on vertebrate gut microbiota. Nat. Commun. 2014, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ni, J.; Li, J.; Wang, C.; Li, X.; Wu, S.; Zhang, T.; Yu, Y.; Yan, Q. Comparative study on gastrointestinal microbiota of eight fish species with different feeding habits. J. Appl. Microbiol. 2014, 117, 750–760. [Google Scholar] [CrossRef]
- Tietjen, M. “You are what you eat”: How diet can influence the gut microbiota of marine invertebrates. Plymouth Study Sci. 2014, 7, 203–211. [Google Scholar]
- Miyake, S.; Ngugi, D.K.; Stingl, U. Diet strongly influences the gut microbiota of surgeonfishes. Mol. Ecol. 2015, 24, 656–672. [Google Scholar] [CrossRef]
- Kashinskaya, E.N.; Belkova, N.L.; Izvekova, G.I.; Simonov, E.P.; Andree, K.B.; Glupov, V.V.; Solovyev, M.M. A comparative study on microbiota from the gut of Prussian carp (Carassius gibelio) and their aquatic environmental compartments, using different molecular methods. J. Appl. Microbiol. 2015, 119, 948–961. [Google Scholar] [CrossRef]
- Liu, H.; Guo, X.; Gooneratne, R.; Lai, R.; Zeng, C.; Zhan, F.; Wang, W. The gut microbiome and degradation enzyme activity of wild freshwater fishes influenced by their trophic levels. Sci. Rep. 2016, 6, 24340. [Google Scholar] [CrossRef] [Green Version]
- Roeselers, G.; Mittge, E.K.; Stephens, W.Z.; Parichy, D.M.; Cavanaugh, C.M.; Guillemin, K.; Rawls, J.F. Evidence for a core gut microbiota in the zebrafish. ISME J. 2011, 5, 1595–1608. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Yu, Y.; Zhang, T.; Gao, L. Comparison of intestinal bacterial communities in grass carp, Ctenopharyngodon idellus, from two different habitats. Chin. J. Oceanol. Limnol. 2012, 30, 757–765. [Google Scholar] [CrossRef]
- Li, X.; Yan, Q.; Xie, S.; Hu, W.; Yu, Y.; Hu, Z. Gut microbiota contributes to the growth of fast-growing transgenic Common Carp (Cyprinus carpio L.). PLoS ONE 2013, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ringø, E.; Zhou, Z.; Vecino, J.G.; Wadsworth, S.; Romero, J.; Krogdahl, A.; Olsen, R.E.; Dimitroglou, A.; Foey, A.; Davies, S.; et al. Effect of dietary components on the gut microbiota of aquatic animals. A never-ending story? Aquacult. Nutr. 2016, 22, 219–282. [Google Scholar] [CrossRef] [Green Version]
- Nelson, J.S. Fishes of the World, 4th ed.; John Wiley and Sons, Inc.: New York, NY, USA, 2006. [Google Scholar]
- Wu, A.Y.; Liang, Z.Q.; Li, C.W.; Liu, L.; Chen, X.Y.; Liu, M.Q. Analysis and evaluation of nutritional components in the muscle of two kinds of mastacembelus. Acta Nutr. Sin. 2010, 5, 499–502. [Google Scholar]
- Li, F.; Chen, Q.P.; He, P.Y.; Huang, X.Q.; Chen, Z.K.; Zhou, H.Q.; Zhong, D.M.; Shu, H. The karyotype pattern and genetic diversity of mitochondrial DNA Cytb gene and D-loop sequence in Mastacembelus armtus Beijiang River, south China. Oceanol. Limnol. Sin. 2019, 50, 449–454. [Google Scholar]
- Yan, Q.; van der Gast, C.J.; Yu, Y. Bacterial community assembly and turnover within the intestines of developing zebrafish. PLoS ONE 2012, 7, e30603. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. SINTAX: A simple non-Bayesian taxonomy classifier for 16S and ITS sequences. BioRxiv. 2016. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing. Foundation for Statistical Computing; R Development Core Team: Vienna, Austria, 2020. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Wagner, H.H. Vegan: Community Ecology Package Software; R Development Core Team: Vienna, Austria, 2016. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.; Alm, E.J. Inferring correlation networks from genomic survey data. PLoS Computat. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; Mcdonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Loudon, A.H.; Woodhams, D.C.; Parfrey, L.W.; Archer, H.; Knight, R.; McKenzie, V.; Harris, R.N. Microbial community dynamics and effect of environmental microbial reservoirs on red-backed salamanders (Plethodon cinereus). ISME J. 2014, 8, 830–840. [Google Scholar] [CrossRef]
- Li, X.H.; Yu, Y.H.; Li, C.; Yan, Q.Y. Comparative study on the gut microbiotas of four economically important Asian carp species. Sci. China Life Sci. 2018, 61, 696–705. [Google Scholar] [CrossRef]
- Zhang, B.; Ren, J.; Yang, D.; Liu, S.; Gong, X. Comparative analysis and characterization of the gut microbiota of four farmed snakes from southern China. Peer J. 2019, 7, e6658. [Google Scholar] [CrossRef]
- Hu, X.; Liu, G.; Shafer, A.B.A.; Wei, Y.; Zhou, J.; Lin, S.; Wu, H.B.; Zhou, M.; Hu, D.F.; Liu, S.Q. Comparative analysis of the gut microbial communities in forest and alpine musk deer using high-throughput sequencing. Front. Microbiol. 2017, 8, 572. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Wu, F.; Zhou, Q.; Zhao, D. Comparative analysis of gut microbiota in captive and wild Oriental White storks: Implications for conservation biology. Front. Microbiol. 2021, 12, 649466. [Google Scholar] [CrossRef]
- Austin, B. Taxonomy of bacterial fish pathogens. Vet. Res. 2011, 42, 20. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Wang, Q.; Li, S.; Ran, C.; Guo, X.; Zhang, Z.; Zhou, Z. Antibiotic growth promoter olaquindox increases pathogen susceptibility in fish by inducing gut microbiota dysbiosis. Sci. China Life Sci. 2017, 60, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.C.; Wafula, D.; Chauhan, A.; Green, S.J.; Gragg, R.; Jagoe, C. A survey of deepwater horizon (DWH) oil-degrading bacteria from the Eastern oyster biome and its surrounding environment. Front. Microbiol. 2014, 5, 149. [Google Scholar] [CrossRef] [PubMed]
- Kaakoush, N.O. Insights into the role of Erysipelotrichaceae in the human host. Front. Cell. Infect. Microbiol. 2015, 5, 84. [Google Scholar] [CrossRef] [PubMed]
- Bernini, L.J.; Simão, A.N.C.; Alfieri, D.F.; Lozovoy, M.A.B.; Mari, N.L.; de Souza, C.H.; Costa, G. Beneficial effects of Bifidobacterium lactis on lipid profile and cytokines in patients with metabolic syndrome: A randomized trial. Effects of probiotics on metabolic syndrome. Nutrition 2016, 32, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Berry, D. The emerging view of Firmicutes as key fibre degraders in the human gut. Environ. Microbiol. 2016, 18, 2081–2083. [Google Scholar] [CrossRef]
- Jami, E.; White, B.A.; Mizrahi, I. Potential role of the bovine rumen microbiome in modulating milk composition and feed efficiency. PLoS ONE 2014, 9, e85423. [Google Scholar] [CrossRef]
- Wu, S.; Tian, J.; Gatesoupe, J.; Li, W.; Zou, H.; Yang, B.; Wang, G. Intestinal microbiota of gibel carp (Carassius auratus gibelio) and its origin as revealed by 454 pyrosequencing. World J. Microbiol. Biot. 2013, 29, 1585–1595. [Google Scholar] [CrossRef]
- Li, T.; Long, M.; Ji, C.; Shen, Z.; Gatesoupe, F.J.; Zhang, X.; Zhang, Q.; Zhang, L.; Zhao, Y.; Liu, X.; et al. Alterations of the gut microbiome of Largemouth Bronze Gudgeon (Coreius guichenoti) suffering from furunculosis. Sci. Rep. 2016, 6, 30606. [Google Scholar]
- Li, T.; Long, M.; Gatesoupe, F.J.; Zhang, Q.; Li, A.; Gong, X. Comparative analysis of the intestinal bacterial communities in different species of carp by pyrosequencing. Microb. Ecol. 2015, 69, 25–36. [Google Scholar] [CrossRef]
- Larsen, A.M.; Mohammed, H.H.; Arias, C.R. Characterization of the gut microbiota of three commercially valuable warmwater fish species. J. Appl. Microbiol. 2014, 116, 1396–1404. [Google Scholar] [CrossRef]
- Romero, J.; Navarrete, P. 16S rDNA-based analysis of dominant bacterial populations associated with early life stages of Coho Salmon (Oncorhynchus kisutch). Microb. Ecol. 2006, 51, 422–430. [Google Scholar] [CrossRef]
- Han, S.; Liu, Y.; Zhou, Z.; He, S.; Cao, Y.; Shi, P.; Yao, B.; Ring, O.E. Analysis of bacterial diversity in the intestine of grass carp (Ctenopharyngodon idellus) based on 16S rDNA gene sequences. Aquacult. Res. 2010, 42, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Ostaff, M.J.; Stange, E.F.; Wehkamp, J. Antimicrobial peptides and gut microbiota in homeostasis and pathology. EMBO Mol. Med. 2013, 5, 1465–1483. [Google Scholar] [CrossRef]
- Chauhan, A.; Wafula, D.; Lewis, D.E.; Pathak, A. Metagenomic assessment of the Eastern oyster-associated microbiota. Genome Announc. 2014, 2, e01083-14. [Google Scholar] [CrossRef]
- Ramírez, C.; Romero, J. Fine Flounder (Paralichthys adspersus) microbiome showed important differences between wild and reared specimens. Front. Microbiol. 2017, 8, 271. [Google Scholar] [CrossRef] [Green Version]
- Ward, N.L.; Steven, B.; Penn, K.; Methe, B.A.; Detrich, W.H. Characterization of the intestinal microbiota of two Antarctic notothenioid fish species. Extremophiles 2009, 13, 679–685. [Google Scholar] [CrossRef]
- Xiong, J.; Zhu, J.; Dai, W.; Dong, C.; Qiu, Q.; Li, C. Integrating gut microbiota immaturity and disease-discriminatory taxa to diagnose the initiation and severity of shrimp disease. Environ. Microbiol. 2017, 19, 1490–1501. [Google Scholar] [CrossRef]
- Yu, W.; Dai, W.; Tao, Z.; Xiong, J. Characterizing the compositional and functional structures of intestinal microflora between healthy and diseased Litopenaeus vannamei. J. Fish. China 2018, 42, 399–409. [Google Scholar]
- Hao, Y.T.; Wu, S.G.; Xiong, F.; Tran, N.T.; Jakovlic, I.; Zou, H.; Li, W.X.; Wang, G. Succession and fermentation products of grass carp (Ctenopharyngodon idellus) hindgut microbiota in response to an extreme dietary shift. Front. Microbiol. 2017, 8, 1585. [Google Scholar] [CrossRef] [Green Version]
- Fang, G.; Rocha, E.P.; Danchin, A. How essential are nonessential genes? Mol. Biol. Evol. 2005, 22, 2147–2156. [Google Scholar] [CrossRef]
- Tong, Q.; Liu, X.N.; Hu, Z.F.; Ding, J.F.; Bie, J.; Wang, H.B.; Zhang, J.T. Effects of captivity and season on the gut microbiota of the brown frog (Rana dybowskii). Front. Microbiol. 2019, 10, 1912. [Google Scholar] [CrossRef] [Green Version]
- Thomas, F.; Hehemann, J.H.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and gut Bacteroidetes: The food connection. Front. Microbiol. 2011, 2, 93. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BF | BM | BR | WF | WM | WR | p Value | |
|---|---|---|---|---|---|---|---|
| Sequences | 43,598 ± 8902 | 62,634 ± 7627 | 45,974 ± 12842 | 46,397 ± 23293 | 55,732 ± 10324 | 49,985 ± 12,155 | 0.54 |
| OTUs | 51± 6 | 89 ± 5 | 70 ± 20 | 498 ± 251 | 441 ± 354 | 728 ± 468 | 0.02 |
| Shannon | 2.30 ± 0.52 | 2.03 ± 0.47 | 1.95 ± 0.27 | 3.74 ± 0.72 | 3.08 ± 1.36 | 3.86 ± 1.74 | 0.08 |
| Chao | 56.64 ± 9.69 | 98.58 ± 11.07 | 101.12 ± 32.71 | 536.03 ± 258.75 | 465.61 ± 371.51 | 747.60 ± 470.24 | 0.02 |
| Ace | 60.93 ± 14.02 | 99.77 ± 13.02 | 97.72 ± 36.01 | 532.51 ± 244.49 | 467.70 ± 368.59 | 737.34 ± 469.79 | 0.02 |
| Simpson | 0.15 ± 0.08 | 0.24 ± 0.14 | 0.21 ± 0.06 | 0.12 ± 0.11 | 0.21 ± 0.21 | 0.17 ± 0.19 | 0.87 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Fan, Y.; Mo, T.; Chen, Q.; Chen, W. Comparative Study of the Gut Microbiota Community between the Farmed and Wild Mastacembelus armatus (Zig-Zag Eel). Metabolites 2022, 12, 1193. https://doi.org/10.3390/metabo12121193
Liu X, Fan Y, Mo T, Chen Q, Chen W. Comparative Study of the Gut Microbiota Community between the Farmed and Wild Mastacembelus armatus (Zig-Zag Eel). Metabolites. 2022; 12(12):1193. https://doi.org/10.3390/metabo12121193
Chicago/Turabian StyleLiu, Xiongjun, Yuqin Fan, Tao Mo, Qingxiu Chen, and Weiting Chen. 2022. "Comparative Study of the Gut Microbiota Community between the Farmed and Wild Mastacembelus armatus (Zig-Zag Eel)" Metabolites 12, no. 12: 1193. https://doi.org/10.3390/metabo12121193