

Echinocystic Acid Bidesmoside Saponins from Microglossa afzelii O. Hoffm and Their Cytotoxic Activity against the CAL-27 Oral Squamous Carcinoma Cell Line
 ,
,
Abstract
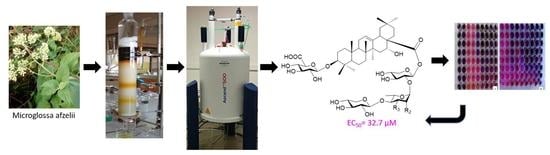
:
1. Introduction
2. Materials and Methods
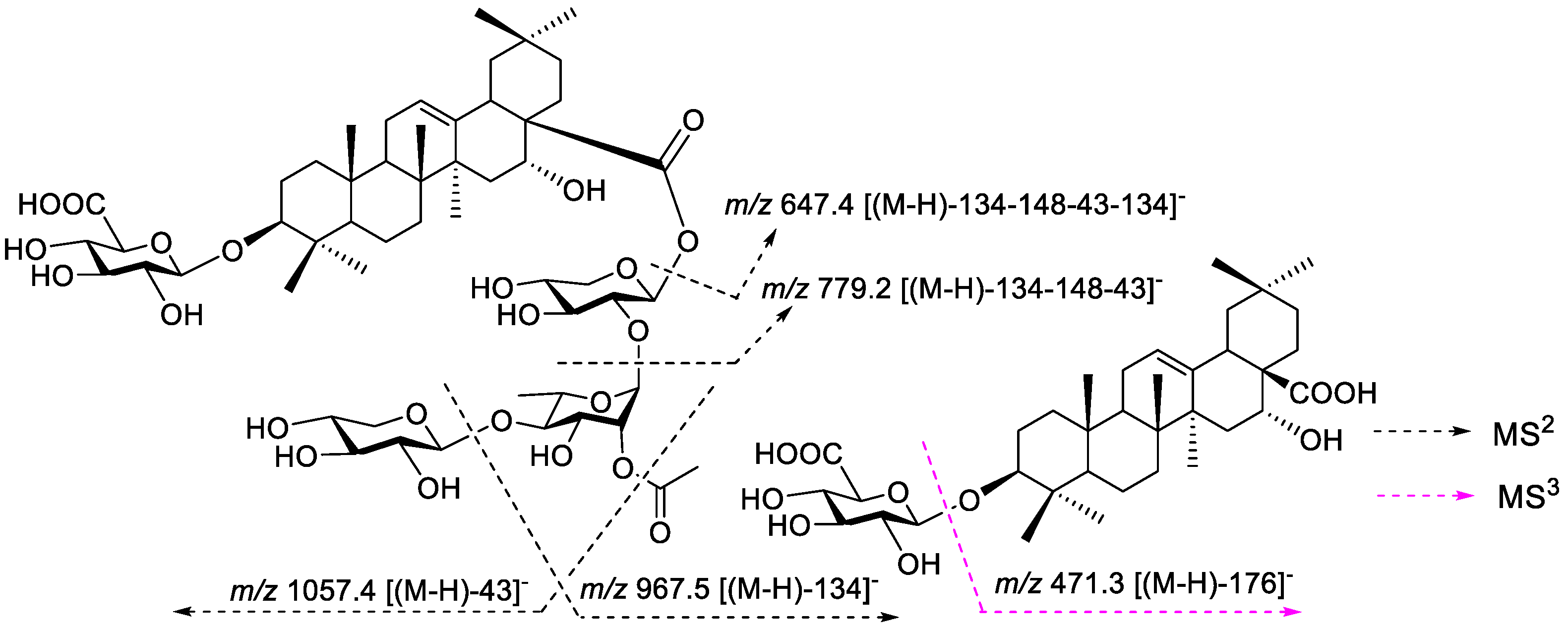
2.1. General Methods
2.2. Plant Material
2.3. Extraction and Isolation
2.4. Acid Hydrolysis and GC-MS Analysis of Saponins 1–9
2.5. Culture Conditions
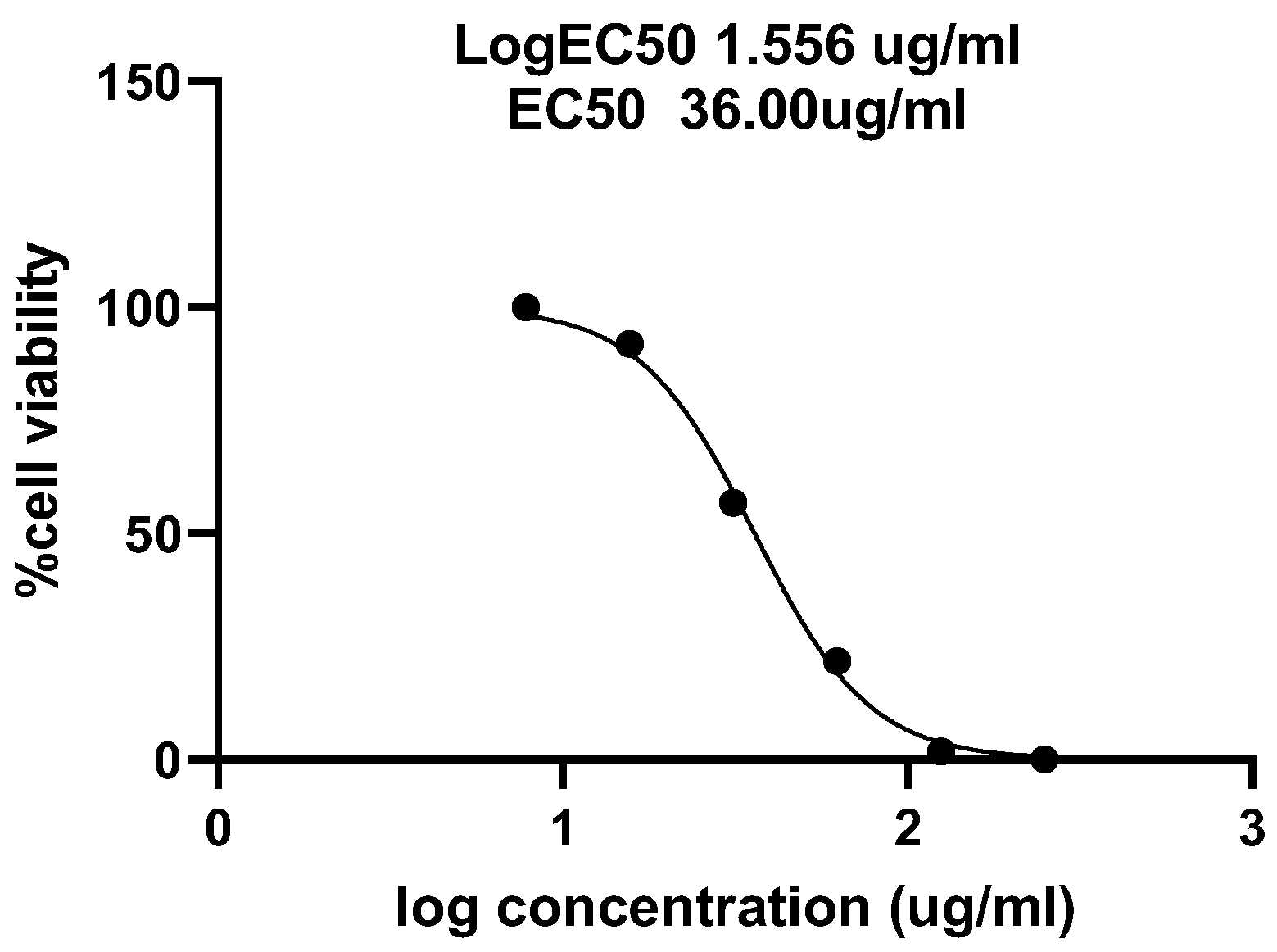
2.6. Cytotoxic Assay
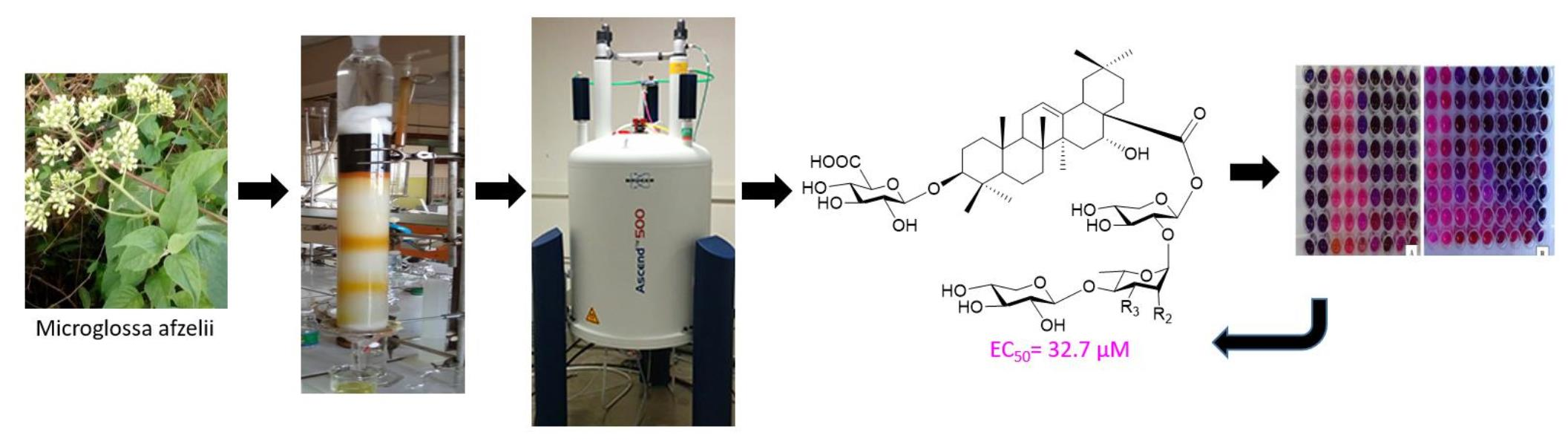
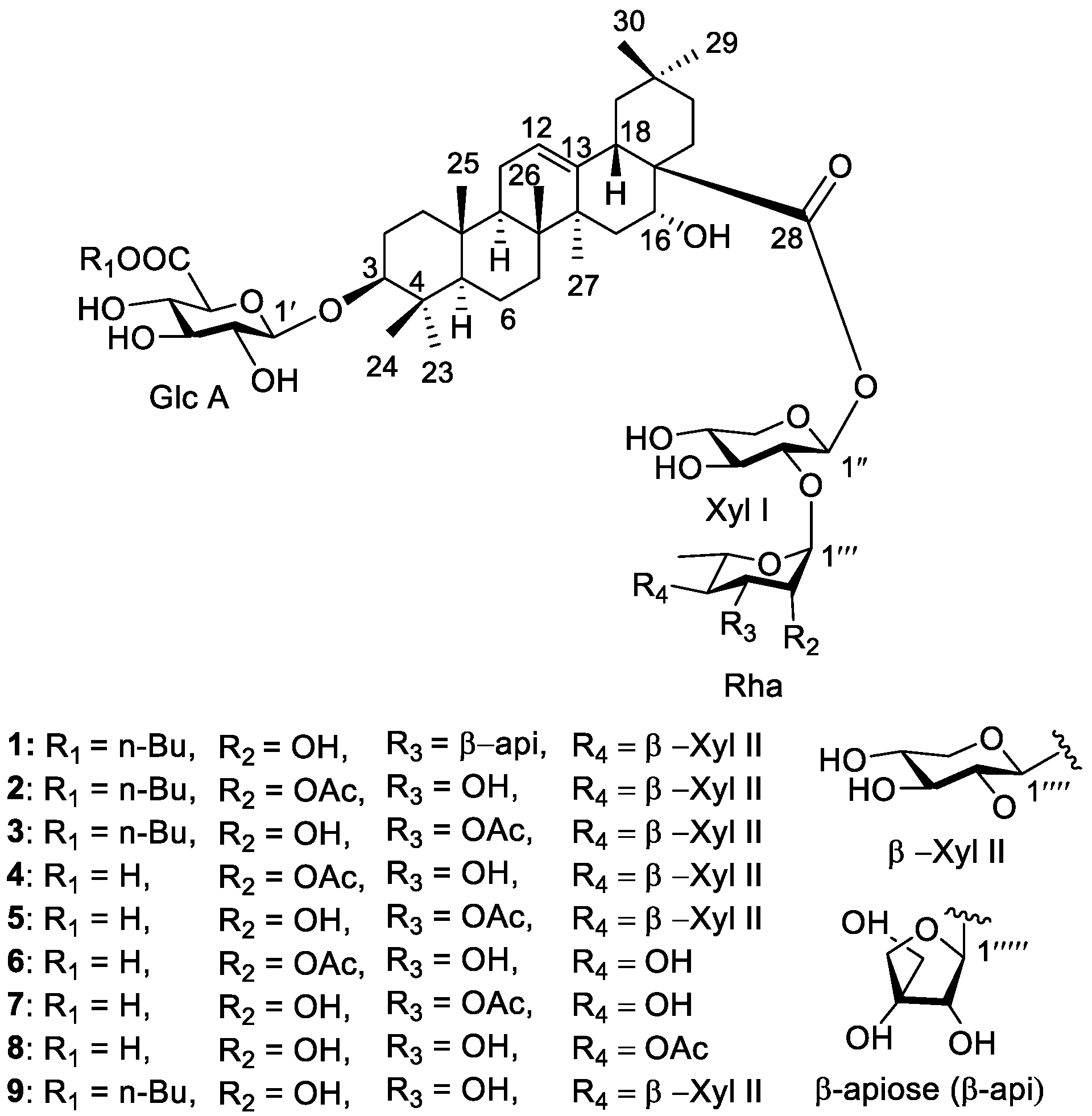
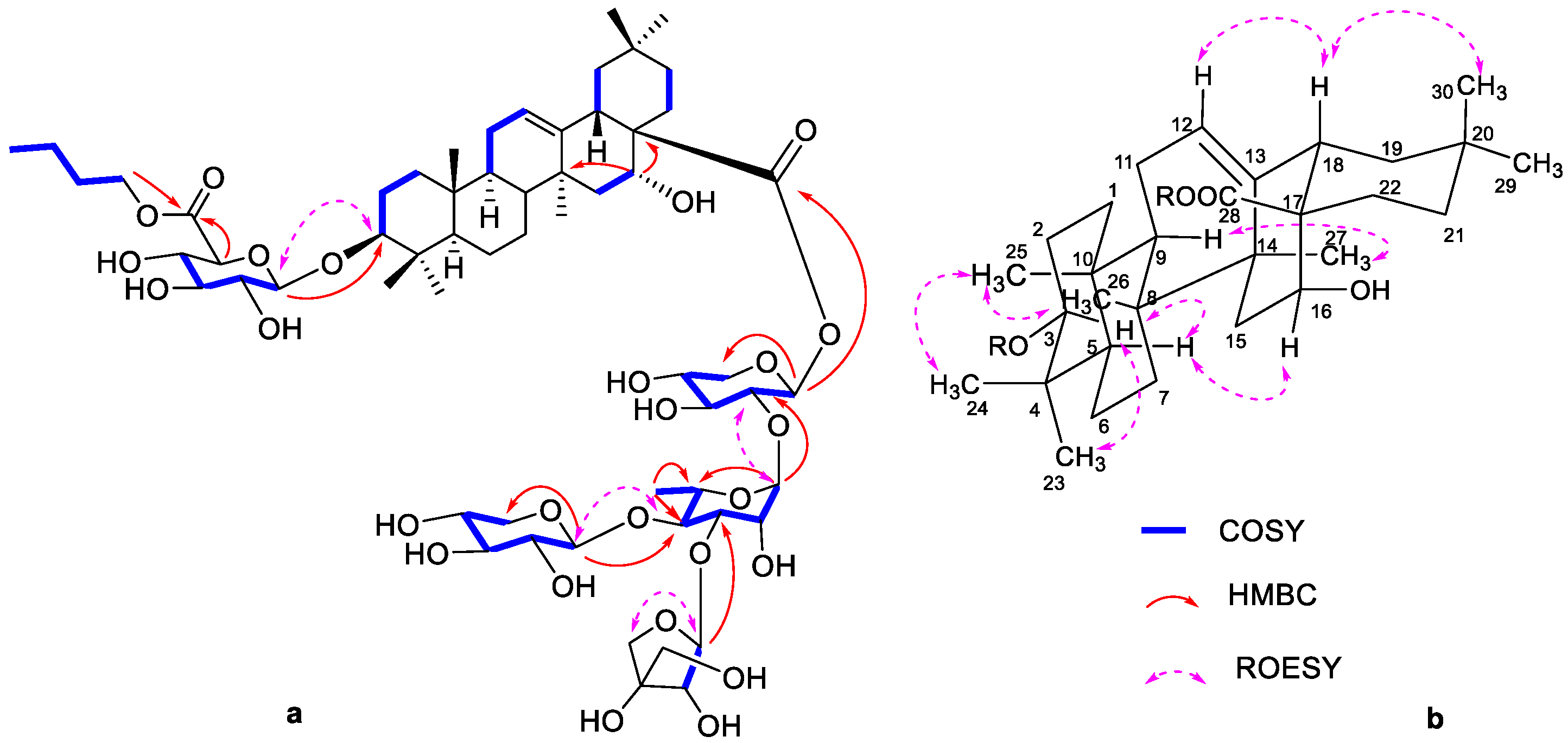
3. Results
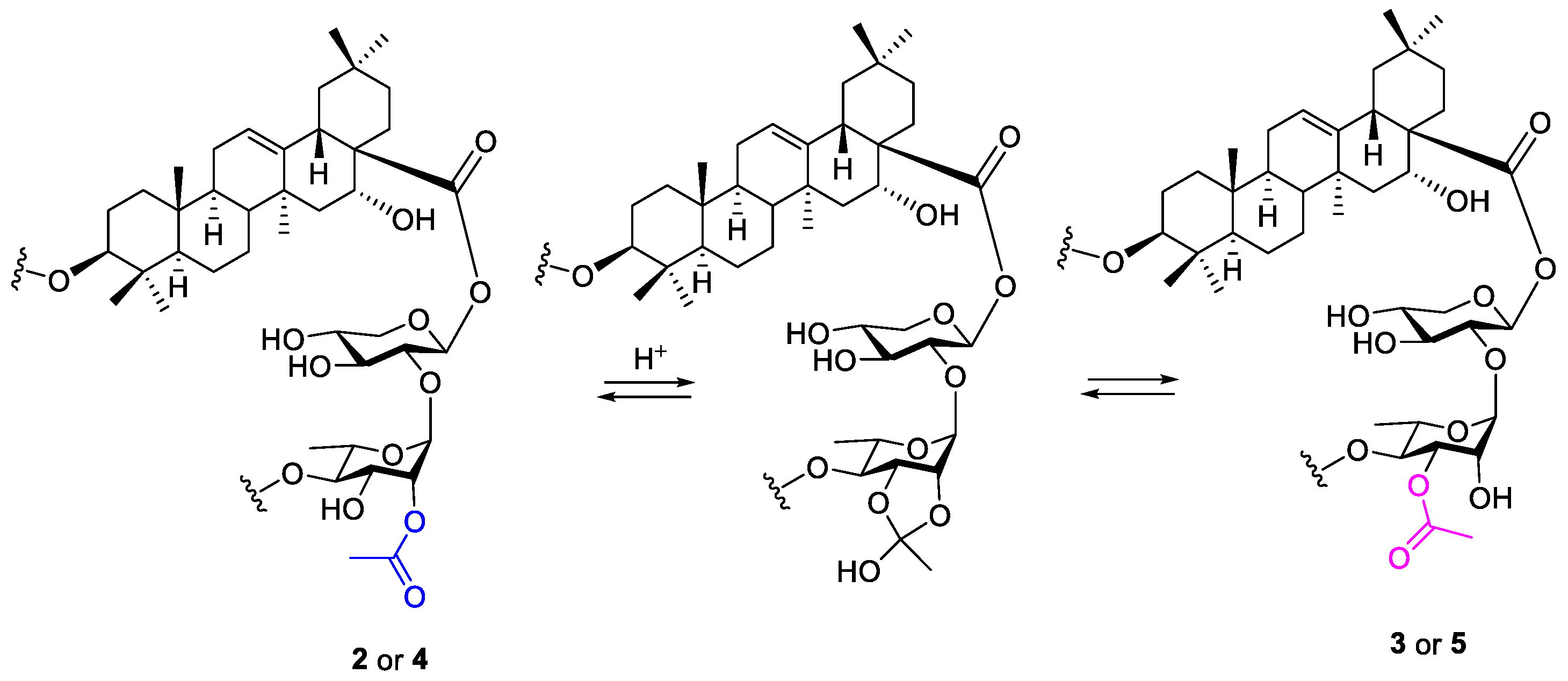
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- El Aziz, M.M.A.; Ashour, A.S.; Melad, A.S.G. A review on saponins from medicinal plants: Chemistry, isolation, and determination. J. Nanomed. Res. 2019, 8, 282–288. [Google Scholar]
- Cammareri, M.; Consiglio, M.F.; Pecchia, P.; Corea, G.; Lanzotti, V.; Ibeas, J.I.; Tava, A.; Conicella, C. Molecular characterization of β-amyrin synthase from Aster sedifolius L. and triterpenoid saponin analysis. Plant Sci. 2008, 175, 255–261. [Google Scholar] [CrossRef]
- Tene, M.; Tane, P.; Sondengam, B.L.; Connolly, J.D. Clerodane diterpenoids from Microglossa angolensis. Tetrahedron 2005, 61, 2655–2658. [Google Scholar] [CrossRef]
- Okafor, J.C. D. Abbiw 1990. Useful plants of Ghana. Intermediate Technology Publications & the Royal Botanic Gardens, Kew; London, xii + 337 pages. Hardback: ISBN 1 85339 080 1. Price: £30.00. Paperback: ISBN 1 85339 043 7. Price: £9.95. J. Trop. Ecol. 2009, 7, 286–287. [Google Scholar]
- Köhler, I.; Jenett-Siems, K.; Kraft, C.; Siems, K.; Abbiw, D.; Bienzle, U.; Eich, E. Herbal remedies traditionally used against malaria in Ghana: Bioassay-guided fractionation of Microglossa pyrifolia (Asteraceae). Z. Nat. C 2002, 57, 1022–1027. [Google Scholar] [CrossRef]
- Tamokou, J.D.; Kuiate, J.R.; Tene, M.; Tane, P. Antimicrobial clerodane diterpenoids from Microglossa angolensis Oliv. et Hiern. Indian J. Pharmacol. 2009, 41, 60–63. [Google Scholar]
- Rücker, G.; Kehrbaum, S.; Sakulas, H.; Lawong, B.; Goeltenboth, F. Acetylenic glucosides from Microglossa pyrifolia. Planta Med. 1992, 58, 266–269. [Google Scholar] [CrossRef]
- Akimanya, A.; Midiwo, J.O.; Matasyoh, J.; Okanga, F.; Masila, V.M.; Walker, L.; Tekwani, B.L.; Muhammad, I.; Omosa, L.K. Two polymethoxylated flavonoids with antioxidant activities and a rearranged clerodane diterpenoid from the leaf exudates of Microglossa pyrifolia. Phytochem. Lett. 2015, 11, 183–187. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Hildebrand, M.R.; Willuhn, G. New Dihydrobenzofurans and Triterpenoids from Roots of Microglossa pyrifolia. Planta Med. 2003, 69, 258–264. [Google Scholar] [CrossRef]
- Calabria, L.M.; Piacente, S.; Kapusta, I.; Dharmawardhane, S.F.; Segarra, F.M.; Pessiki, P.J.; Mabry, T.J. Triterpene saponins from Silphium radula. Phytochemistry 2008, 69, 961–972. [Google Scholar] [CrossRef]
- Fang, Z.; Li, J.; Yang, R.; Fang, L.; Zhang, Y. A Review: The triterpenoid saponins and biological activities of Lonicera Linn. Molecules 2020, 25, 3773. [Google Scholar] [CrossRef] [PubMed]
- Tapondjou, L.A.; Ponou, K.B.; Teponno, R.B.; Mbiantcha, M.; Djoukeng, J.D.; Nguelefack, T.B.; Watcho, P.; Cadenas, A.G.; Park, H.-J. In vivo anti-inflammatory effect of a new steroidal saponin, mannioside A, and its derivatives isolated from Dracaena mannii. Arch. Pharm. Res. 2008, 31, 653–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nzowa, L.K.; Barboni, L.; Teponno, R.B.; Ricciutelli, M.; Lupidi, G.; Quassinti, L.; Bramucci, M.; Tapondjou, L.A. Rheediinosides A and B, two antiproliferative and antioxidant triterpene saponins from Entada rheedii. Phytochemistry 2010, 71, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Fouedjou, R.T.; Teponno, R.B.; Quassinti, L.; Bramucci, M.; Petrelli, D.; Vitali, L.A.; Fiorini, D.; Tapondjou, L.A.; Barboni, L. Steroidal saponins from the leaves of Cordyline fruticosa (L.) A. Chev. and their cytotoxic and antimicrobial activity. Phytochem. Lett. 2014, 7, 62–68. [Google Scholar] [CrossRef]
- Tchegnitegni, B.T.; Teponno, R.B.; Jenett-Siems, K.; Melzig, M.F.; Miyamoto, T.; Tapondjou, L.A. A dihydrochalcone derivative and further steroidal saponins from Sansevieria trifasciata Prain. Z. Nat. C 2017, 72, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, N.; Adhikari, A.; Hameed, A.; Hafizur, R.M.; Musharraf, S.G. Isolation and characterization of non-sulfated and sulfated triterpenoid saponins from Fagonia indica. Phytochemistry 2017, 143, 151–159. [Google Scholar] [CrossRef]
- Farooq, U.; Naz, S.; Zehra, B.; Khan, A.; Ali, S.A.; Ahmed, A.; Sarwar, R.; Bukhari, S.M.; Rauf, A.; Ahmad, I.; et al. Isolation and characterization of three new anti-proliferative Sesquiterpenes from Polygonum barbatum and their mechanism via apoptotic pathway. BMC Cancer 2017, 17, 694. [Google Scholar] [CrossRef]
- Zhou, X.-F.; Zhao, X.-Y.; Tang, L.; Ruan, H.-L.; Zhang, Y.-H.; Pi, H.-F.; Xiao, W.-L.; Sun, H.-D.; Wu, J.-Z. Three new triterpenoid saponins from the rhizomes of Impatiens pritzellii var. hupehensis. J. Asian Nat. Prod. Res. 2007, 9, 379–385. [Google Scholar] [CrossRef]
- Teponno, R.B.; Tanaka, C.; Jie, B.; Tapondjou, L.A.; Miyamoto, T. Trifasciatosides A–J, Steroidal Saponins from Sansevieria trifasciata. Chem. Pharm. Bull. 2016, 64, 1347–1355. [Google Scholar] [CrossRef] [Green Version]
- Fujihara, K.; Shimoyama, T.; Kawazu, R.; Sasaki, H.; Koyama, K.; Takahashi, K.; Kinoshita, K. Amyloid β aggregation inhibitory activity of triterpene saponins from the cactus Stenocereus pruinosus. J. Nat. Med. 2020, 75, 284–298. [Google Scholar] [CrossRef]
- Mahato, S.B.; Kundu, A.P. 13C NMR Spectra of pentacyclic triterpenoids—A compilation and some salient features. Phytochemistry 1994, 37, 1517–1575. [Google Scholar] [CrossRef]
- Koz, O.; Bedir, E.; Masullo, M.; Alankus-Caliskan, O.; Piacente, S. Triterpene glycosides from Agrostemma gracilis. Phytochemistry 2010, 71, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Tapondjou, L.A.; Nyaa, L.B.T.; Tane, P.; Ricciutelli, M.; Quassinti, L.; Bramucci, M.; Lupidi, G.; Ponou, B.K.; Barboni, L. Cytotoxic and antioxidant triterpene saponins from Butyrospermum parkii (Sapotaceae). Carbohydr. Res. 2011, 346, 2699–2704. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, T.; Li, X.; Nishida, E.; Nakamura, S.; Ninomiya, K.; Matsuda, H.; Hamao, M.; Muraoka, O.; Hayakawa, T.; Yoshikawa, M. Medicinal Flowers. XXXII. Structures of Oleanane-Type Triterpene Saponins, Perennisosides VIII, IX, X, XI, and XII, from the Flowers of Bellis perennis. Chem. Pharm. Bull. 2011, 59, 889–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.-T.; Choi, J.; Jung, W.-T.; Nam, J.-H.; Jung, H.-J.; Park, H.-J. Structure of a new echinocystic Acid bisdesmoside isolated from Codonopsis lanceolata roots and the cytotoxic activity of prosapogenins. J. Agric. Food Chem. 2002, 50, 4190–4193. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zhu, M.; Zhong, J.; Yan, W. Structural stability of acetyl saponins in different solvents and separation materials. Phytochem. Lett. 2015, 11, 368–372. [Google Scholar] [CrossRef]
- Wang, F.; Hua, H.; Pei, Y.; Chen, D.; Jing, Y. Triterpenoids from the resin of Styrax t onkinensis and their antiproliferative and differentiation effects in human leukemia HL-60 cells. J. Nat. Prod. 2006, 69, 807–810. [Google Scholar] [CrossRef]
- Miyase, T.; Melek, F.R.; Ghaly, N.S.; Warashina, T.; El-Kady, M.; Nabil, M. Echinocystic acid 3,16-O-bisglycosides from Albizia procera. Phytochemistry 2010, 71, 1375–1380. [Google Scholar] [CrossRef]
- Zhang, Q.; Lu, Y.-Y.; Yang, L.; Tang, H.-F. New triterpenoid saponins from the whole plants of Clematis heracleifolia. Fitoterapia 2022, 159, 105179. [Google Scholar] [CrossRef]
- Yadava, R.N.; Jharbade, J. New antibacterial triterpenoid saponin from Lactuca scariola. Fitoterapia 2008, 79, 245–249. [Google Scholar] [CrossRef]
- Alvarez, L.; Zamilpa, A.; Marquina, S.; González, M. Two new oleanolic acid saponins from the roots of Viguiera hypargyrea. Rev. Soc. Quím. Mex. 2003, 47, 173–177. [Google Scholar]
- Chabani, S.; Lavaud, C.; Benkhaled, M.; Harakat, D.; Long, C.; Haba, H. Three new oleanane-type triterpene saponins from Atractylis flava. Phytochem. Lett. 2016, 15, 88–93. [Google Scholar] [CrossRef]
- Kırmızıbekmez, H.; Bassarello, C.; Piacente, S.; Pizza, C.; Çalış, I. Triterpene saponins from Calendula arvensis. Z. Nat. B 2006, 61b, 1170–1173. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | ||||
|---|---|---|---|---|---|
| δC | δH | δC | δH | ||
| 1 | 39.9 | 0.97 1.61 | 3-O-Glc A | ||
| 2 | 27.1 | 1.67 1.81 | 1′ | 107.0 | 4.38, d (7.8) |
| 3 | 91.1 | 3.15 | 2′ | 75.3 | 3.22, dd (7.8; 2) |
| 4 | 40.2 | 3′ | 77.9 | 3.84 | |
| 5 | 57.1 | 0.76, d (3.7) | 4′ | 73.0 | 3.52 |
| 6 | 19.3 | 1.60 | 5′ | 76.7 | 3.78, d (9.8) |
| 7 | 34.3 | 1.38 1.52 | 6′ | 170.9 | |
| 8 | 40.8 | Ester | |||
| 9 | 48.1 | 1.62 | 1″ | 66.1 | 4.18, t (6.4) |
| 10 | 37.9 | 2″ | 31.7 | 1.64 | |
| 11 | 24.5 | 1.88, d (5.8) | 3″ | 20.1 | 1.42 |
| 12 | 123.5 | 5.31, br t (3.7) | 4″ | 14.0 | 0.95 |
| 13 | 144.7 | 28-O-Xyl I | |||
| 14 | 42.7 | 1‴ | 95.4 | 5.46, d (5.6) | |
| 15 | 36.5 | 1.41, d (4.1) 1.74, dd (4.0; 15.0) | 2‴ | 76.5 | 3.53 |
| 16 | 74.8 | 4.45 | 3‴ | 77.6 | 3.58, t (7.2) |
| 17 | 50.2 | 4‴ | 70.7 | 3.52 | |
| 18 | 42.3 | 2.95, dd (4.5; 14.3) | 5‴ | 66.3 | 3.33, dd (3.7; 7.9) 3.94, dd (4.5; 11.6) |
| 19 | 47.9 | 1.03 2.26, t (13.6) | Rha | ||
| 20 | 31.3 | 1⁗ | 101.1 | 5.16, d (1.8) | |
| 21 | 36.4 | 1.15, d (11.7) 1.91, d (10.3) | 2⁗ | 71.7 | 4.04, dd (1.8; 3.3) |
| 22 | 31.7 | 1.78 | 3⁗ | 81.4 | 3.87, dd (3.3; 9.4) |
| 23 | 28.5 | 1.04, s | 4⁗ | 78.4 | 3.69, t (9.5) |
| 24 | 16.9 | 0.84, s | 5⁗ | 69.1 | 3.76, dd (6.2; 0.5) |
| 25 | 16.1 | 0.95, s | 6⁗ | 18.4 | 1.26, d (6.2) |
| 26 | 17.9 | 0.77, s | Xyl II | ||
| 27 | 27.2 | 1.36, s | 1′′′′′ | 105.1 | 4.62, d (7.8) |
| 28 | 177.2 | 2′′′′′ | 75.6 | 3.16 | |
| 29 | 33.3 | 0.86, s | 3′′′′′ | 78.1 | 3.85 |
| 30 | 25.0 | 0.94, s | 4′′′′′ | 71.4 | 3.47 |
| 5′′′′′ | 67.0 | 3.16 3.84, dd (5.5; 11.4) | |||
| Api | |||||
| 1′′′′′′ | 111.9 | 5.26, d (3.8) | |||
| 2′′′′′′ | 78.2 | 4.02, d (3.8) | |||
| 3′′′′′′ | 80.0 | ||||
| 4′′′′′′ | 74.9 | 3.74, d (9.5) 4.08, d (9.6) | |||
| 5′′′′′′ | 64.9 | 3.56, d (1.4) | |||
| Position | 2 | 3 | 4 | 5 |
|---|---|---|---|---|
| 1 | 39.9 | 39.9 | 39.9 | 39.9 |
| 2 | 27.0 | 27.1 | 26.9 | 26.9 |
| 3 | 91.1 | 91.1 | 90.9 | 90.9 |
| 4 | 40.2 | 40.2 | 40.8 | 40.8 |
| 5 | 57.0 | 57.1 | 57.1 | 57.1 |
| 6 | 19.3 | 19.3 | 18.5 | 19.4 |
| 7 | 34.1 | 34.2 | 34.3 | 34.3 |
| 8 | 40.8 | 40.8 | 40.8 | 40.8 |
| 9 | 48.0 | 48.1 | 48.1 | 48.1 |
| 10 | 37.9 | 37.9 | 37.8 | 37.8 |
| 11 | 24.4 | 24.4 | 24.5 | 24.5 |
| 12 | 123.5 | 123.5 | 123.6 | 123.6 |
| 13 | 144.6 | 144.6 | 144.7 | 144.6 |
| 14 | 42.7 | 42.8 | 42.7 | 42.7 |
| 15 | 36.2 | 36.3 | 36.5 | 36.5 |
| 16 | 74.8 | 74.9 | 74.6 | 74.6 |
| 17 | 50.2 | 50.3 | 50.2 | 50.2 |
| 18 | 42.2 | 42.3 | 42.2 | 42.2 |
| 19 | 47.9 | 47.9 | 47.9 | 47.9 |
| 20 | 31.3 | 31.3 | 31.3 | 31.9 |
| 21 | 36.4 | 36.5 | 36.4 | 36.5 |
| 22 | 33.3 | 33.3 | 34.3 | 34.3 |
| 23 | 28.5 | 28.5 | 28.6 | 28.5 |
| 24 | 16.8 | 16.9 | 17.0 | 17.0 |
| 25 | 16.1 | 16.2 | 16.2 | 16.2 |
| 26 | 17.7 | 17.8 | 17.8 | 17.8 |
| 27 | 27.1 | 27.1 | 27.1 | 27.1 |
| 28 | 177.0 | 177.1 | 177.0 | 177.0 |
| 29 | 33.3 | 33.3 | 33.3 | 33.3 |
| 30 | 24.9 | 25.0 | 24.9 | 25.0 |
| Position | 2 | 3 | 4 | 5 | ||||
|---|---|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | δC | δH | |
| 3-O-GlcA | ||||||||
| 1′ | 107.0 | 4.38, d (7.8) | 107.0 | 4.38, d (7.8) | 106.9 | 4.38, d (7.7) | 106.9 | 4.38, d (7.7) |
| 2′ | 75.9 | 3.23 | 75.9 | 3.23 | 76.0 | 3.23 | 76.0 | 3.23 |
| 3′ | 77.5 | 3.35 | 77.0 | 3.34 | 77.9 | 3.34 | 77.9 | 3.34 |
| 4′ | 72.8 | 3.58 | 73.0 | 3.60 | 72.3 | 3.90 | 72.3 | 3.90 |
| 5′ | 76.6 | 3.78, d (9.9) | 76.2 | 3.78, d (9.9) | 76.1 | 3.79 | 76.1 | 3.79 |
| 6′ | 170.9 | 170.9 | nd | nd | ||||
| Ester | ||||||||
| 1″ | 66.1 | 4.18, td (2.2; 6.4) | 66.1 | 4.18, td (2.2; 6.4) | ||||
| 2″ | 31.7 | 1.65 | 31.7 | 1.65 | ||||
| 3″ | 20.0 | 1.42 | 20.0 | 1.42 | ||||
| 4″ | 14.0 | 0.92, t (3.0) | 14.0 | 0.92, t (3.0) | ||||
| 28-O-XylI | ||||||||
| 1‴ | 95.5 | 5.47, d (6.0) | 95.4 | 5.41, d (6.0) | 95.4 | 5.47, d (6.0) | 95.2 | 5.41, d (6.0) |
| 2‴ | 76.0 | 3.56, dd (5.9; 9.1) | 76.6 | 3.53 | 76.6 | 3.56 | 75.5 | 3.53 |
| 3‴ | 77.8 | 3.89 | 77.5 | 3.90 | 77.5 | 3.40 | 76.0 | 3.24 |
| 4‴ | 70.9 | 3.44 | 70.8 | 3.47 | 70.8 | 3.46 | 71.1 | 3.48 |
| 5‴ | 66.7 | 3.92, dd (4.7; 11.7) 3.33 | 66.5 | 3.89 | 66.5 | 3.91 | 66.6 | 3.92 |
| Rha | ||||||||
| 1⁗ | 98.4 | 5.22, d (1.8) | 101.0 | 5.23, d (1.8) | 98.4 | 5.24, d (2.0) | 101.1 | 5.23, d (1.8) |
| 2⁗ | 73.6 | 5.12, dd (1.8; 3.5) | 69.7 | 4.08, dd (1.8; 3.2) | 70.1 | 4.09, dd (1.8; 3.2) | 73.7 | 5.12, dd (1.7; 3.4) |
| 3⁗ | 70.3 | 4.06, dd (3.5; 9.5) | 75.8 | 5.04, dd (3.5; 9.5) | 75.9 | 5.05, dd (3.2; 9.1) | 70.8 | 4.06, dd (3.4; 9.3) |
| 4⁗ | 83.2 | 3.52 | 77.3 | 3.84 | 77.9 | 3.83 | 83.8 | 4.53 |
| 5⁗ | 69.0 | 3.76 | 69.2 | 3.82 | 69.2 | 3.81 | 68.9 | 3.74 |
| 6⁗ | 18.2 | 1.29, d (6.2) | 18.3 | 1.30, d (6.2) | 18.2 | 1.31, d (6.2) | 18.2 | 1.29, d (6.2) |
| Xyl II | ||||||||
| 1′′′′′ | 106.5 | 4.52, d (7.6) | 105.6 | 4.41, d (7.6) | 105.5 | 4.42, d (7.6) | 106.7 | 4.53, d (7.6) |
| 2′′′′′ | 75.3 | 3.22 | 75.8 | 3.15 | 75.8 | 3.15 | 76.0 | 3.23 |
| 3′′′′′ | 78.0 | 3.32 | 77.8 | 3.31 | 78.1 | 3.39 | 78.1 | 3.37 |
| 4′′′′′ | 71.1 | 3.50 | 71.3 | 3.51 | 71.1 | 3.46 | 71.8 | 3.45 |
| 5′′′′′ | 67.3 | 3.85 3.17 | 67.0 | 3.83 3.19 | 67.2 | 3.87 3.32 | 67.2 | 3.87 3.32 |
| OAc | 172.2 | 172.5 | 172.1 | 172.0 | ||||
| 20.9 | 2.09, s | 21.3 | 2.10, s | 21.3 | 2.11, s | 21.3 | 2.10, s | |
| Position | 6 | 7 | 8 |
|---|---|---|---|
| 1 | 39.9 | 39.9 | 39.9 |
| 2 | 26.7 | 26.8 | 26.9 |
| 3 | 90.8 | 90.8 | 90.7 |
| 4 | 40.2 | 40.2 | 40.2 |
| 5 | 57.1 | 57.1 | 57.2 |
| 6 | 19.3 | 19.3 | 19.4 |
| 7 | 34.2 | 34.3 | 34.5 |
| 8 | 40.8 | 40.9 | 40.8 |
| 9 | 48.1 | 48.2 | 48.2 |
| 10 | 37.9 | 37.9 | 37.9 |
| 11 | 24.5 | 24.5 | 24.5 |
| 12 | 123.6 | 123.6 | 123.7 |
| 13 | 145.5 | 145.7 | 145.6 |
| 14 | 42.8 | 42.8 | 42.8 |
| 15 | 36.4 | 36.5 | 36.6 |
| 16 | 74.9 | 74.6 | 74.7 |
| 17 | 50.2 | 50.3 | 50.4 |
| 18 | 42.2 | 42.2 | 42.3 |
| 19 | 47.9 | 47.8 | 47.8 |
| 20 | 31.3 | 31.3 | 31.3 |
| 21 | 36.4 | 36.4 | 36.4 |
| 22 | 31.8 | 31.8 | 32.0 |
| 23 | 28.6 | 28.5 | 28.5 |
| 24 | 17.1 | 17.0 | 17.0 |
| 25 | 16.2 | 16.2 | 16.2 |
| 26 | 17.9 | 17.8 | 17.8 |
| 27 | 27.2 | 27.2 | 27.2 |
| 28 | 177.0 | 177.1 | 177.2 |
| 29 | 33.4 | 33.4 | 33.4 |
| 30 | 24.9 | 25.0 | 25.1 |
| Position | 6 | 7 | 8 | |||
|---|---|---|---|---|---|---|
| δC | δH | δC | δH | δC | δH | |
| 3-O-Glc A | ||||||
| 1′ | 106.7 | 4.34, d (7.8) | 106.7 | 4.34, d (7.8) | 106.7 | 4.34, d (7.8) |
| 2′ | 75.2 | 3.22–3.24 | 75.5 | 3.22–3.24 | 75.4 | 3.22–3.24 |
| 3′ | 78.0 | 3.37 | 78.0 | 3.37 | 78.0 | 3.37 |
| 4′ | 73.7 | 3.47, t (9.3) | 73.7 | 3.47, t (9.3) | 73.8 | 3.47, t (9.3) |
| 5′ | 76.8 | 3.54–3.58 | 76.8 | 3.54–3.58 | 76.8 | 3.54–3.58 |
| 6′ | nd | nd | nd | |||
| 28-O-Xyl | ||||||
| 1″ | 95.5 | 5.52, d (6.0) | 95.3 | 5.48, d (6.0) | 95.4 | 5.44, d (6.0) |
| 2″ | 76.9 | 3.56 | 76.9 | 3.56 | 76.8 | 3.54–3.58 |
| 3″ | 75.8 | 3.56–3.58 | 75.8 | 3.56–3.58 | 75.8 | 3.56–3.58 |
| 4″ | 70.8 | 3.51–3.53 | 70.8 | 3.51–3.53 | 70.8 | 3.51–3.53 |
| 5″ | 66.5 | 3.31–3.33 3.92–3.94 | 66.5 | 3.31–3.33 3.92–3.94 | 66.6 | 3.31–3.33 3.92–3.94 |
| Rha | ||||||
| 1‴ | 101.5 | 5.33, d (2.0) | 101.5 | 5.23, d (1.9) | 101.6 | 5.15, d (1.8) |
| 2‴ | 73.9 | 5.12, d (2.1) | 70.2 | 4.07, dd (1.9; 3.3) | 70.2 | 3.89 |
| 3‴ | 70.8 | 3.51–3.53 | 75.8 | 4.87 | 72.2 | 3.95 |
| 4‴ | 70.9 | 3.59 | 70.9 | 3.59 | 75.4 | 4.93 |
| 5‴ | 70.5 | 3.65–3.68 | 70.5 | 3.65–3.68 | 68.2 | 3.35 |
| 6‴ | 18.0 | 1.26, d (6.5) | 18.3 | 1.29, d (6.7) | 18.2 | 1.27, d (6.7) |
| OAc | 172.4 | 172.7 | 172.6 | |||
| 20.5 | 2.08, s | 21.1 | 2.10, s | 21.2 | 2.09, s | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tchegnitegni, B.T.; Ahmed, T.; Ngouafong, F.T.; Kamso, V.F.K.; Teponno, R.B.; Lenta, B.N.; Tapondjou, L.A.; Ali, A.; Musharraf, S.G. Echinocystic Acid Bidesmoside Saponins from Microglossa afzelii O. Hoffm and Their Cytotoxic Activity against the CAL-27 Oral Squamous Carcinoma Cell Line. Metabolites 2022, 12, 1018. https://doi.org/10.3390/metabo12111018
Tchegnitegni BT, Ahmed T, Ngouafong FT, Kamso VFK, Teponno RB, Lenta BN, Tapondjou LA, Ali A, Musharraf SG. Echinocystic Acid Bidesmoside Saponins from Microglossa afzelii O. Hoffm and Their Cytotoxic Activity against the CAL-27 Oral Squamous Carcinoma Cell Line. Metabolites. 2022; 12(11):1018. https://doi.org/10.3390/metabo12111018
Chicago/Turabian StyleTchegnitegni, Billy Toussie, Tehmina Ahmed, Francis Tatong Ngouafong, Viviane Flore Kamlo Kamso, Rémy Bertrand Teponno, Bruno Ndjakou Lenta, Léon Azefack Tapondjou, Arslan Ali, and Syed Ghulam Musharraf. 2022. "Echinocystic Acid Bidesmoside Saponins from Microglossa afzelii O. Hoffm and Their Cytotoxic Activity against the CAL-27 Oral Squamous Carcinoma Cell Line" Metabolites 12, no. 11: 1018. https://doi.org/10.3390/metabo12111018