

Cheminformatics Bioprospection of Broad Spectrum Plant Secondary Metabolites Targeting the Spike Proteins of Omicron Variant and Wild-Type SARS-CoV-2
 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Library of Plant Secondary Metabolites
2.2. Collection and Preparation of LOCM and Reference Standards
2.3. Collection and Preparation of SC-2WT and Omicron SPs
2.4. SC-2WT and Omicron SP Active Sites Identification and Molecular Docking of Ligands
2.5. Docking Protocol Validation
2.6. Molecular Dynamics Simulation
2.7. Post-MD Simulation
2.8. Pharmacokinetic Analysis and Molecular Fingerprinting of the Top-Ranked LOCM
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mokhtari, T.; Hassani, F.; Ghaffari, N.; Ebrahimi, B.; Yarahmadi, A.; Hassanzadeh, G. COVID-19 and multiorgan failure: A narrative review on potential mechanisms. J. Mol. Histol. 2020, 51, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Renu, K.; Prasanna, P.L.; Valsala, G.A. Coronaviruses pathogenesis, comorbidities and multi-organ damage—A review. Life Sci. 2020, 255, 117839. [Google Scholar] [CrossRef] [PubMed]
- Islam, T.; Hasan, M.; Rahman, M.S.; Islam, M.R. Comparative evaluation of authorized drugs for treating Covid-19 patients. Health Sci. Rep. 2022, 5, e671. [Google Scholar] [CrossRef] [PubMed]
- Şenay, Ş. Coronavirus pandemic and cardiovascular issues. Turk. Gogus Kalp Damar Cerrahisi Derg. 2020, 28, 227–228. [Google Scholar] [CrossRef]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Vardhan, S.; Sahoo, S.K. Computational studies on the interaction of SARS-CoV-2 Omicron SGp RBD with human receptor ACE2, limonin and glycyrrhizic acid. Comput. Biol. Med. 2022, 144, 105367. [Google Scholar] [CrossRef]
- Gu, H.; Krishnan, P.; Ng, D.Y.M.; Chang, L.D.J.; Liu, G.Y.Z.; Cheng, S.S.M.; Hui, M.M.Y.; Fan, M.C.Y.; Wan, J.H.L.; Lau, L.H.K.; et al. Probable transmission of SARS-CoV-2 omicron variant in quarantine hotel, Hong Kong, China, November 2021. Emerg. Infect. Dis. 2022, 28, 460–462. [Google Scholar] [CrossRef]
- Ahmad, S.; Zahiruddin, S.; Parveen, B.; Basist, P.; Parveen, A.; Gautam, G.; Parveen, R.; Ahmad, M. Indian medicinal plants and formulations and their potential against COVID-19-preclinical and clinical research. Front Pharm. 2021, 11, 578970. [Google Scholar] [CrossRef]
- Rasool, N.; Bakht, A.; Hussain, W. Analysis of inhibitor binding combined with reactivity studies to discover the potentially inhibiting phytochemicals targeting chikungunya viral replication. Curr. Drug Discov. Technol. 2021, 18, 437–450. [Google Scholar] [CrossRef]
- Sabiu, S.; O’Neill, F.H.; Ashafa, A.O.T. Toxicopathological evaluation of a 28-day repeated dose administration of Zea mays L. (Poaceae), Stigma maydis aqueous extract on key metabolic markers of Wistar rats. Trans. R. Soc. S. Afr. 2017, 72, 225–233. [Google Scholar] [CrossRef]
- Volpato, G.; God´ınez, D.; Beyra, A.; Barreto, A. Uses of medicinal plants by Haitian immigrants and their descendants in the Province of Camag¨uey, Cuba. J. Ethnobiol. Ethnomed. 2009, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Balogun, F.O.; Sabiu, S. A Review of the Phytochemistry, Ethnobotany, Toxicology, and Pharmacological Potentials of Crescentia cujete L. (Bignoniaceae). Evid. Based Complement. Altern. Med. 2021, 2021, e6683708. [Google Scholar] [CrossRef]
- Shode, F.O.; Idowu, A.S.K.; Uhomoibhi, O.J.; Sabiu, S. Repurposing drugs and identification of inhibitors of integral proteins (spike protein and main protease) of SARS-CoV-2. J. Biomol. Struct. Dyn. 2021, 2, 1–16. [Google Scholar] [CrossRef]
- Aribisala, J.O.; Abdulsalam, R.A.; Dweba, Y.; Madonsela, K.; Sabiu, S. Identification of secondary metabolites from Crescentia cujete as promising antibacterial therapeutics targeting type 2A topoisomerases through molecular dynamics simulation. Comput. Biol. Med. 2022, 145, 105432. [Google Scholar] [CrossRef]
- Delijewski, M.; Haneczok, J. AI drug discovery screening for COVID-19 reveals zafirlukast as a repurposing candidate. Med. Drug Discov. 2021, 9, 100077. [Google Scholar] [CrossRef]
- Farhat, N.; Khan, A.U. Repurposing drug molecule against SARS-Cov-2 (COVID-19) through molecular docking and dynamics: A quick approach to pick FDA-approved drugs. J. Mol. Model. 2021, 27, 312. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Sabiu, S.; Balogun, F.O.; Amoo, S.O. Phenolics profiling of Carpobrotus edulis (L.) N.E.Br. and insights into molecular dynamics of their significance in type 2 diabetes therapy and Its retinopathy complication. Molecules 2021, 26, 4867. [Google Scholar] [CrossRef]
- BIOVIA; Dassault Systèmes. Discovery Studio, version 21.1.0; Dassault Systèmes: San Diego, CA, USA, 2021. [Google Scholar]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein–ACE2 complex. Sciences 2022, 375, 760–764. [Google Scholar] [CrossRef]
- Aribisala, J.O.; Sabiu, S. Cheminformatics Identification of Phenolics as Modulators of Penicillin-Binding Protein 2a of Staphylococcus aureus: A Structure–Activity-Relationship-Based Study. Pharmaceutics 2022, 14, 1818. [Google Scholar] [CrossRef]
- Acharya, A.; Agarwal, R.; Baker, M.B.; Baudry, J.; Bhowmik, D.; Boehm, S.; Byler, K.G.; Chen, S.Y.; Coates, L.; Cooper, C.J.; et al. Supercomputer-Based Ensemble Docking Drug Discovery Pipeline with Application to Covid-19. Chem. Inf. Model. 2020, 60, 5832–5852. [Google Scholar] [CrossRef]
- Uhomoibhi, J.O.; Shode, F.O.; Idowu, K.A.; Sabiu, S. Molecular modelling identification of phytocompounds from selected African botanicals as promising therapeutics against druggable human host cell targets of SARS-CoV-2. J. Mol. Graph. Model. 2022, 114, 108185. [Google Scholar] [CrossRef]
- Ramirex, D.; Caballero, J. Is it reliable to use common molecular docking methods for comparing the binding affinities of Enantiomer pairs for their protein target? Int. J. Mol. Sci. 2016, 17, 525. [Google Scholar] [CrossRef] [Green Version]
- Nasution, F.; Toepak, M.A.; Alkaff, E.P.; Tambunan, U.S.F. Flexible docking-based molecular dynamics simulation of natural product compounds and Ebola virus Nucleocapsid (EBOV NP): A computational approach to discover new drug for combating Ebola. BMC Bioinform. 2018, 19, 419–436. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, X.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into Protein-Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef] [Green Version]
- Kufareva, I.; Abagyan, R. Methods of protein structure comparison. Methods Mol. Biol. 2012, 857, 231–257. [Google Scholar] [CrossRef] [Green Version]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Fahad, M.; Al-Khodairy, M.; Kalim, A.; Khan, M.K.; Manogaran, S.P.; Salman, A.; Jamal, M.A. In Silico Prediction of Mechanism of Erysolin-induced Apoptosis in Human Breast Cancer Cell Lines, American. J. Bioinform. 2013, 3, 62–71. [Google Scholar] [CrossRef]
- Mousavi, S.S.; Karami, A.; Haghighi, T.M.; Tumilaar, S.G.; Fatimawali, I.R.; Mahmud, S.; Celik, I.A.; Agagündüz, D.; Tallei, T.E.; Emran, T.B.; et al. In Silico Evaluation of Iranian Medicinal Plant Phytoconstituents as Inhibitors against Main Protease and the Receptor-Binding Domain of SARS-CoV-2. Molecules 2021, 26, 5724. [Google Scholar] [CrossRef] [PubMed]
- Izadi, H.; Stewart, K.M.E.; Penlidis, A. Role of contact electrification and electrostatic interactions in gecko adhesion. J. R. Soc. Interface 2014, 11, 371–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kretschmer, R.; Kinzel, D.; Gonza´ Lez, L. The Role of Hydrogen Bonds in Protein—Ligand Interactions. DFT Calculations in 1,3-Dihydrobenzimidazole-2 Thione Derivatives with Glycinamide as Model HIV RT Inhibitors. Int. J. Quantum Chem. 2010, 112, 1787–1795. [Google Scholar] [CrossRef]
- Yamashita, F.; Hashida, M. In silico approaches for predicting ADME properties of drugs. Drug Metab. Pharmacokinet. 2004, 19, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Remko, M.; Boháč, A.; Kováčiková, L. Molecular structure, p K a, lipophilicity, solubility, absorption, polar surface area, and blood brain barrier penetration of some antiangiogenic agents. Struct. Chem. 2011, 22, 635–648. [Google Scholar] [CrossRef]
- Khumbulani, M.; Alayande, K.A.; Sabiu, S. Orientin Enhances Colistin-Mediated Bacterial Lethality through Oxidative Stress Involvement". Evid. Based Complement. Altern. Med. 2022, 2022, 3809232. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2009, 3, 935–949. [Google Scholar] [CrossRef]
- Bissantz, C.; Kuhn, B.; Stahl, M.A. Medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 14, 5061–5084. [Google Scholar] [CrossRef]
- Stella, L.; Melchionna, S. Equilibration and sampling in molecular dynamics simulations of biomolecules. Chem. Phys. 1998, 109, 10115. [Google Scholar] [CrossRef]
- Al-Karmalawy, A.A.; Dahab, M.A.; Metwaly, A.M.; Elhady, S.S.; Elkaeed, E.B.; Eissa, I.H.; Darwish, K.M. Molecular Docking and Dynamics Simulation Revealed the Potential Inhibitory Activity of ACEIs Against SARS-CoV-2 Targeting the hACE2 Receptor. Front. Chem. 2021, 9, 661230. [Google Scholar] [CrossRef]
- Dalke, A. The FPS fingerprint format and chemfp toolkit. J. Cheminform. 2013, 5, 36–60. [Google Scholar] [CrossRef] [Green Version]
- Verma, A.K.; Ahmed, S.F.; Hossain, M.S.; Bhojiya, A.A.; Mathur, A.; Upadhyay, S.K.; Srivastava, A.K.; Vishvakarma, N.K.; Barik, M.; Rahaman, M.M.; et al. Molecular docking and simulation studies of flavonoid compounds against PBP-2a of methicillin-resistant. Staphylococcus Aureus J. Biomol. Struct. Dyn. 2021, 1–17. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligands | Binding Affinity (kcal/mol) |
|---|---|
| Omicron variant SP | |
| Zafirlukast | −7.4 |
| Cefoperazone | −6.2 |
| Maysin | −7.5 |
| 6-Hydroxylcyanidin-3-rutinoside | −7.2 |
| Kaempferol-7-glucoside | −7.2 |
| Geraniin | −7.5 |
| Epigallocatechin gallate | −7.2 |
| SC-2WT SP | |
| Zafirlukast | −7.9 |
| Cefoperazone | −6.7 |
| Maysin | −8.4 |
| Geraniin | −7.1 |
| Catalposide | −7.4 |
| Kaempferol-7-glucoside | −7.3 |
| 6-Hydroxylcyanidin-3 | −7.2 |
| Energy Components (kcal/mol) | |||||
|---|---|---|---|---|---|
| Complex | ΔEvdW | ΔEelec | ΔGgas | ΔGsolv | ΔGbind |
| Omicron SP | |||||
| 6-Hydroxycyanidin 3-rutinoside | −26.63 ± 9.10 | −137.98 ± 33.21 | −164.62 ± 29.55 | 121.65 ± 23.84 | −42.97 ± 8.34 |
| Epigallocatechin gallate | −22.16 ± 6.10 | −58.77 ± 18.02 | −80.93 ± 15.53 | 53.06 ± 11.68 | −27.86 ± 5.98 |
| Geraniin | −34.44 ± 4.42 | −31.07 ± 10.71 | −65.52 ± 11.96 | 34.23 ± 8.83 | −31.28 ± 7.25 |
| Kaempferol-7-glucoside | −21.98 ± 5.45 | −36.20 ± 20.45 | −58.19 ± 21.36 | 38.87 ± 17.02 | −19.31 ± 5.33 |
| Maysin | −43.96 ± 6.71 | −59.86 ± 17.91 | −103.83 ± 20.81 | 64.94 ± 12.05 | −38.88 ± 10.21 |
| Zafirlukast | −36.22 ± 6.65 | −23.79 ± 13.96 | −60.02 ± 15.90 | 37.64 ± 13.87 | −22.38 ± 5.95 |
| SC-2WT SP | |||||
| 6-Hydroxycyanidin 3-rutinoside | −37.69 ± 5.97 | 26.06 ± 28.31 | −11.63 ± 29.43 | −17.20 ± 19.81 | −28.84 ± 10.87 |
| Catalposide | −37.96 ± 3.84 | −22.18 ± 9.58 | −60.14 ± 11.44 | 31.23 ± 7.65 | −28.90 ± 4.95 |
| Geraniin | −36.41 ± 4.51 | −45.64 ± 11.29 | −82.07 ± 11.43 | 45.16 ± 8.53 | −36.90 ± 4.55 |
| Kaempferol-7-glucoside | −47.06 ± 6.40 | −20.68 ± 8.10 | −67.75 ± 10.97 | 30.64 ± 5.15 | −37.11 ± 7.01 |
| Maysin | −35.97 ± 7.15 | −38.54 ± 10.88 | −74.51 ± 10.98 | 41.66 ± 6.49 | −34.85 ± 6.01 |
| Zafirlukast | −44.23 ± 5.55 | −14.70 ± 8.92 | −58.94 ± 11.24 | 25.21 ± 7.82 | −33.73 ± 4.99 |
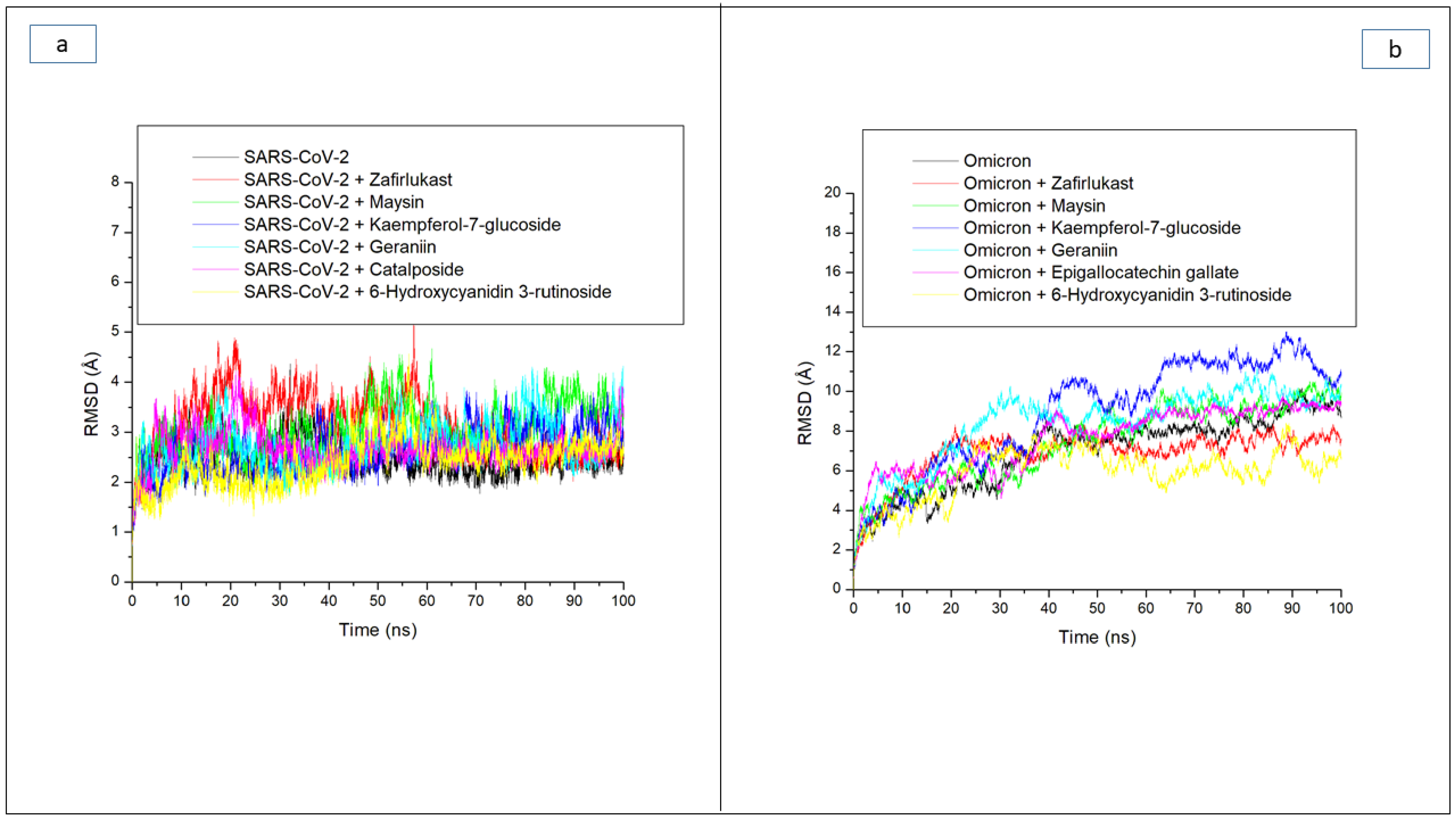
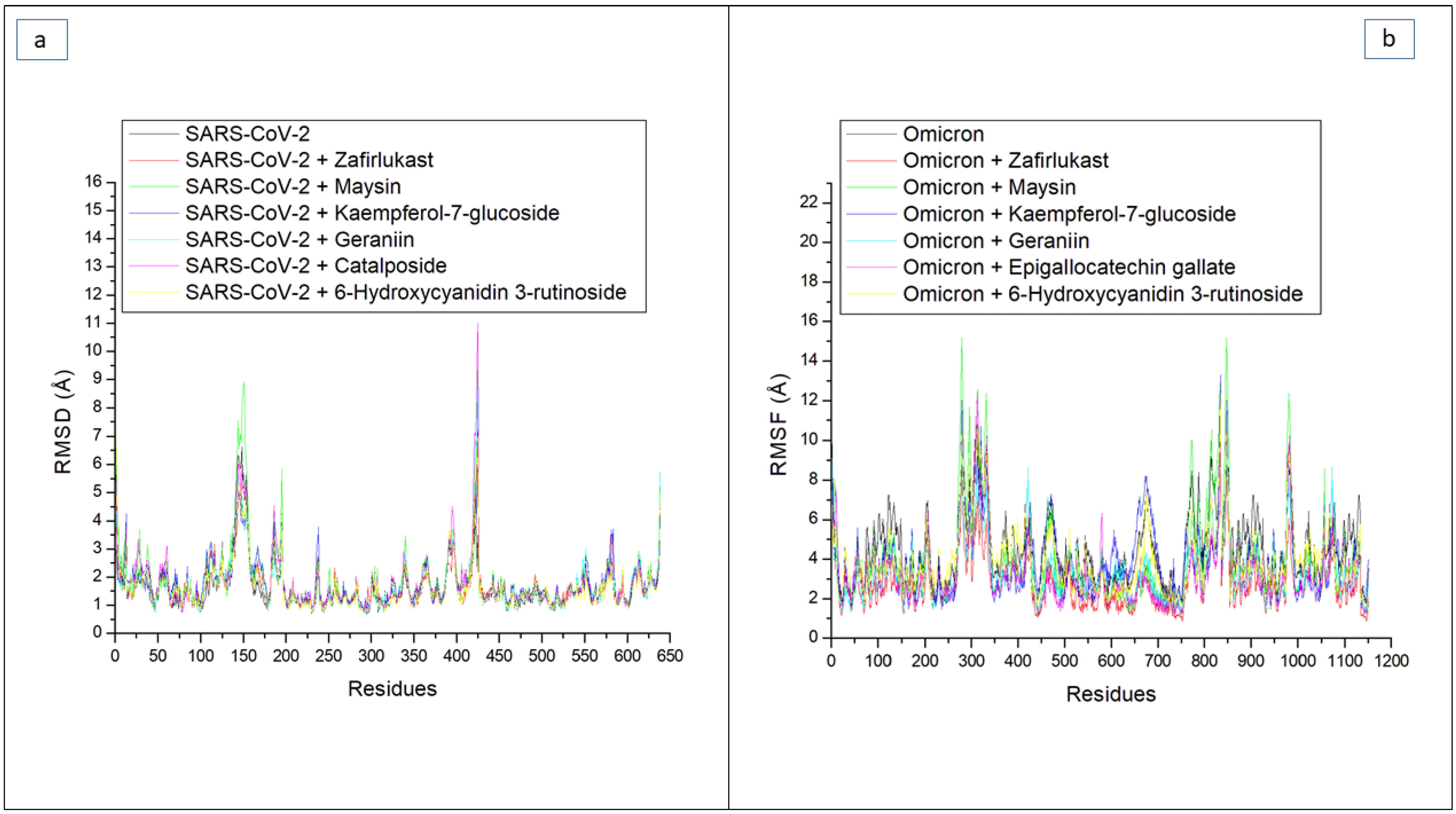
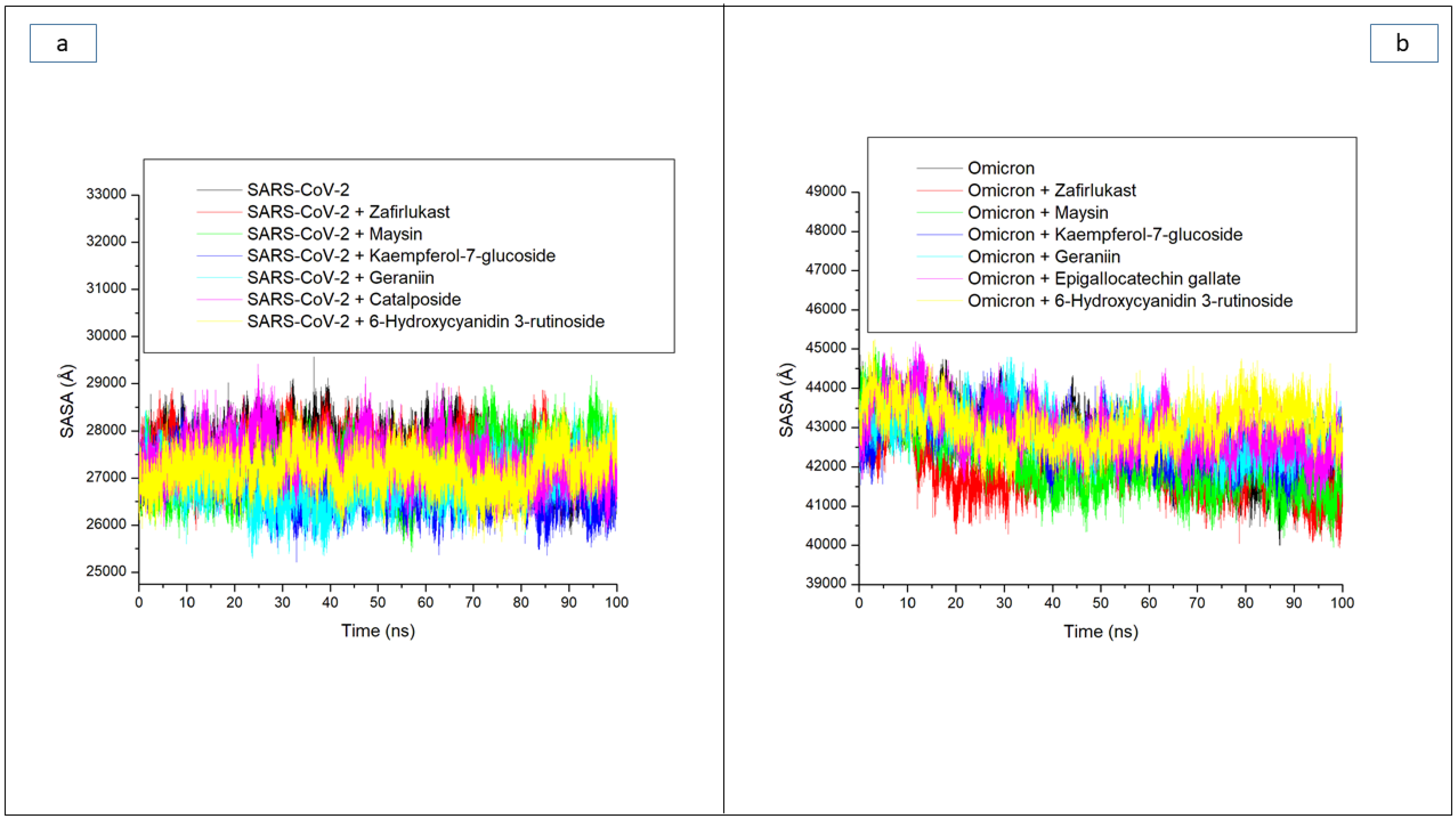
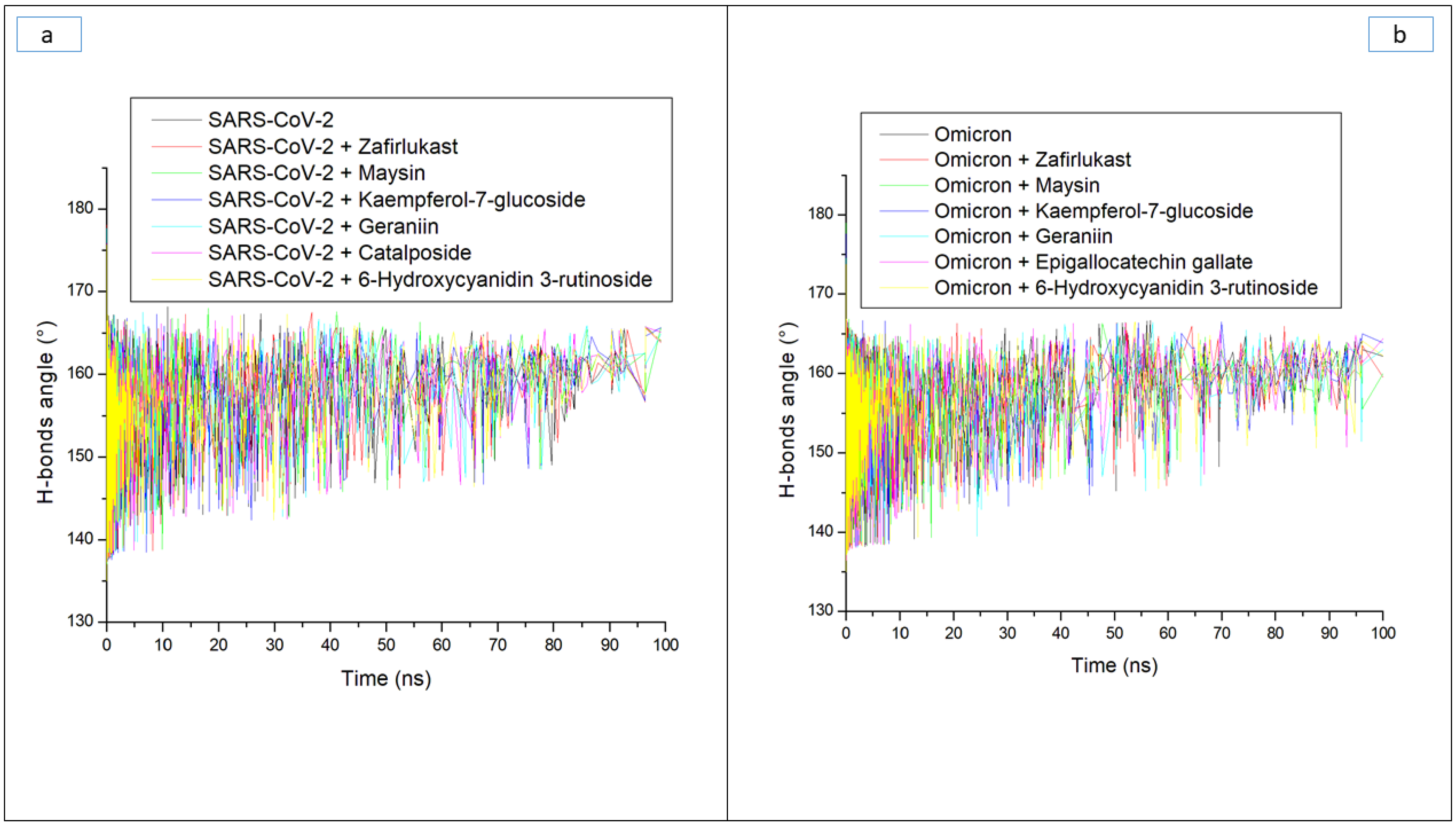
| Systems | Average RMSD (Å) | Average RMSF (Å) | Average ROG (Å) | Average SASA (Å) | Average Number of H-Bonds | Average Distance (Å) of H-Bonds | Average Angle (°) of H-Bonds |
|---|---|---|---|---|---|---|---|
| Omicron SP | |||||||
| 6-Hydroxycyanidin 3-rutinoside | 5.94 ± 1.32 | 3.91 ± 1.75 | 39.50 ± 0.42 | 43,070.18 ± 533 | 176.16 ± 8.4 | 2.87 ± 0.04 | 151.73 ± 7.8 |
| Epigallocatechin gallate | 7.60 ± 1.61 | 3.28 ± 1.78 | 39.12 ± 0.67 | 42,933.95 ± 612 | 162.78 ± 8.4 | 2.87 ± 0.04 | 151.66 ± 7.4 |
| Geraniin | 8.71 ± 1.70 | 3.26 ± 1.48 | 38.58 ± 0.44 | 42,988.52 ± 552 | 172.74 ± 8.3 | 2.87 ± 0.04 | 151.89 ± 7.7 |
| Kaempferol-7-glucoside | 9.44 ± 2.50 | 3.91 ± 1.82 | 38.12 ± 0.71 | 42,578.88 ± 587 | 168.86 ± 8.3 | 2.87 ± 0.04 | 152.02 ± 7.6 |
| Maysin | 8.25 ± 2.14 | 3.91 ± 2.41 | 38.20 ± 0.68 | 41,966.76 ± 761 | 174.83 ± 8.4 | 2.87 ± 0.04 | 151.84 ± 7.3 |
| Zafirlukast | 6.99 ± 1.00 | 2.70 ± 1.30 | 38.41 ± 0.45 | 41,942.55 ± 578 | 171.01 ± 8.6 | 2.87 ± 0.04 | 151.66 ± 7.4 |
| Apo omicron | 8.56 ± 2.14 | 4.43 ± 1.82 | 38.07 ± 0.69 | 42,060.34 ± 865 | 163.40 ± 7.8 | 2.87 ± 0.04 | 151.93 ± 7.6 |
| SC-2WT SP | |||||||
| 6-Hydroxycyanidin 3-rutinoside | 2.43 ± 0.45 | 1.66 ± 0.85 | 30.48 ± 0.28 | 27,171.49 ± 396 | 162.74 ± 8.3 | 2.85 ± 0.06 | 164.74 ± 8.4 |
| Catalposide | 2.67 ± 0.36 | 1.77 ± 1.02 | 30.18 ± 0.29 | 27,394.87 ± 439 | 161.02 ± 9.3 | 2.85 ± 0.06 | 161.02 ± 9.33 |
| Geraniin | 2.75 ± 0.44 | 1.66 ± 0.89 | 30.09 ± 0.29 | 26,914.74 ± 433 | 165.32 ± 7.3 | 2.85 ± 0.05 | 158.31 ± 8.7 |
| Kaempferol-7-glucoside | 2.64 ± 0.43 | 1.78 ± 0.81 | 30.28 ± 0.33 | 26,835.58 ± 418 | 166.81 ± 8.7 | 2.85 ± 0.06 | 165.21 ± 8.7 |
| Maysin | 3.07 ± 0.49 | 1.92 ± 1.14 | 30.39 ± 0.33 | 27,277.10 ± 491 | 163.32 ± 6.4 | 2.85 ± 0.06 | 157.95 ± 8.3 |
| Zafirlukast | 3.15 ± 0.58 | 1.70 ± 0.76 | 30.16 ± 0.26 | 27,473.62 ± 390 | 161.21 ± 7.3 | 2.85 ± 0.05 | 160.26 ± 8.2 |
| Apo SC-2WT | 2.48 ± 0.33 | 1.64 ± 0.83 | 30.27 ± 0.34 | 27,541.98 ± 449 | 159.24 ± 8.3 | 2.85 ± 0.06 | 161.56 ± 8.7 |
| RBD Residues of Omicron SP | 6-Hydroxycyanidin 3-Rutinoside | Epigallocatechin Gallate | Geraniin | Kaempferol-7-Glucoside | Maysin | Zafirlukast | Apo-Omicron SP |
|---|---|---|---|---|---|---|---|
| 353 | 3.64 | 2.10 | 2.94 | 2.45 | 2.17 | 2.12 | 3.17 |
| 493 | 2.56 | 1.51 | 1.89 | 2.53 | 2.23 | 1.60 | 3.17 |
| 496 | 2.83 | 2.13 | 2.36 | 2.25 | 2.53 | 1.95 | 3.47 |
| 498 | 2.50 | 1.95 | 1.88 | 1.77 | 2.13 | 2.27 | 2.89 |
| 500 | 2.64 | 2.22 | 2.31 | 2.01 | 2.42 | 2.61 | 3.15 |
| 501 | 3.00 | 2.76 | 2.85 | 2.16 | 2.78 | 2.78 | 3.60 |
| 505 | 2.89 | 3.76 | 4.16 | 3.02 | 3.21 | 2.45 | 3.88 |
| Total RMSF | 2.86 | 2.34 | 2.62 | 2.31 | 2.49 | 2.25 | 3.33 |
| RBD Residues of SC-2WT SP | 6-Hydroxycyanidin 3-Rutinoside | Catalposide | Geraniin | Kaempferol-7-Glucoside | Maysin | Zafirlukast | Apo-SC-2WT SP |
| 473 | 1.12 | 1.22 | 1.03 | 1.34 | 1.18 | 1.14 | 1.08 |
| 475 | 1.45 | 1.44 | 1.35 | 1.59 | 1.58 | 1.41 | 1.26 |
| 478 | 1.44 | 1.36 | 1.25 | 1.63 | 1.40 | 1.51 | 1.07 |
| 484 | 1.32 | 1.24 | 1.21 | 1.37 | 1.36 | 1.39 | 1.14 |
| 486 | 1.29 | 1.19 | 1.17 | 1.34 | 1.36 | 1.36 | 1.08 |
| 487 | 1.35 | 1.11 | 1.24 | 1.27 | 1.43 | 1.36 | 1.21 |
| 489 | 1.23 | 1.25 | 1.16 | 1.42 | 1.42 | 1.34 | 1.08 |
| Total RMSF | 1.31 | 1.26 | 1.20 | 1.42 | 1.39 | 1.35 | 1.13 |
| Ligands | MW < 500 (g/mol) | HB- A ≤ 10 | HB- D ≤ 5 | Log P o/w ≤ 5 | WS | GI Absorption | BBB Permeant | Pgp | Inhibitor of CYP 450 s | LV (N) | BS | H | C | IM | M | CY | LD50 (mg/kg) | TC | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CYP 1A2 | CYP 2C19 | CYP 2C9 | CYP 2D6 | CYP 3A4 | ||||||||||||||||||
| 6-Hydroxycyanidin 3-rutinoside | 611.53 | 16 | 11 | −2.45 | S | L | N | N | N | N | N | N | N | Y (3) | 0.17 | I | I | A | I | I | 5000 | 5 |
| Epigallocatechin gallate | 458.3 | 11 | 8 | 1.01 | S | L | N | N | N | N | N | N | N | Y (2) | 0.17 | I | I | I | I | I | 1000 | 4 |
| Catalposide | 482.4 | 12 | 6 | −0.90 | S | L | N | Y | N | N | N | N | N | Y (2) | 0.17 | I | I | I | I | I | 2000 | 4 |
| Geraniin | 952.6 | 27 | 14 | −1.70 | S | L | N | Y | N | N | N | N | N | Y (2) | 0.17 | I | I | A | I | I | 300 | 3 |
| Kaempferol-7-glucoside | 952.6 | 27 | 14 | −1.70 | S | L | N | Y | N | N | N | N | N | Y (2) | 0.17 | I | I | I | I | I | 5000 | 5 |
| Maysin | 952.6 | 27 | 14 | −1.70 | S | L | N | Y | N | N | N | N | N | Y (3) | 0.17 | I | I | A | I | I | 5000 | 5 |
| Zafirlukast | 448.3 | 11 | 7 | −0.04 | S | L | N | N | N | N | N | N | N | Y (2) | 0.17 | A | I | A | I | I | 300 | 3 |
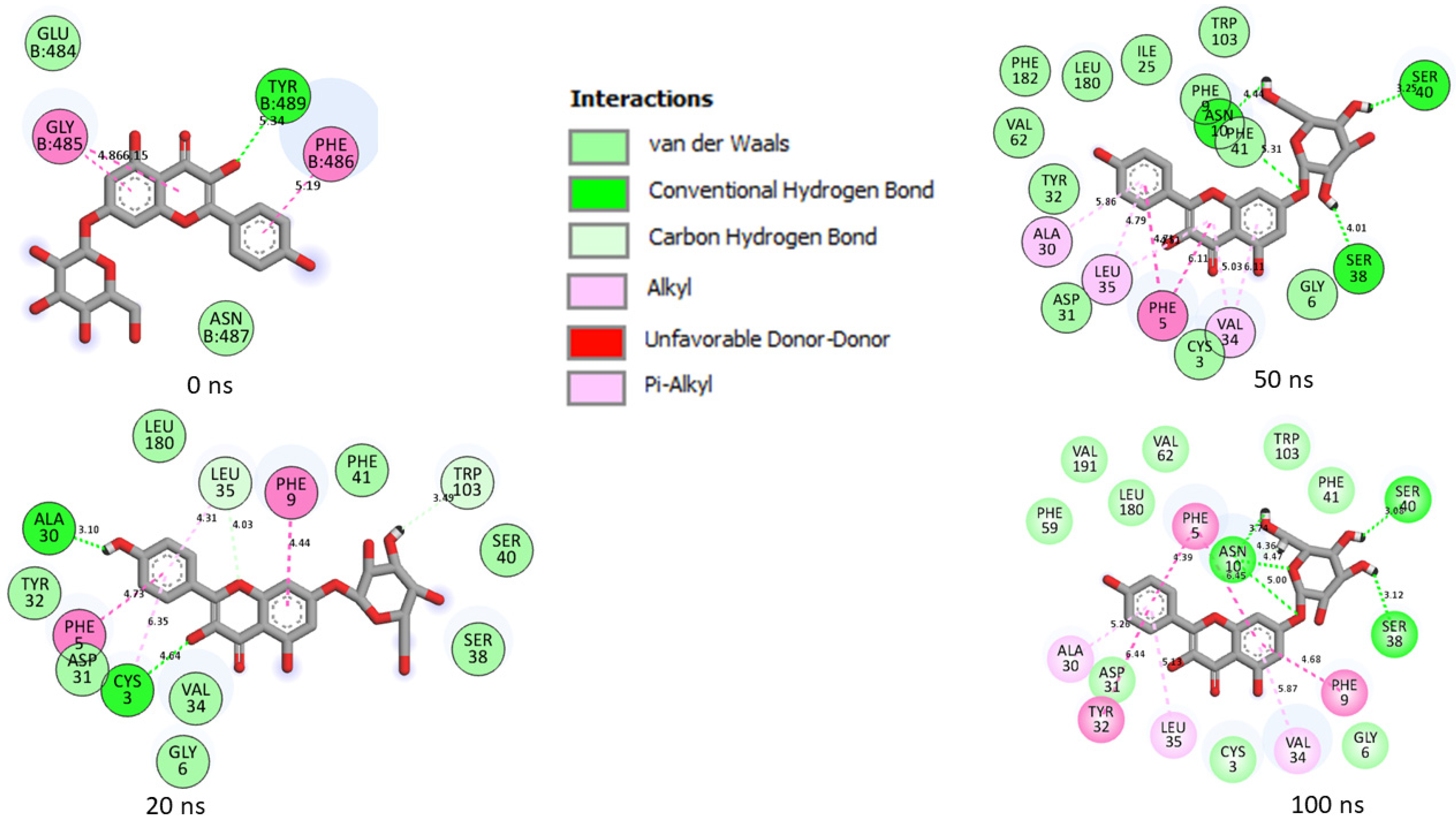
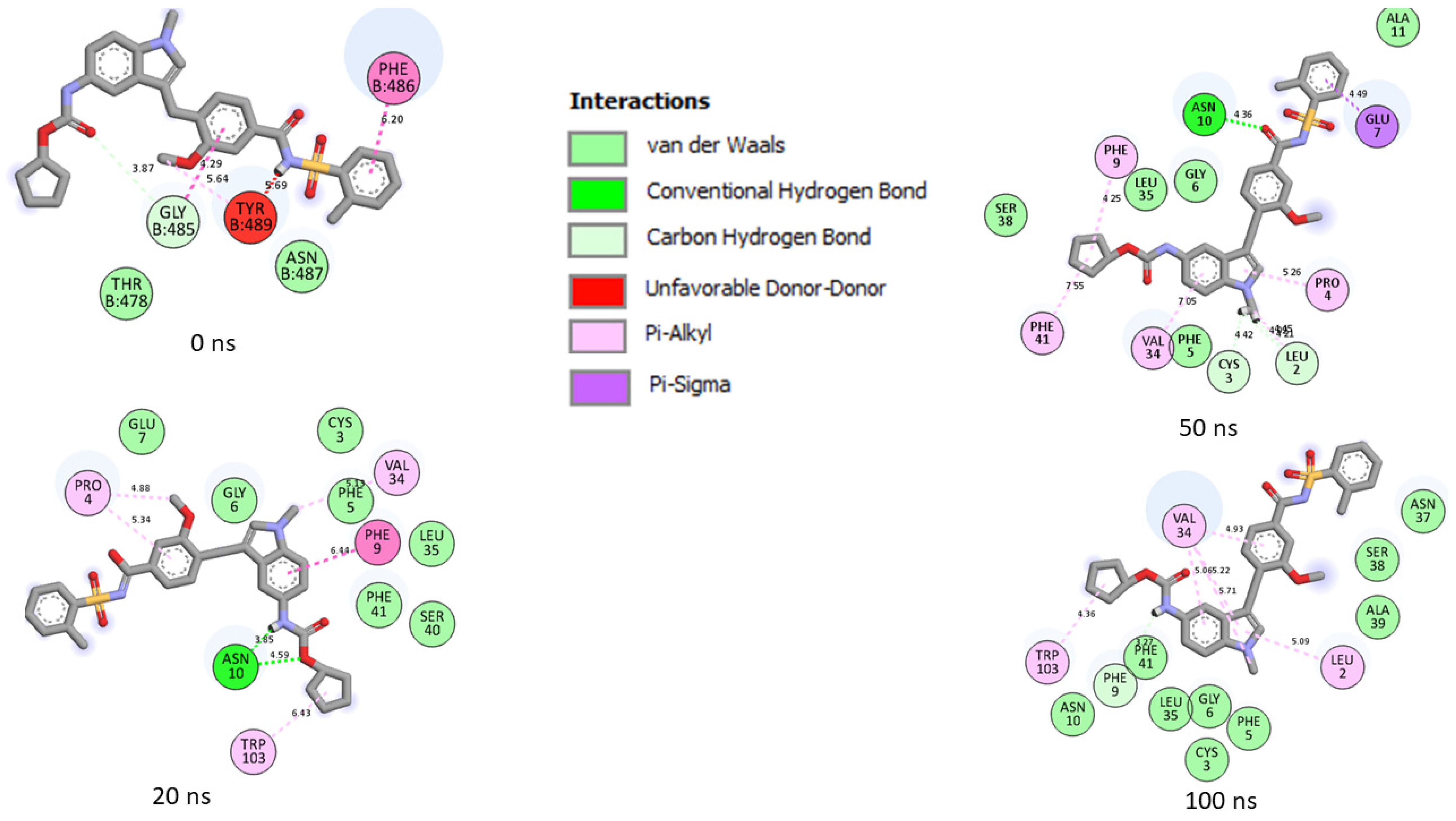
| Top Five LOCM Compounds | Total Number of Interactions (Average Distance) | Number of Hydrogen Bonds (Average Distance) and Interaction Residues | Other Important Interactions and Residues | Unfavorable Bonds |
|---|---|---|---|---|
| Omicron SP | ||||
| 6-Hydroxycyanidin 3-rutinoside | 19 (4.65 Å) | 9 (4.42 Å) [Gln39, Asp40, Lys156, Asp667 (2), Tyr38, Hie36, Asp27 (2)] | 2 (4.94 Å) [Ile158 (2)] | None |
| Epigallocatechin gallate | 14 (4.29 Å) | 7 (4.16 Å) [Tyr161, Asp667 (2), Ser662 (2), Arg671, Asp185] | 2 (4.76 Å) [Tyr161(2)] | None |
| Geraniin | 15 (4.69 Å) | 4 (4.33 Å) [Asn157, Asp40, Pro220, Lys652] | 4 (5.05 Å) [Asp40, Leu41, Ile158, Pro220] | None |
| Kaempferol-7-glucoside | 2 (4.52 Å) | 1 (4.52) [Lys28] | None | None |
| Maysin | 26 (4.89 Å) | 9 (4.26 Å) [Asp461, Ser655 (2), Val651 (2), Asp40 (2), Lys156 (2) | 7 (5.55 Å) [Asp461, Asp185, Ile158(3), Asp40, Lys156] | 2 [Leu41, Lys156] |
| Zafirlukast | 12 (5.05 Å) | 3 (3.83 Å) [Asn666, Asp667, Asp185] | 4 (5.96 Å) [Val664, Pro187, Phe137, Leu186] | None |
| SC-2WT SP | ||||
| 6-Hydroxycyanidin 3-rutinoside | 18 (4.60 Å) | 6 (4.08 Å) [Asp31 (3), Ala39, Asn10, Ser40] | 5 (5.45 Å) [Leu180, Tyr32, Phe9, Val32 (2)] | None |
| Catalposide | 15 (5.07 Å) | 4 (4.33 Å) [Asn10, Gly6, Asp31 (2)] | 3 (6.28 Å) [Val34, Phe9, Trp103] | None |
| Geraniin | 17 (4.67 Å) | 8 (4.21 Å) [Tyr12, Arg13, Ser66, Ala64, Asn21 (2), Glu7, Asn10] | 5 (5.48 Å) [Ala11 (3), Lys23, Val8] | 1 [Lys23] |
| Kaempferol-7-glucoside | 21 (4.62 Å) | 5 (3.88 Å) [Asn10 (3), Ser40, Ser38] | 7 (5.46 Å) [Tyr32, Phe9, Val34, Leu35, Phe5, Ala30, Val34] | None |
| Maysin | 13 (4.72 Å) | 4 (3.49 Å) [Asn10, Glu7 (2), Gly6] | 4 (5.96 Å) [Trp103, Phe41, Phe9, Pro4] | None |
| Zafirlukast | 16 (4.80 Å) | 1 (3.27 Å) [Phe9] | 6 (5.22 Å) [Val34 (4), Trp103, Leu2] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aribisala, J.O.; Aruwa, C.E.; Uthman, T.O.; Nurain, I.O.; Idowu, K.; Sabiu, S. Cheminformatics Bioprospection of Broad Spectrum Plant Secondary Metabolites Targeting the Spike Proteins of Omicron Variant and Wild-Type SARS-CoV-2. Metabolites 2022, 12, 982. https://doi.org/10.3390/metabo12100982
Aribisala JO, Aruwa CE, Uthman TO, Nurain IO, Idowu K, Sabiu S. Cheminformatics Bioprospection of Broad Spectrum Plant Secondary Metabolites Targeting the Spike Proteins of Omicron Variant and Wild-Type SARS-CoV-2. Metabolites. 2022; 12(10):982. https://doi.org/10.3390/metabo12100982
Chicago/Turabian StyleAribisala, Jamiu Olaseni, Christiana Eleojo Aruwa, Taofik Olatunde Uthman, Ismaila Olanrewaju Nurain, Kehinde Idowu, and Saheed Sabiu. 2022. "Cheminformatics Bioprospection of Broad Spectrum Plant Secondary Metabolites Targeting the Spike Proteins of Omicron Variant and Wild-Type SARS-CoV-2" Metabolites 12, no. 10: 982. https://doi.org/10.3390/metabo12100982