
Identification of a Variant in APOB Gene as a Major Cause of Hypobetalipoproteinemia in Lebanese Families

 , ,
, ,
Abstract
:1. Introduction
2. Results
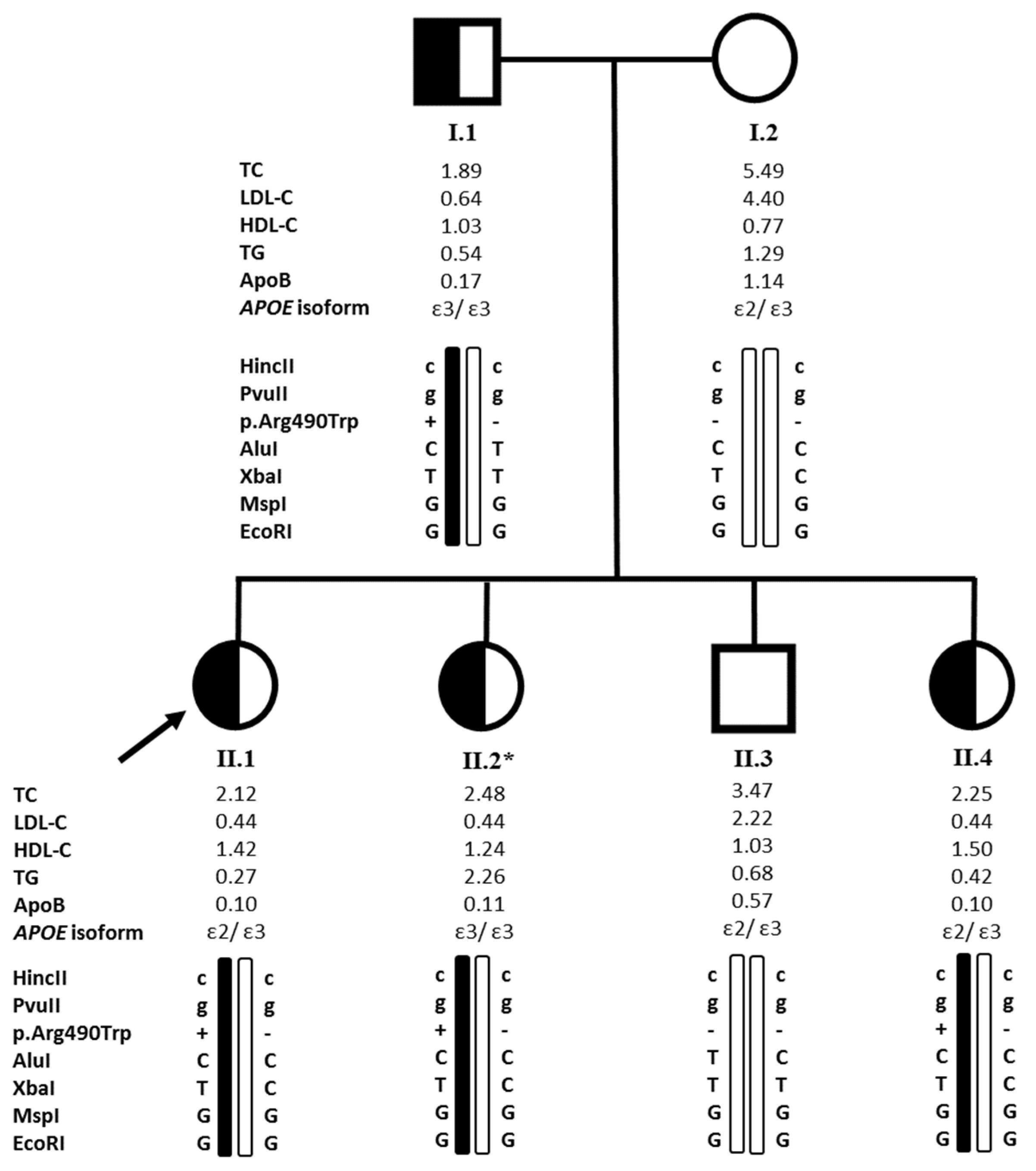
2.1. Clinical Characteristics and Biochemical Analysis of the Probands and Their Relatives
2.2. Genetic Analysis
2.3. The Founder p.(Arg490Trp) Variant
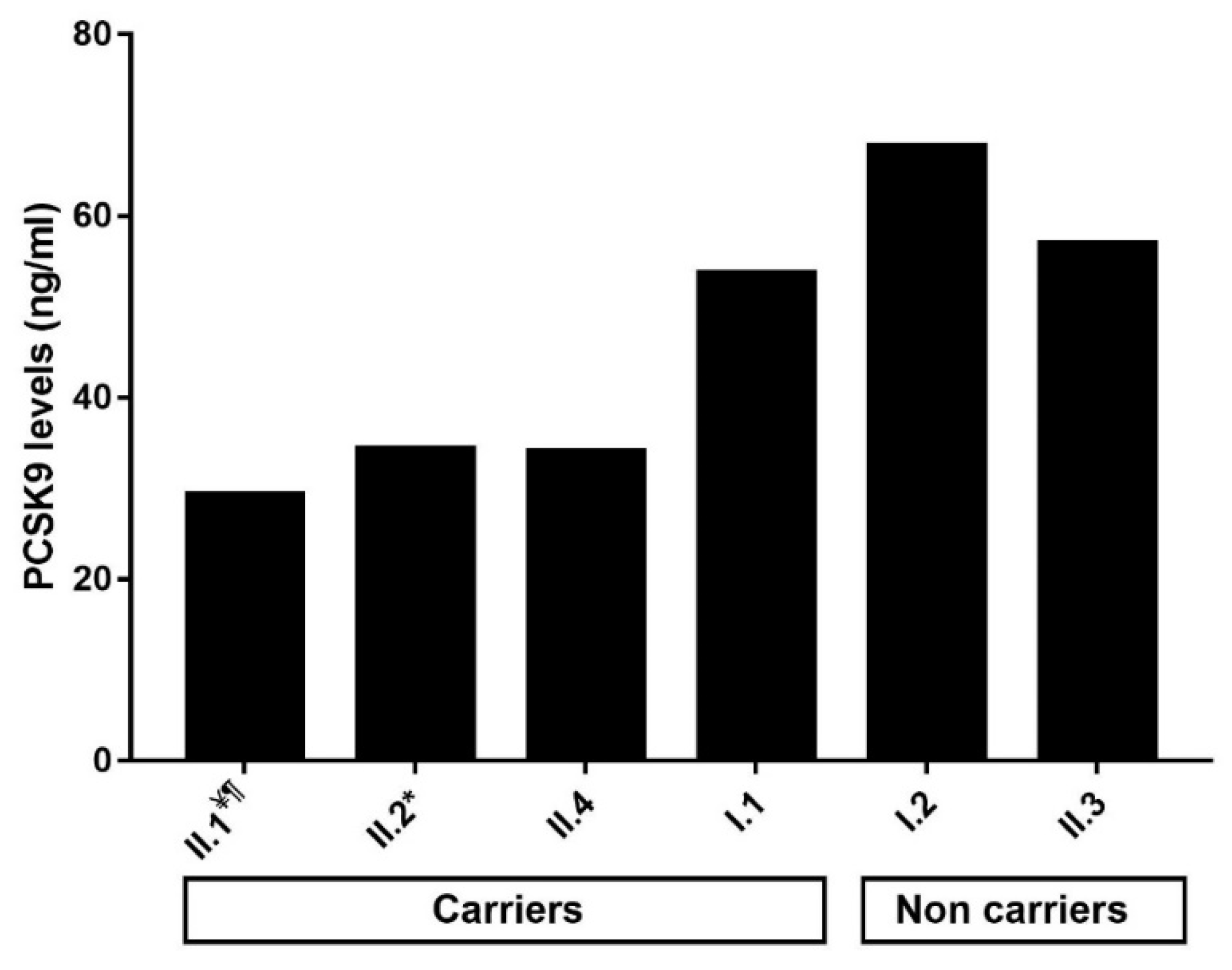
2.4. Impact of the p.(Arg490Trp) Mutation on Plasma PCSK9 Levels
3. Discussion
4. Materials and Methods
4.1. Study Participants
4.2. Laboratory and Biochemical Tests
4.3. DNA Analysis and Variant Detection
4.4. In Silico Analysis of the Variant
4.5. Haplotype Analysis
4.6. PCSK9 Measurements
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Di Costanzo, A.; Di Leo, E.; Noto, D.; Cefalù, A.B.; Minicocci, I.; Polito, L.; D’Erasmo, L.; Cantisani, V.; Spina, R.; Tarugi, P.; et al. Clinical and Biochemical Characteristics of Individuals with Low Cholesterol Syndromes: A Comparison between Familial Hypobetalipoproteinemia and Familial Combined Hypolipidemia. J. Clin. Lipidol. 2017, 11, 1234–1242. [Google Scholar] [CrossRef]
- Cariou, B.; Challet-Bouju, G.; Bernard, C.; Marrec, M.; Hardouin, J.-B.; Authier, C.; Bach-Ngohou, K.; Leux, C.; Pichelin, M.; Grall-Bronnec, M. Prevalence of Hypobetalipoproteinemia and Related Psychiatric Characteristics in a Psychiatric Population: Results from the Retrospective HYPOPSY Study. Lipids Health Dis. 2018, 17, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarugi, P.; Averna, M.; Di Leo, E.; Cefalù, A.B.; Noto, D.; Magnolo, L.; Cattin, L.; Bertolini, S.; Calandra, S. Molecular Diagnosis of Hypobetalipoproteinemia: An ENID Review. Atherosclerosis 2007, 195, e19–e27. [Google Scholar] [CrossRef] [PubMed]
- Hooper, A.J.; Burnett, J.R. Update on Primary Hypobetalipoproteinemia. Curr. Atheroscler. Rep. 2014, 16, 423. [Google Scholar] [CrossRef] [Green Version]
- Samson-Bouma, M.-E.; Berriot-Varoqueaux, N.; Aparicio, T.; Schmitz, J.; Aggerbeck, L. Hypocholestérolémies Génétiques: Abêtalipoprotéinémie, Hypobêtalipoprotéinémie Familiale, Maladie d’Anderson. EMC Gastro-Entérol. 2009, 4, 1–8. [Google Scholar] [CrossRef]
- Rimbert, A.; Pichelin, M.; Lecointe, S.; Marrec, M.; Le Scouarnec, S.; Barrak, E.; Croyal, M.; Krempf, M.; Le Marec, H.; Redon, R.; et al. Identification of Novel APOB Mutations by Targeted Next-Generation Sequencing for the Molecular Diagnosis of Familial Hypobetalipoproteinemia. Atherosclerosis 2016, 250, 52–56. [Google Scholar] [CrossRef] [Green Version]
- APOB Gene: MedlinePlus Genetics. Available online: https://medlineplus.gov/genetics/gene/apob/ (accessed on 30 March 2021).
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 Cause Autosomal Dominant Hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Ramasamy, I. Update on the Molecular Biology of Dyslipidemias. Clin. Chim. Acta 2016, 454, 143–185. [Google Scholar] [CrossRef]
- OMIM Entry—+107730—−APOLIPOPROTEIN B; APOB. Available online: https://www.omim.org/entry/107730 (accessed on 20 July 2018).
- Hooper, A.J.; van Bockxmeer, F.M.; Burnett, J.R. Monogenic Hypocholesterolaemic Lipid Disorders and Apolipoprotein B Metabolism. Crit. Rev. Clin. Lab. Sci. 2005, 42, 515–545. [Google Scholar] [CrossRef]
- Welty, F.K. Hypobetalipoproteinemia and Abetalipoproteinemia. Curr. Opin. Lipidol. 2014, 25, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonfeld, G.; Lin, X.; Yue, P. Familial Hypobetalipoproteinemia: Genetics and Metabolism. Cell. Mol. Life Sci. 2005, 62, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- Elbitar, S.; Susan-Resiga, D.; Ghaleb, Y.; El Khoury, P.; Peloso, G.; Stitziel, N.; Rabès, J.-P.; Carreau, V.; Hamelin, J.; Ben-Djoudi-Ouadda, A.; et al. New Sequencing Technologies Help Revealing Unexpected Mutations in Autosomal Dominant Hypercholesterolemia. Sci. Rep. 2018, 8, 1943. [Google Scholar] [CrossRef]
- Burnett, J.R.; Bell, D.A.; Hooper, A.J.; Hegele, R.A. Clinical Utility Gene Card for: Familial Hypobetalipoproteinaemia (APOB)—Update 2014. Eur. J. Hum. Genet. 2015, 23, 890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, J.R.; Shan, J.; Miskie, B.A.; Whitfield, A.J.; Yuan, J.; Tran, K.; McKnight, C.J.; Hegele, R.A.; Yao, Z. A Novel Nontruncating APOB Gene Mutation, R463W, Causes Familial Hypobetalipoproteinemia. J. Biol. Chem. 2003, 278, 13442–13452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, J.R.; Proos, A.L.; Koutts, J.; Burnett, L. Familial Hypobetalipoproteinaemia: A Rare Presentation to the Lipid Clinic. Med. J. Aust. 1993, 159, 272–274. [Google Scholar] [CrossRef] [PubMed]
- Burnett, J.R.; Hooper, A.J. Vitamin E and Oxidative Stress in Abetalipoproteinemia and Familial Hypobetalipoproteinemia. Free Radic. Biol. Med. 2015, 88, 59–62. [Google Scholar] [CrossRef]
- Heeks, L.V.; Hooper, A.J.; Adams, L.A.; Robbins, P.; Barrett, P.H.R.; van Bockxmeer, F.M.; Burnett, J.R. Non-Alcoholic Steatohepatitis-Related Cirrhosis in a Patient with APOB L343V Familial Hypobetalipoproteinaemia. Clin. Chim. Acta 2013, 421, 121–125. [Google Scholar] [CrossRef]
- Al Rasadi, K.; Almahmeed, W.; AlHabib, K.F.; Abifadel, M.; Farhan, H.A.; AlSifri, S.; Jambart, S.; Zubaid, M.; Awan, Z.; Al-Waili, K.; et al. Dyslipidaemia in the Middle East: Current Status and a Call for Action. Atherosclerosis 2016, 252, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Abifadel, M.; Rabès, J.-P.; Jambart, S.; Halaby, G.; Gannagé-Yared, M.-H.; Sarkis, A.; Beaino, G.; Varret, M.; Salem, N.; Corbani, S.; et al. The Molecular Basis of Familial Hypercholesterolemia in Lebanon: Spectrum of LDLR Mutations and Role of PCSK9 as a Modifier Gene. Hum. Mutat. 2009, 30, E682–E691. [Google Scholar] [CrossRef]
- Abifadel, M.; Jambart, S.; Allard, D.; Rabès, J.-P.; Varret, M.; Derré, A.; Chouery, E.; Salem, N.; Junien, C.; Aydénian, H.; et al. Identification of the First Lebanese Mutation in the LPL Gene and Description of a Rapid Detection Method. Clin. Genet. 2004, 65, 158–161. [Google Scholar] [CrossRef]
- El Khoury, P.; Couvert, P.; Elbitar, S.; Ghaleb, Y.; Abou-Khalil, Y.; Azar, Y.; Ayoub, C.; Superville, A.; Guérin, M.; Rabès, J.-P.; et al. Identification of the First Tangier Disease Patient in Lebanon Carrying a New Pathogenic Variant in ABCA1. J. Clin. Lipidol. 2018, 12, 1374–1382. [Google Scholar] [CrossRef]
- Abifadel, M.; Elbitar, S.; El Khoury, P.; Ghaleb, Y.; Chémaly, M.; Moussalli, M.-L.; Rabès, J.-P.; Varret, M.; Boileau, C. Living the PCSK9 Adventure: From the Identification of a New Gene in Familial Hypercholesterolemia towards a Potential New Class of Anticholesterol Drugs. Curr. Atheroscler. Rep. 2014, 16, 439. [Google Scholar] [CrossRef]
- Noto, D.; Cefalù, A.B.; Cannizzaro, A.; Minà, M.; Fayer, F.; Valenti, V.; Barbagallo, C.M.; Tuttolomondo, A.; Pinto, A.; Sciumè, C.; et al. Familial Hypobetalipoproteinemia due to Apolipoprotein B R463W Mutation Causes Intestinal Fat Accumulation and Low Postprandial Lipemia. Atherosclerosis 2009, 206, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Yue, P.; Isley, W.L.; Harris, W.S.; Rosipal, S.; Akin, C.D.; Schonfeld, G. Genetic Variants of ApoE Account for Variability of Plasma Low-Density Lipoprotein and Apolipoprotein B Levels in FHBL. Atherosclerosis 2005, 178, 107–113. [Google Scholar] [CrossRef]
- Ludwig, E.H.; McCarthy, B.J. Haplotype Analysis of the Human Apolipoprotein B Mutation Associated with Familial Defective Apolipoprotein B100. Am. J. Hum. Genet. 1990, 47, 712–720. [Google Scholar] [PubMed]
- Al-Bustan, S.A.; Alnaqeeb, M.A.; Annice, B.G.; Ebrahim, G.A.; Refai, T.M. Genetic Association of APOB Polymorphisms with Variation in Serum Lipid Profile among the Kuwait Population. Lipids Health Dis. 2014, 13, 157. [Google Scholar] [CrossRef] [Green Version]
- Dementieva, Y.; Green, T.L.; Primerano, D.A.; Wei, L.; Denvir, J.; Wehner, P.; Dodson, S.; Flood, M.R.; Pollock, B.A.; Huff, M.; et al. Identification of Genes Contributing to Cardiovascular Disease in Overweight and Obese Individuals from West Virginia. W. Va. Med. J. 2012, 108, 23–30. [Google Scholar]
- Fouchier, S.W.; Sankatsing, R.R.; Peter, J.; Castillo, S.; Pocovi, M.; Alonso, R.; Kastelein, J.J.P.; Defesche, J.C. High Frequency of APOB Gene Mutations Causing Familial Hypobetalipoproteinaemia in Patients of Dutch and Spanish Descent. J. Med. Genet. 2005, 42, e23. [Google Scholar] [CrossRef] [Green Version]
- Lam, M.C.W.; Singham, J.; Hegele, R.A.; Riazy, M.; Hiob, M.A.; Francis, G.; Steinbrecher, U.P. Familial Hypobetalipoproteinemia-Induced Nonalcoholic Steatohepatitis. Case Rep. Gastroenterol. 2012, 6, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Fasano, T.; Cefalù, A.B.; Di Leo, E.; Noto, D.; Pollaccia, D.; Bocchi, L.; Valenti, V.; Bonardi, R.; Guardamagna, O.; Averna, M.; et al. A Novel Loss of Function Mutation of PCSK9 Gene in White Subjects with Low-Plasma Low-Density Lipoprotein Cholesterol. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 677–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, S.; Magnolo, A.L.; Sundaram, M.; Zhou, H.; Yao, E.F.; Leo, E.D.; Loria, P.; Wang, S.; Bamji-Mirza, M.; Wang, L.; et al. Nonsynonymous Mutations within APOB in Human Familial Hypobetalipoproteinemia Evidence for feedback inhibition of lipogenesis and postendoplasmic reticulum degradation of apolipoprotein B. J. Biol. Chem. 2010, 285, 6453–6464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welty, F.K.; Lahoz, C.; Tucker, K.L.; Ordovas, J.M.; Wilson, P.W.F.; Schaefer, E.J. Frequency of ApoB and ApoE Gene Mutations as Causes of Hypobetalipoproteinemia in the Framingham Offspring Population. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1745–1751. [Google Scholar] [CrossRef] [Green Version]
- Fazio, S.; Minnier, J.; Shapiro, M.D.; Tsimikas, S.; Tarugi, P.; Averna, M.R.; Arca, M.; Tavori, H. Threshold Effects of Circulating Angiopoietin-Like 3 Levels on Plasma Lipoproteins. J. Clin. Endocrinol. Metab. 2017, 102, 3340–3348. [Google Scholar] [CrossRef]
- Noto, D.; Arca, M.; Tarugi, P.; Cefalù, A.B.; Barbagallo, C.M.; Averna, M.R. Association between Familial Hypobetalipoproteinemia and the Risk of Diabetes. Is This the Other Side of the Cholesterol-Diabetes Connection? A Systematic Review of Literature. Acta Diabetol. 2017, 54, 111–122. [Google Scholar] [CrossRef]
- Amaro, A.; Fabbrini, E.; Kars, M.; Yue, P.; Schechtman, K.; Schonfeld, G.; Klein, S. Dissociation between Intrahepatic Triglyceride Content and Insulin Resistance in Familial Hypobetalipoproteinemia. Gastroenterology 2010, 139, 149–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonfeld, G.; Yue, P.; Lin, X.; Chen, Z. Fatty Liver and Insulin Resistance: Not Always Linked. Trans. Am. Clin. Clim. Assoc. 2008, 119, 217–224. [Google Scholar]
- Tanoli, T.; Yue, P.; Yablonskiy, D.; Schonfeld, G. Fatty Liver in Familial Hypobetalipoproteinemia: Roles of the APOB Defects, Intra-Abdominal Adipose Tissue, and Insulin Sensitivity. J. Lipid Res. 2004, 45, 941–947. [Google Scholar] [CrossRef] [Green Version]
- Members. Available online: https://idf.org/our-network/regions-members/middle-east-and-north-africa/members/39-lebanon.html (accessed on 3 April 2021).
- Welty, F.K. Hypobetalipoproteinemia and Abetalipoproteinemia: Liver Disease and Cardiovascular Disease. Curr. Opin. Lipidol. 2020, 31, 49–55. [Google Scholar] [CrossRef]
- Welty, F.K.; Ordovas, J.; Schaefer, E.J.; Wilson, P.W.; Young, S. Identification and Molecular Analysis of Two ApoB Gene Mutations Causing Low Plasma Cholesterol Levels. Circulation 1995, 92, 2036–2040. [Google Scholar] [CrossRef]
- Musialik, J.; Boguszewska-Chachulska, A.; Pojda-Wilczek, D.; Gorzkowska, A.; Szymańczak, R.; Kania, M.; Kujawa-Szewieczek, A.; Wojcieszyn, M.; Hartleb, M.; Więcek, A. A Rare Mutation in The APOB Gene Associated with Neurological Manifestations in Familial Hypobetalipoproteinemia. Int. J. Mol. Sci. 2020, 21, 1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutzouri, E.; Elisaf, M.; Liberopoulos, E.N. Hypocholesterolemia. Curr. Vasc. Pharmacol. 2011, 9, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Xie, Y.-Z.; Cao, T.-T.; Wang, Z.; Wang, T.; Li, X.; Shen, R.-C.; Xu, H.; Bu, G.; Chen, X.-F. A Rapid and Cost-Effective Method for Genotyping Apolipoprotein E Gene Polymorphism. Mol. Neurodegener. 2016, 11, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Khoury, P.; Roussel, R.; Fumeron, F.; Abou-Khalil, Y.; Velho, G.; Mohammedi, K.; Jacob, M.-P.; Steg, P.G.; Potier, L.; Ghaleb, Y.; et al. Plasma Proprotein-Convertase-Subtilisin/Kexin Type 9 (PCSK9) and Cardiovascular Events in Type 2 Diabetes. Diabetes Obes. Metab. 2018, 20, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Barbour, B.; Salameh, P. Consanguinity in Lebanon: Prevalence, Distribution and Determinants. J. Biosoc. Sci. 2009, 41, 505–517. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Subject | Age | Gender | TC | HDL-C | TG | LDL-C § | LDL-C # | ApoB (g/L) | Plasma PCSK9 Levels (ng/mL) | Fatty Liver | Diabetes | Neurological Problem | HBL | APOE Isoforms | p.Leu21dup in PCSK9 | p.(Arg490Trp) in APOB |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (mmol/L) | ||||||||||||||||
| Family 1 | ||||||||||||||||
| I.1 | 61 | M | 1.89 | 1.03 | 0.54 | 0.64 | 0.17 | 53.70 | Yes | No | Parkinson’s disease | Yes | ε3/ε3 | No | Yes | |
| I.2 | 54 | F | 5.49 | 0.77 | 1.29 | 4.40 | 1.14 | 67.74 | No | No | No | ε2/ε3 | No | No | ||
| II.1 ¥ | 28 | F | 2.12 | 1.42 | 0.27 | 0.44 | 0.10 | 29.36 ¶ | Yes | No | Yes | ε2/ε3 | No | Yes | ||
| II.2 * | 25 | F | 2.48 | 1.24 | 2.26 | 0.44 | 0.11 | 34.41 | No | No | Yes | ε3/ε3 | No | Yes | ||
| II.3 | 21 | M | 3.47 | 1.03 | 0.68 | 2.22 | 0.57 | 57.02 | No | No | No | ε2/ε3 | No | No | ||
| II.4 | 16 | F | 2.25 | 1.50 | 0.42 | 0.44 | 0.10 | 34.01 | No | No | Yes | ε2/ε3 | No | Yes | ||
| Family 2 | ||||||||||||||||
| I.1 | 93 | M | 4.19 | 0.98 | 2.21 | 2.21 | N/A | N/A | Yes | No | N/A | N/A | N/A | |||
| I.2 | 73 | F | 1.84 | 0.93 | 0.47 | 0.70 | N/A | N/A | Yes | Yes | N/A | N/A | N/A | |||
| II.1 | 53 | M | 4.16 | 1.73 | 0.71 | 2.11 | N/A | N/A | Yes | No | N/A | N/A | N/A | |||
| II.2 | 52 | F | 2.59 | 1.14 | 1.69 | 0.68 | 0.45 | 68.35 | No | Yes | Yes | ε3/ε3 | Yes | Yes | ||
| II.3 ¥ | 47 | F | 2.72 | 0.88 | 0.75 | 1.50 | 0.40 | 34.16 | No | No | Yes | ε3/ε3 | No | Yes | ||
| Family 3 | ||||||||||||||||
| I.1 | 90 | M | No | Yes | Multiple System Atrophy | Yes | ε3/ε3 | No | Yes | |||||||
| II.1 ¥ | 60 | F | 2.69 | 1.08 | 0.93 | 1.08 | 0.41 | 68.98 | No | Yes | Yes | ε3/ε3 | No | Yes | ||
| Proband 4 | 49 | M | 1.60 | 0.88 | 0.59 | 0.45 | N/A | Yes | Yes | Yes | ε3/ε3 | No | Yes | |||
| Proband 5 | 63 | F | 2.38 | 0.96 | 1.41 | 0.78 | N/A | Yes | Yes | Neuropathies | Yes | ε3/ε3 | Yes | Yes | ||
| Proband 6 | 33 | F | 3.96 | 2.43 | 0.34 | 1.38 | N/A | N/A | No | Yes | ε3/ε3 | No | No | |||
| Proband 7 | 23 | F | 3.21 | 1.47 | 0.67 | 1.44 | 0.66 | 69.39 | N/A | No | Yes | ε3/ε3 | No | No | ||
| Mean ± SD p.(Arg490Trp)carriers(n = 9) | 2.30 ± 0.38 | 1.13 ± 0.22 | 0.83 ± 0.50 | 0.72 ± 0.36 | 0.25 ± 0.16 | 48.94 ± 17.05 | ||||||||||
| Haplotype of the APOB-p.(Arg490Trp) Carriers | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Haplotype Marker | Reference SNP [29,30] | Nucleotide Change (NM_000384.3) | Amino Acid Change (Original/Present Nomenclature NP_000375.3) | Prevalence in Non-Finnish European (GnomAD) | Proband (II.1) and Affected Members of Family 1 | Proband (II.3) and Member (II.2) of Family 2 | Proband (II.1) and Member (I.1) of Family 3 | Proband 4 | Proband 5 |
| HincII | Intron 4 | c | c | c | c | c | |||
| PvuII | Intron 4 | g | g | g | g | g | |||
| Heterozygous for p.(Arg490Trp) | rs771541567 | c.1468C>T | p.(Arg463Trp)/p.(Arg490Trp) | 0.000017 | T | T | T | T | T |
| AluI | rs679899 | c.1853C>T | p.(Ala591Val)/p.(Ala618Val) | 0.47 | C | C | C | C | C |
| XbaI | rs693 | c.7545C>T | p.(Thr2488=)/p.(Thr2515=) | 0.499 | T | T | T | T | T |
| MspI | rs1801701 | c.10913G>A | p.(Arg3611Gln)/p.(Arg3638Gln) | 0.091 | G | G | G | G | G |
| EcoRI | rs1042031 | c.12541G>A | p.(Glu4154Lys)/p.(Glu4181Lys) | 0.182 | G | G | G | G | G |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ayoub, C.; Azar, Y.; Abou-Khalil, Y.; Ghaleb, Y.; Elbitar, S.; Halaby, G.; Jambart, S.; Gannagé-Yared, M.-H.; Yaghi, C.; Saade Riachy, C.; et al. Identification of a Variant in APOB Gene as a Major Cause of Hypobetalipoproteinemia in Lebanese Families. Metabolites 2021, 11, 564. https://doi.org/10.3390/metabo11090564
Ayoub C, Azar Y, Abou-Khalil Y, Ghaleb Y, Elbitar S, Halaby G, Jambart S, Gannagé-Yared M-H, Yaghi C, Saade Riachy C, et al. Identification of a Variant in APOB Gene as a Major Cause of Hypobetalipoproteinemia in Lebanese Families. Metabolites. 2021; 11(9):564. https://doi.org/10.3390/metabo11090564
Chicago/Turabian StyleAyoub, Carine, Yara Azar, Yara Abou-Khalil, Youmna Ghaleb, Sandy Elbitar, Georges Halaby, Selim Jambart, Marie-Hélène Gannagé-Yared, Cesar Yaghi, Carole Saade Riachy, and et al. 2021. "Identification of a Variant in APOB Gene as a Major Cause of Hypobetalipoproteinemia in Lebanese Families" Metabolites 11, no. 9: 564. https://doi.org/10.3390/metabo11090564