
Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its Discovery to Clinical Trials and the Genomics Era


Abstract
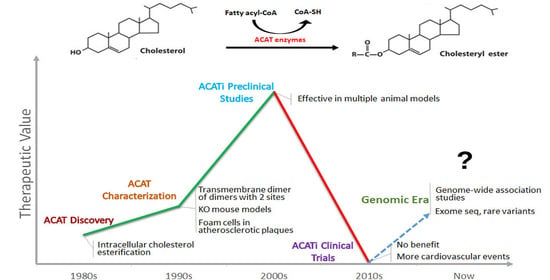
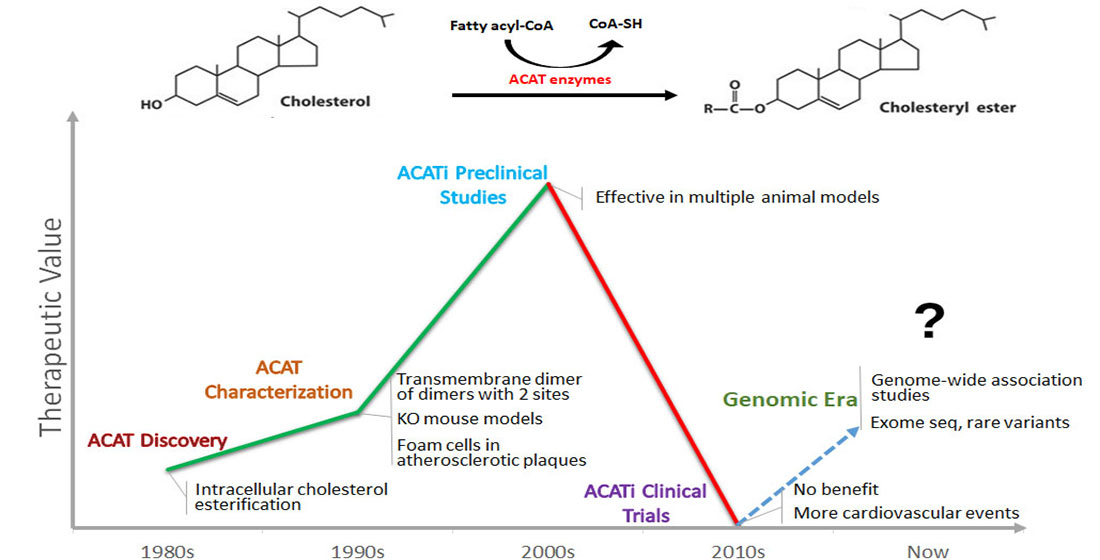
:
1. Introduction
2. The Structure of ACAT1
3. The Role of ACAT in Lipid Metabolism and Atherosclerosis Derived from Knockout Mouse Studies
4. ACAT Inhibitors in Pre-Clinical Studies
- Rabbit models
- Mouse models
- Adverse effects of ACAT inhibitors
5. Clinical Trials of ACAT Inhibitors
6. ACAT Genetics and Genomics
6.1. Mouse Soat1 Genetics
6.2. Human SOAT1/2 Genes and Expression
6.3. Human SOAT1/2 Common and Rare Genetic Variants and Disease Association
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chang, T.Y.; Li, B.L.; Chang, C.C.; Urano, Y. Acyl-coenzyme A:cholesterol acyltransferases. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1–E9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadigan, K.M.; Heider, J.G.; Chang, T.Y. Isolation and characterization of Chinese hamster ovary cell mutants deficient in acyl-coenzyme A:cholesterol acyltransferase activity. J. Biol. Chem. 1988, 263, 274–282. [Google Scholar] [CrossRef]
- Pavanello, C.; Calabresi, L. Genetic, biochemical, and clinical features of LCAT deficiency: Update for 2020. Curr. Opin. Lipidol. 2020, 31, 232–237. [Google Scholar] [CrossRef]
- Anderson, R.A.; Joyce, C.; Davis, M.; Reagan, J.W.; Clark, M.; Shelness, G.S.; Rudel, L.L. Identification of a form of acyl-CoA:cholesterol acyltransferase specific to liver and intestine in nonhuman primates. J. Biol. Chem. 1998, 273, 26747–26754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.Y.; Chang, C.C.; Lin, S.; Yu, C.; Li, B.L.; Miyazaki, A. Roles of acyl-coenzyme A:cholesterol acyltransferase-1 and -2. Curr. Opin. Lipidol. 2001, 12, 289–296. [Google Scholar] [CrossRef] [Green Version]
- Cases, S.; Novak, S.; Zheng, Y.W.; Myers, H.M.; Lear, S.R.; Sande, E.; Welch, C.B.; Lusis, A.J.; Spencer, T.A.; Krause, B.R.; et al. ACAT-2, a second mammalian acyl-CoA:cholesterol acyltransferase. Its cloning, expression, and characterization. J. Biol. Chem. 1998, 273, 26755–26764. [Google Scholar] [CrossRef] [Green Version]
- Cases, S.; Smith, S.J.; Zheng, Y.W.; Myers, H.M.; Lear, S.R.; Sande, E.; Novak, S.; Collins, C.; Welch, C.B.; Lusis, A.J.; et al. Identification of a gene encoding an acyl CoA:diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc. Natl. Acad. Sci. USA 1998, 95, 13018–13023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, K. A superfamily of membrane-bound O-acyltransferases with implications for wnt signaling. Trends Biochem. Sci. 2000, 25, 111–112. [Google Scholar] [CrossRef]
- Rogers, M.A.; Liu, J.; Song, B.L.; Li, B.L.; Chang, C.C.; Chang, T.Y. Acyl-CoA:cholesterol acyltransferases (ACATs/SOATs): Enzymes with multiple sterols as substrates and as activators. J. Steroid Biochem. Mol. Biol. 2015, 151, 102–107. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Dong, R.; Miyazaki, A.; Sakashita, N.; Zhang, Y.; Liu, J.; Guo, M.; Li, B.L.; Chang, T.Y. Human acyl-CoA:cholesterol acyltransferase (ACAT) and its potential as a target for pharmaceutical intervention against atherosclerosis. Acta Biochim. Biophys. Sin. 2006, 38, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Cadigan, K.M.; Chang, C.C.; Chang, T.Y. Isolation of Chinese hamster ovary cell lines expressing human acyl-coenzyme A/cholesterol acyltransferase activity. J. Cell Biol. 1989, 108, 2201–2210. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Huh, H.Y.; Cadigan, K.M.; Chang, T.Y. Molecular cloning and functional expression of human acyl-coenzyme A:cholesterol acyltransferase cDNA in mutant Chinese hamster ovary cells. J. Biol. Chem. 1993, 268, 20747–20755. [Google Scholar] [CrossRef]
- Chang, C.C.; Noll, W.W.; Nutile-McMenemy, N.; Lindsay, E.A.; Baldini, A.; Chang, W.; Chang, T.Y. Localization of acyl coenzyme A:cholesterol acyltransferase gene to human chromosome 1q25. Somat. Cell Mol. Genet. 1994, 20, 71–74. [Google Scholar] [CrossRef]
- Chang, C.C.; Lee, C.Y.; Chang, E.T.; Cruz, J.C.; Levesque, M.C.; Chang, T.Y. Recombinant acyl-CoA:cholesterol acyltransferase-1 (ACAT-1) purified to essential homogeneity utilizes cholesterol in mixed micelles or in vesicles in a highly cooperative manner. J. Biol. Chem. 1998, 273, 35132–35141. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yu, C.; Liu, J.; Spencer, T.A.; Chang, C.C.Y.; Chang, T.-Y. Cholesterol is superior to 7-ketocholesterol or 7 alpha-hydroxycholesterol as an allosteric activator for acyl-coenzyme A:cholesterol acyltransferase 1. J. Biol. Chem. 2003, 278, 11642–11647. [Google Scholar] [CrossRef] [Green Version]
- Rogers, M.A.; Liu, J.; Kushnir, M.M.; Bryleva, E.; Rockwood, A.L.; Meikle, A.W.; Shapiro, D.; Vaisman, B.L.; Remaley, A.T.; Chang, C.C.Y.; et al. Cellular pregnenolone esterification by acyl-CoA:cholesterol acyltransferase. J. Biol. Chem. 2012, 287, 17483–17492. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Miyazaki, A.; Dong, R.; Kheirollah, A.; Yu, C.; Geng, Y.; Higgs, H.N.; Chang, T.Y. Purification of recombinant acyl-coenzyme A:cholesterol acyltransferase 1 (ACAT1) from H293 cells and binding studies between the enzyme and substrates using difference intrinsic fluorescence spectroscopy. Biochemistry 2010, 49, 9957–9963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, T.; Oelkers, P.M.; Giattina, M.R.; Worgall, T.S.; Sturley, S.L.; Deckelbaum, R.J. Differential modulation of ACAT1 and ACAT2 transcription and activity by long chain free fatty acids in cultured cells. Biochemistry 2001, 40, 4756–4762. [Google Scholar] [CrossRef] [PubMed]
- Temel, R.E.; Gebre, A.K.; Parks, J.S.; Rudel, L.L. Compared with Acyl-CoA:cholesterol O-acyltransferase (ACAT) 1 and lecithin:cholesterol acyltransferase, ACAT2 displays the greatest capacity to differentiate cholesterol from sitosterol. J. Biol. Chem. 2003, 278, 47594–47601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, H.; Zhao, X.; Yan, R.; Yao, X.; Gao, S.; Sun, X.; Du, X.; Yang, H.; Wong, C.C.L.; Yan, N. Structural basis for catalysis and substrate specificity of human ACAT1. Nature 2020, 581, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Meiner, V.L.; Cases, S.; Myers, H.M.; Sande, E.R.; Bellosta, S.; Schambelan, M.; Pitas, R.E.; McGuire, J.; Herz, J.; Farese, R.V.J. Disruption of the acyl-CoA:cholesterol acyltransferase gene in mice: Evidence suggesting multiple cholesterol esterification enzymes in mammals. Proc. Natl. Acad. Sci. USA 1996, 93, 14041–14046. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.H.; Gui, J.; Artinger, E.; Craig, R.; Berwin, B.L.; Ernst, P.A.; Chang, C.C.; Chang, T.Y. Acat1 gene ablation in mice increases hematopoietic progenitor cell proliferation in bone marrow and causes leukocytosis. Arter. Thromb. Vasc. Biol. 2013, 33, 2081–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buhman, K.K.; Accad, M.; Novak, S.; Choi, R.S.; Wong, J.S.; Hamilton, R.L.; Turley, S.; Farese, R.V.J. Resistance to diet-induced hypercholesterolemia and gallstone formation in ACAT2-deficient mice. Nat. Med. 2000, 6, 1341–1347. [Google Scholar] [CrossRef]
- Repa, J.J.; Buhman, K.K.; Farese, R.V.J.; Dietschy, J.M.; Turley, S.D. ACAT2 deficiency limits cholesterol absorption in the cholesterol-fed mouse: Impact on hepatic cholesterol homeostasis. Hepatology 2004, 40, 1088–1097. [Google Scholar] [CrossRef] [PubMed]
- Accad, M.; Smith, S.J.; Newland, D.L.; Sanan, D.A.; King, L.E.J.; Linton, M.F.; Fazio, S.; Farese, R.V.J. Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1. J. Clin. Investig. 2000, 105, 711–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazio, S.; Major, A.S.; Swift, L.L.; Gleaves, L.A.; Accad, M.; Linton, M.F.; Farese, R.V.J. Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J. Clin. Investig. 2001, 107, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Yagyu, H.; Kitamine, T.; Osuga, J.I.; Tozawa, R.I.; Chen, Z.; Kaji, Y.; Oka, T.; Perrey, S.; Tamura, Y.; Ohashi, K.; et al. Absence of ACAT-1 attenuates atherosclerosis but ’causes dry eye and cutaneous xanthomatosis mice with congenital hyperlipidemia. J. Biol. Chem. 2000, 275, 21324–21330. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.H.; Melton, E.M.; Li, H.; Sohn, P.; Rogers, M.A.; Mulligan-Kehoe, M.J.; Fiering, S.N.; Hickey, W.F.; Chang, C.C.; Chang, T.Y. Myeloid Acyl-CoA:Cholesterol Acyltransferase 1 Deficiency Reduces Lesion Macrophage Content and Suppresses Atherosclerosis Progression. J. Biol. Chem. 2016, 291, 6232–6244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melton, E.M.; Li, H.; Benson, J.; Sohn, P.; Huang, L.-H.; Song, B.-L.; Li, B.-L.; Chang, C.C.Y.; Chang, T.-Y. Myeloid Acat1/Soat1 KO attenuates pro-inflammatory responses in macrophages and protects against atherosclerosis in a model of advanced lesions. J. Biol. Chem. 2019, 294, 15836–15849. [Google Scholar] [CrossRef]
- Willner, E.L.; Tow, B.; Buhman, K.K.; Wilson, M.; Sanan, D.A.; Rudel, L.L.; Farese, R.V.J. Deficiency of acyl CoA:cholesterol acyltransferase 2 prevents atherosclerosis in apolipoprotein E-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 1262–1267. [Google Scholar] [CrossRef] [Green Version]
- Kusunoki, J.; Aragane, K.; Kitamine, T.; Higashinakagawa, S.; Kase, N.; Yamaura, T.; Ohnishi, H. Hypocholesterolemic action and prevention of cholesterol absorption via the gut by F-1394, a potent acyl-CoA:cholesterol acyltransferase (ACAT) inhibitor, in cholesterol diet-fed rats. Jpn. J. Pharmacol. 1995, 69, 53–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiwata, T.; Aragane, K.; Fujinami, K.; Kojima, K.; Ishibashi, S.; Yamada, N.; Kusunoki, J. Direct effect of an acyl-CoA:cholesterol acyltransferase inhibitor, F-1394, on atherosclerosis in apolipoprotein E and low density lipoprotein receptor double knockout mice. Br. J. Pharmacol. 2001, 133, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Aragane, K.; Fujinami, K.; Kojima, K.; Kusunoki, J. ACAT inhibitor F-1394 prevents intimal hyperplasia induced by balloon injury in rabbits. J. Lipid Res. 2001, 42, 480–488. [Google Scholar] [CrossRef]
- Kusunoki, J.; Hansoty, D.K.; Aragane, K.; Fallon, J.T.; Badimon, J.J.; Fisher, E.A. Acyl-CoA:cholesterol acyltransferase inhibition reduces atherosclerosis in apolipoprotein E-deficient mice. Circulation 2001, 103, 2604–2609. [Google Scholar] [CrossRef] [Green Version]
- Rong, J.X.; Blachford, C.; Feig, J.E.; Bander, I.; Mayne, J.; Kusunoki, J.; Miller, C.; Davis, M.; Wilson, M.; Dehn, S.; et al. ACAT inhibition reduces the progression of preexisting, advanced atherosclerotic mouse lesions without plaque or systemic toxicity. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amengual, J.; Ogando, Y.; Nikain, C.; Quezada, A.; Qian, K.; Vaisar, T.; Fisher, E.A. Short-Term Acyl-CoA:Cholesterol Acyltransferase Inhibition, Combined with Apoprotein A1 Overexpression, Promotes Atherosclerosis Inflammation Resolution in Mice. Mol. Pharmacol. 2021, 99, 175–183. [Google Scholar] [CrossRef]
- Ramharack, R.; Spahr, M.A.; Sekerke, C.S.; Stanfield, R.L.; Bousley, R.F.; Lee, H.T.; Krause, B.K. CI-1011 lowers lipoprotein(a) and plasma cholesterol concentrations in chow-fed cynomolgus monkeys. Atherosclerosis 1998, 136, 79–87. [Google Scholar] [CrossRef]
- Burnett, J.R.; Wilcox, L.J.; Telford, D.E.; Kleinstiver, S.J.; Barrett, P.H.; Newton, R.S.; Huff, M.W. Inhibition of ACAT by avasimibe decreases both VLDL and LDL apolipoprotein B production in miniature pigs. J. Lipid Res. 1999, 40, 1317–1327. [Google Scholar] [CrossRef]
- Bocan, T.M.; Krause, B.R.; Rosebury, W.S.; Mueller, S.B.; Lu, X.; Dagle, C.; Major, T.; Lathia, C.; Lee, H. The ACAT inhibitor avasimibe reduces macrophages and matrix metalloproteinase expression in atherosclerotic lesions of hypercholesterolemic rabbits. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 70–79. [Google Scholar] [CrossRef] [Green Version]
- Robertson, D.G.; Breider, M.A.; Milad, M.A. Preclinical safety evaluation of avasimibe in beagle dogs: An ACAT inhibitor with minimal adrenal effects. Toxicol. Sci. 2001, 59, 324–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delsing, D.J.; Offerman, E.H.; van Duyvenvoorde, W.; van Der Boom, H.; de Wit, E.C.; Gijbels, M.J.; van Der Laarse, A.; Jukema, J.W.; Havekes, L.M.; Princen, H.M. Acyl-CoA:cholesterol acyltransferase inhibitor avasimibe reduces atherosclerosis in addition to its cholesterol-lowering effect in ApoE*3-Leiden mice. Circulation 2001, 103, 1778–1786. [Google Scholar] [CrossRef]
- Burnett, J.R.; Telford, D.E.; Barrett, P.H.R.; Huff, M.W. The ACAT inhibitor avasimibe increases the fractional clearance rate of postprandial triglyceride-rich lipoproteins in miniature pigs. Biochim. Biophys. Acta 2005, 1738, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Miyazaki, A.; Kasanuki, N.; Ito, K.; Ubukata, N.; Koieyama, T.; Kitayama, K.; Tanimoto, T.; Maeda, N.; Inaba, T. ACAT inhibitor pactimibe sulfate (CS-505) reduces and stabilizes atherosclerotic lesions by cholesterol-lowering and direct effects in apolipoprotein E-deficient mice. Atherosclerosis 2007, 190, 239–247. [Google Scholar] [CrossRef]
- Worthley, S.G.; Helft, G.; Corti, R.; Worthley, M.I.; Chew, D.P.; Fayad, Z.A.; Zaman, A.G.; Fallon, J.T.; Fuster, V.; Badimon, J.J. Statin therapy alone and in combination with an acyl-CoA:cholesterol O-acyltransferase inhibitor on experimental atherosclerosis. Pathophysiol. Haemost. Thromb. 2007, 36, 9–17. [Google Scholar] [CrossRef]
- Kitayama, K.; Koga, T.; Maeda, N.; Inaba, T.; Fujioka, T. Pactimibe stabilizes atherosclerotic plaque through macrophage acyl-CoA:cholesterol acyltransferase inhibition in WHHL rabbits. Eur. J. Pharmacol. 2006, 539, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, K.; Tanimoto, T.; Koga, T.; Terasaka, N.; Fujioka, T.; Inaba, T. Importance of acyl-coenzyme A:cholesterol acyltransferase 1/2 dual inhibition for anti-atherosclerotic potency of pactimibe. Eur. J. Pharmacol. 2006, 540, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, K.; Koga, T.; Inaba, T.; Fujioka, T. Multiple mechanisms of hypocholesterolemic action of pactimibe, a novel acyl-coenzyme A:cholesterol acyltransferase inhibitor. Eur. J. Pharmacol. 2006, 543, 123–132. [Google Scholar] [CrossRef]
- Yoshinaka, Y.; Shibata, H.; Kobayashi, H.; Kuriyama, H.; Shibuya, K.; Tanabe, S.; Watanabe, T.; Miyazaki, A. A selective ACAT-1 inhibitor, K-604, stimulates collagen production in cultured smooth muscle cells and alters plaque phenotype in apolipoprotein E-knockout mice. Atherosclerosis 2010, 213, 85–91. [Google Scholar] [CrossRef]
- Bocan, T.M.; Mueller, S.B.; Uhlendorf, P.D.; Newton, R.S.; Krause, B.R. Comparison of CI-976, an ACAT inhibitor, and selected lipid-lowering agents for antiatherosclerotic activity in iliac-femoral and thoracic aortic lesions. A biochemical, morphological, and morphometric evaluation. Arterioscler. Thromb. J. Vasc. Biol. 1991, 11, 1830–1843. [Google Scholar] [CrossRef] [Green Version]
- Krause, B.R.; Bousley, R.F.; Kieft, K.A.; Stanfield, R.L. Effect of the ACAT inhibitor CI-976 on plasma cholesterol concentrations and distribution in hamsters fed zero- and low-cholesterol diets. Clin. Biochem. 1992, 25, 371–377. [Google Scholar] [CrossRef]
- Krause, B.R.; Anderson, M.; Bisgaier, C.L.; Bocan, T.; Bousley, R.; DeHart, P.; Essenburg, A.; Hamelehle, K.; Homan, R.; Kieft, K. In vivo evidence that the lipid-regulating activity of the ACAT inhibitor CI-976 in rats is due to inhibition of both intestinal and liver ACAT. J. Lipid Res. 1993, 34, 279–294. [Google Scholar] [CrossRef]
- Bocan, T.M.; Mueller, S.B.; Uhlendorf, P.D.; Brown, E.Q.; Mazur, M.J.; Black, A.E. Inhibition of acyl-CoA cholesterol O-acyltransferase reduces the cholesteryl ester enrichment of atherosclerotic lesions in the Yucatan micropig. Atherosclerosis 1993, 99, 175–186. [Google Scholar] [CrossRef]
- Krause, B.R.; Pape, M.E.; Kieft, K.; Auerbach, B.; Bisgaier, C.L.; Homan, R.; Newton, R.S. ACAT inhibition decreases LDL cholesterol in rabbits fed a cholesterol-free diet. Marked changes in LDL cholesterol without changes in LDL receptor mRNA abundance. Arterioscler. Thromb. J. Vasc. Biol. 1994, 14, 598–604. [Google Scholar] [CrossRef] [Green Version]
- Bocan, T.M.; Mueller, S.B.; Brown, E.Q.; Lee, P.; Bocan, M.J.; Rea, T.; Pape, M.E. HMG-CoA reductase and ACAT inhibitors act synergistically to lower plasma cholesterol and limit atherosclerotic lesion development in the cholesterol-fed rabbit. Atherosclerosis 1998, 139, 21–30. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Liang, K.H. Acyl-coenzyme A:cholesterol acyltransferase inhibition ameliorates proteinuria, hyperlipidemia, lecithin-cholesterol acyltransferase, SRB-1, and low-denisty lipoprotein receptor deficiencies in nephrotic syndrome. Circulation 2004, 110, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Ikenoya, M.; Yoshinaka, Y.; Kobayashi, H.; Kawamine, K.; Shibuya, K.; Sato, F.; Sawanobori, K.; Watanabe, T.; Miyazaki, A. A selective ACAT-1 inhibitor, K-604, suppresses fatty streak lesions in fat-fed hamsters without affecting plasma cholesterol levels. Atherosclerosis 2007, 191, 290–297. [Google Scholar] [CrossRef]
- Krause, B.R.; Black, A.; Bousley, R.; Essenburg, A.; Cornicelli, J.; Holmes, A.; Homan, R.; Kieft, K.; Sekerke, C.; Shaw-Hes, M.K. Divergent pharmacologic activities of PD 132301-2 and CL 277,082, urea inhibitors of acyl-CoA:cholesterol acyltransferase. J. Pharmacol. Exp. Ther. 1993, 267, 734–743. [Google Scholar] [PubMed]
- Azuma, Y.; Date, K.; Ohno, K.; Matsushiro, S.; Nobuhara, Y.; Yamada, T. NTE-122, an acyl-coa:cholesterol acyltransferase inhibitor, prevents the progression of atherogenesis in cholesterol-fed rabbits. Jpn. J. Pharmacol. 2001, 86, 120–123. [Google Scholar] [CrossRef] [Green Version]
- Aragane, K.; Kojima, K.; Fujinami, K.; Kamei, J.; Kusunoki, J. Effect of F-1394, an acyl-CoA:cholesterol acyltransferase inhibitor, on atherosclerosis induced by high cholesterol diet in rabbits. Atherosclerosis 2001, 158, 139–145. [Google Scholar] [CrossRef]
- Dominick, M.A.; McGuire, E.J.; Reindel, J.F.; Bobrowski, W.F.; Bocan, T.M.; Gough, A.W. Subacute toxicity of a novel inhibitor of acyl-CoA: Cholesterol acyltransferase in beagle dogs. Fundam. Appl. Toxicol. 1993, 20, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Dominick, M.A.; Bobrowski, W.A.; MacDonald, J.R.; Gough, A.W. Morphogenesis of a zone-specific adrenocortical cytotoxicity in guinea pigs administered PD 132301-2, an inhibitor of acyl-CoA:cholesterol acyltransferase. Toxicol. Pathol. 1993, 21, 54–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vernetti, L.A.; MacDonald, J.R.; Wolfgang, G.H.; Dominick, M.A.; Pegg, D.G. ATP depletion is associated with cytotoxicity of a novel lipid regulator in guinea pig adrenocortical cells. Toxicol. Appl. Pharmacol. 1993, 118, 30–38. [Google Scholar] [CrossRef]
- Warner, G.J.; Stoudt, G.; Bamberger, M.; Johnson, W.J.; Rothblat, G.H. Cell toxicity induced by inhibition of acyl coenzyme A:cholesterol acyltransferase and accumulation of unesterified cholesterol. J. Biol. Chem. 1995, 270, 5772–5778. [Google Scholar] [CrossRef] [Green Version]
- Kellner-Weibel, G.; Jerome, W.G.; Small, D.M.; Warner, G.J.; Stoltenborg, J.K.; Kearney, M.A.; Corjay, M.H.; Phillips, M.C.; Rothblat, G.H. Effects of intracellular free cholesterol accumulation on macrophage viability: A model for foam cell death. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 423–431. [Google Scholar] [CrossRef] [Green Version]
- Kellner-Weibel, G.; Yancey, P.G.; Jerome, W.G.; Walser, T.; Mason, R.P.; Phillips, M.C.; Rothblat, G.H. Crystallization of free cholesterol in model macrophage foam cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1891–1898. [Google Scholar] [CrossRef]
- Yao, P.M.; Tabas, I. Free cholesterol loading of macrophages induces apoptosis involving the fas pathway. J. Biol. Chem. 2000, 275, 23807–23813. [Google Scholar] [CrossRef] [Green Version]
- Yao, P.M.; Tabas, I. Free cholesterol loading of macrophages is associated with widespread mitochondrial dysfunction and activation of the mitochondrial apoptosis pathway. J. Biol. Chem. 2001, 276, 42468–42476. [Google Scholar] [CrossRef] [Green Version]
- Devries-Seimon, T.; Li, Y.; Yao, P.M.; Stone, E.; Wang, Y.; Davis, R.J.; Flavell, R.; Tabas, I. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J. Cell Biol. 2005, 171, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.-C.; Grégoire, J.; L’Allier, P.L.; Anderson, T.J.; Bertrand, O.; Reeves, F.; Title, L.M.; Alfonso, F.; Schampaert, E.; Hassan, A.; et al. Effects of the acyl coenzyme A:cholesterol acyltransferase inhibitor avasimibe on human atherosclerotic lesions. Circulation 2004, 110, 3372–3377. [Google Scholar] [CrossRef] [Green Version]
- Nissen, S.E.; Tuzcu, E.M.; Brewer, H.B.; Sipahi, I.; Nicholls, S.J.; Ganz, P.; Schoenhagen, P.; Waters, D.D.; Pepine, C.J.; Crowe, T.D.; et al. Effect of ACAT inhibition on the progression of coronary atherosclerosis. N. Engl. J. Med. 2006, 354, 1253–1263. [Google Scholar] [CrossRef] [PubMed]
- Meuwese, M.C.; de Groot, E.; Duivenvoorden, R.; Trip, M.D.; Ose, L.; Maritz, F.J.; Basart, D.C.G.; Kastelein, J.J.P.; Habib, R.; Davidson, M.H.; et al. ACAT inhibition and progression of carotid atherosclerosis in patients with familial hypercholesterolemia: The CAPTIVATE randomized trial. JAMA 2009, 301, 1131–1139. [Google Scholar] [CrossRef]
- Farese, R.V.J. The nine lives of ACAT inhibitors. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1684–1686. [Google Scholar] [CrossRef] [Green Version]
- Sahi, J.; Milad, M.A.; Zheng, X.; Rose, K.A.; Wang, H.; Stilgenbauer, L.; Gilbert, D.; Jolley, S.; Stern, R.H.; LeCluyse, E.L. Avasimibe induces CYP3A4 and multiple drug resistance protein 1 gene expression through activation of the pregnane X receptor. J. Pharmacol. Exp. Ther. 2003, 306, 1027–1034. [Google Scholar] [CrossRef]
- Sahi, J.; Stern, R.H.; Milad, M.A.; Rose, K.A.; Gibson, G.; Zheng, X.; Stilgenbauer, L.; Sadagopan, N.; Jolley, S.; Gilbert, D.; et al. Effects of avasimibe on cytochrome P450 2C9 expression in vitro and in vivo. Drug Metab. Dispos. 2004, 32, 1370–1376. [Google Scholar] [CrossRef] [Green Version]
- ARNESEN, K. Constitutional difference in lipid content of adrenals in two strains of mice and their hybrids. Acta Endocrinol. 1955, 18, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Meiner, V.L.; Welch, C.L.; Cases, S.; Myers, H.M.; Sande, E.; Lusis, A.J.; Farese, R.V.J. Adrenocortical lipid depletion gene (ald) in AKR mice is associated with an acyl-CoA:cholesterol acyltransferase (ACAT) mutation. J. Biol. Chem. 1998, 273, 1064–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hai, Q.; Ritchey, B.; Robinet, P.; Alzayed, A.M.; Brubaker, G.; Zhang, J.; Smith, J.D. Quantitative Trait Locus Mapping of Macrophage Cholesterol Metabolism and CRISPR/Cas9 Editing Implicate an ACAT1 Truncation as a Causal Modifier Variant. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Opoku, E.; Traughber, C.A.; Hai, Q.; Robinet, P.; Berisha, S.; Smith, J.D. Soat1 mediates the mouse strain effects on cholesterol loading-induced endoplasmic reticulum stress and CHOP expression in macrophages. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158825. [Google Scholar] [CrossRef] [PubMed]
- Li, B.L.; Li, X.L.; Duan, Z.J.; Lee, O.; Lin, S.; Ma, Z.M.; Chang, C.C.; Yang, X.Y.; Park, J.P.; Mohandas, T.K.; et al. Human acyl-CoA:cholesterol acyltransferase-1 (ACAT-1) gene organization and evidence that the 4.3-kilobase ACAT-1 mRNA is produced from two different chromosomes. J. Biol. Chem. 1999, 274, 11060–11071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, G.-J.; Chen, J.; Zhao, X.-N.; Xu, J.-J.; Guo, D.-Q.; Lu, M.; Zhu, M.; Xiong, Y.; Li, Q.; Chang, C.C.; et al. Production of ACAT1 56-kDa isoform in human cells via trans-splicing involving the ampicillin resistance gene. Cell Res. 2013, 23, 1007–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, Q.; Li, C.; Zuo, Z.; Huang, C.; Cheng, H.; Zhou, R. Evolutionary Insights into RNA trans-Splicing in Vertebrates. Genome Biol. Evol. 2016, 8, 562–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Yuan, C.; Chen, L.; Lei, M.; Zellmer, L.; Huang, H.; Liao, D.J. Transcriptional-Readthrough RNAs Reflect the Phenomenon of “A Gene Contains Gene(s)” or “Gene(s) within a Gene” in the Human Genome, and Thus Are Not Chimeric RNAs. Genes 2018, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannidis, J.P.; Ntzani, E.E.; Trikalinos, T.A.; Contopoulos-Ioannidis, D.G. Replication validity of genetic association studies. Nat. Genet. 2001, 29, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Takata, K.; Katsuren, K.; Fukuyama, S. The influence of the acyl-CoA:cholesterol acyltransferase-1 gene (-77G-->A) polymorphisms on plasma lipid and apolipoprotein levels in normolipidemic and hyperlipidemic subjects. Biochim. Biophys. Acta 2004, 1682, 56–62. [Google Scholar] [CrossRef]
- Chen, S.N.; Cilingiroglu, M.; Todd, J.; Lombardi, R.; Willerson, J.T.; Gotto, A.M.J.; Ballantyne, C.M.; Marian, A.J. Candidate genetic analysis of plasma high-density lipoprotein-cholesterol and severity of coronary atherosclerosis. BMC Med. Genet. 2009, 10, 111. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.-F.; Yin, R.-X.; Aung, L.H.H.; Hu, X.-J.; Cao, X.-L.; Miao, L.; Li, Q.; Yan, T.-T.; Wu, J.-Z.; Pan, S.-L. Polymorphism of rs1044925 in the acyl-CoA:cholesterol acyltransferase-1 gene and serum lipid levels in the Guangxi Bai Ku Yao and Han populations. Lipids Health Dis. 2010, 9, 139. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.-F.; Yin, R.-X.; Aung, L.H.H.; Li, Q.; Yan, T.-T.; Zeng, X.-N.; Huang, K.-K.; Huang, P.; Wu, J.-Z.; Pan, S.-L. Sex-specific association of ACAT-1 rs1044925 SNP and serum lipid levels in the hypercholesterolemic subjects. Lipids Health Dis. 2012, 11, 9. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.-F.; Yin, R.-X.; Cao, X.-L.; Chen, W.-X. Association between single nucleotide polymorphism rs1044925 and the risk of coronary artery disease and ischemic stroke. Int. J. Mol. Sci. 2014, 15, 3546–3559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.-T.; Wang, Y.-H.; Ma, Y.-T.; Fu, Z.-Y.; Yang, Y.-N.; Ma, X.; Li, X.-M.; Adi, D.; Liu, F.; Chen, B.-D. ACAT-1 gene polymorphism is associated with increased susceptibility to coronary artery disease in Chinese Han population: A case-control study. Oncotarget 2017, 8, 89055–89063. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-T.; Maitusong, B.; Ma, Y.-T.; Fu, Z.-Y.; Yang, Y.-N.; Ma, X.; Li, X.-M.; Liu, F.; Chen, B.-D. Acyl-CoA: Cholesterol acyltransferases-2 gene polymorphism is associated with increased susceptibility to coronary artery disease in Uygur population in Xinjiang, China. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, T.B.; Padovani, C.R.; de Oliveira Junior, L.R.; Latini, A.C.P.; Kurokawa, C.S.; Pereira, P.C.M.; Dos Santos, R.M. ACAT-1 gene rs1044925 SNP and its relation with different clinical forms of chronic Chagas disease. Parasitol. Res. 2019, 118, 2343–2351. [Google Scholar] [CrossRef]
- Huang, L.-H.; Nishi, K.; Li, S.; Ho, T.; Dong, R.; Chang, C.C.Y.; Chang, T.-Y. Acyl-coenzyme A:cholesterol acyltransferase 1—significance of single-nucleotide polymorphism at residue 526 and the role of Pro347 near the fifth transmembrane domain. FEBS J. 2014, 281, 1773–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klos, K.L.E.; Sing, C.F.; Boerwinkle, E.; Hamon, S.C.; Rea, T.J.; Clark, A.; Fornage, M.; Hixson, J.E. Consistent effects of genes involved in reverse cholesterol transport on plasma lipid and apolipoprotein levels in CARDIA participants. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1828–1836. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, A.C.; Braund, P.S.; Stylianou, I.M.; Khera, A.V.; Nelson, C.P.; Wolfe, M.L.; Derohannessian, S.L.; Keating, B.J.; Qu, L.; He, J.; et al. Dense genotyping of candidate gene loci identifies variants associated with high-density lipoprotein cholesterol. Circ. Cardiovasc. Genet. 2011, 4, 145–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollmer, M.A.; Streffer, J.R.; Tsolaki, M.; Grimaldi, L.M.E.; Lütjohann, D.; Thal, D.; von Bergmann, K.; Nitsch, R.M.; Hock, C.; Papassotiropoulos, A. Genetic association of acyl-coenzyme A: Cholesterol acyltransferase with cerebrospinal fluid cholesterol levels, brain amyloid load, and risk for Alzheimer’s disease. Mol. Psychiatry 2003, 8, 635–638. [Google Scholar] [CrossRef] [Green Version]
- Papassotiropoulos, A.; Wollmer, M.A.; Tsolaki, M.; Brunner, F.; Molyva, D.; Lütjohann, D.; Nitsch, R.M.; Hock, C. A cluster of cholesterol-related genes confers susceptibility for Alzheimer’s disease. J. Clin. Psychiatry 2005, 66, 940–947. [Google Scholar] [CrossRef]
- Huttunen, H.J.; Greco, C.; Kovacs, D.M. Knockdown of ACAT-1 reduces amyloidogenic processing of APP. FEBS Lett. 2007, 581, 1688–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharyya, R.; Kovacs, D.M. ACAT inhibition and amyloid beta reduction. Biochim. Biophys. Acta 2010, 1801, 960–965. [Google Scholar] [CrossRef] [Green Version]
- Murphy, S.R.; Chang, C.C.; Dogbevia, G.; Bryleva, E.Y.; Bowen, Z.; Hasan, M.T.; Chang, T.-Y. Acat1 knockdown gene therapy decreases amyloid-β in a mouse model of Alzheimer’s disease. Mol. Ther. 2013, 21, 1497–1506. [Google Scholar] [CrossRef]
- Bryleva, E.Y.; Rogers, M.A.; Chang, C.C.Y.; Buen, F.; Harris, B.T.; Rousselet, E.; Seidah, N.G.; Oddo, S.; LaFerla, F.M.; Spencer, T.A.; et al. ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc. Natl. Acad. Sci. USA 2010, 107, 3081–3086. [Google Scholar] [CrossRef] [Green Version]
- Naelitz, B.D.; Sharifi, N. Through the Looking-Glass: Reevaluating DHEA Metabolism Through HSD3B1 Genetics. Trends Endocrinol. Metab. 2020, 31, 680–690. [Google Scholar] [CrossRef]
- Jansen, P.R.; Watanabe, K.; Stringer, S.; Skene, N.; Bryois, J.; Hammerschlag, A.R.; de Leeuw, C.A.; Benjamins, J.S.; Muñoz-Manchado, A.B.; Nagel, M.; et al. Genome-wide analysis of insomnia in 1,331,010 individuals identifies new risk loci and functional pathways. Nat. Genet. 2019, 51, 394–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruth, K.S.; Day, F.R.; Tyrrell, J.; Thompson, D.J.; Wood, A.R.; Mahajan, A.; Beaumont, R.N.; Wittemans, L.; Martin, S.; Busch, A.S.; et al. Using human genetics to understand the disease impacts of testosterone in men and women. Nat. Med. 2020, 26, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Vujkovic, M.; Keaton, J.M.; Lynch, J.A.; Miller, D.R.; Zhou, J.; Tcheandjieu, C.; Huffman, J.E.; Assimes, T.L.; Lorenz, K.; Zhu, X.; et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat. Genet. 2020, 52, 680–691. [Google Scholar] [CrossRef]
- Zhao, B.; Zhang, J.; Ibrahim, J.G.; Luo, T.; Santelli, R.C.; Li, Y.; Li, T.; Shan, Y.; Zhu, Z.; Zhou, F.; et al. Large-scale GWAS reveals genetic architecture of brain white matter microstructure and genetic overlap with cognitive and mental health traits (n = 17,706). Mol. Psychiatry 2019, 1–13. [Google Scholar] [CrossRef]
- Yan, Q.; Ding, Y.; Liu, Y.; Sun, T.; Fritsche, L.G.; Clemons, T.; Ratnapriya, R.; Klein, M.L.; Cook, R.J.; Liu, Y.; et al. Genome-wide analysis of disease progression in age-related macular degeneration. Hum. Mol. Genet. 2018, 27, 929–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Name | Animal | Plasma Cholesterol | Atherosclerotic Lesion Effect | Other Effects | Reference |
|---|---|---|---|---|---|
| F-1394 | Rats | ↓ | N.D. | ↓ cholesterol absorption | [31] |
| Apoe−/− ldlr−/− mice | N.S. | ↓ | [32] | ||
| Rabbits (balloon injury) | N.S. | N.D. | ↓ neointimal thickening | [33] | |
| Apoe−/− mice | ↓ | ↓ | [34] | ||
| Apoe−/− mice | N.S. | ↓ progression | ↓ lesion tissue factor | [35] | |
| Apoe−/− mice | N.S. | N.S. | [36] | ||
| Apoe−/− and human APO A1-transgenic mice | N.S. | N.S. | [36] | ||
| CI-1011 (avasimibe) | Male monkeys | ↓ | N.D. | ↓ Lp(a) | [37] |
| Miniature pigs | ↓ | N.D. | [38] | ||
| Male rabbits | N.S. | ↓ progression | ↓ lesion MMP expression | [39] | |
| Beagle dogs | N.D. | N.D. | ↑ Emesis, saliva, hepatic toxicity, | [40] | |
| ApoE *3-Leiden mice | ↓ | ↓ | [41] | ||
| Miniature pigs | ↓ | N.D. | [42] | ||
| Apoe−/− mice | ↓ | ↓ progression | [43] | ||
| Rabbits | N.D. | ↓ progression | [44] | ||
| CS-505 (pactimibe sulfate) | Apoe−/− mice | ↓ | ↓ progression | ↓ MMP expression | [43] |
| Rabbits | N.S. | N.S. | ↑ lesion fibers and smooth muscle cells | [45] | |
| Hamsters | ↓ | ↓ | [46] | ||
| Hamsters | ↓ | N.D. | ↓ hepatic lipids; ↓ lipid absorption; ↑ fecal lipid excretion | [47] | |
| Monkeys | ↓ | N.D. | [47] | ||
| Diabetic rats | N.D. | N.D. | ↓ postprandial fat loading | [47] | |
| Apoe−/− mice | ↓ | ↓ | [48] | ||
| CI-976 | Rabbits | N.S. | ↓ progression | [49] | |
| Hamsters | ↓ | N.D. | [50] | ||
| Rats | ↓ | N.D. | ↓ liver CE | [51] | |
| Micropigs | N.S. | N.D. | ↓ liver CE | [52] | |
| Endogenous hypercholesterolemia rabbits | ↓ | N.D. | [53] | ||
| Rabbits | ↓ | ↓ | [54] | ||
| Nephrotic syndrome (NS) rats | ↓ | N.D. | restore LDL and HDL receptors | [55] | |
| K-604 (ACAT-1 selective) | Hamsters | ↓ (at high dose) | ↓ | [56] | |
| Apoe−/− mice | ↓ (at high dose) | N.S. | ↑ lesion collagen | [48] | |
| PD 132301-2 | Rats | ↓ | N.D. | [57] | |
| Guinea pigs | ↓ | N.D. | [57] | ||
| Rabbits | ↓ | N.D. | [57] | ||
| CL 277,082 | Rats | ↓ | N.D. | [57] | |
| Guinea pigs | ↓ (at high dose) | N.D. | [57] | ||
| Rabbits | ↓ | N.D. | [57] | ||
| Rats | ↓ | N.D. | [51] |
| Gene | Variant | p-Value | Reported Trait | Reference |
|---|---|---|---|---|
| SOAT1 | rs13306728 | 6 × 10−14 | Morningness | [102] |
| rs2152318 | 2 × 10−27 | Bioavailable testosterone levels | [103] | |
| rs2248979 | 2 × 10−12 | Bioavailable testosterone levels | [103] | |
| rs569421885 | 1 × 10−62 | Total testosterone levels | [103] | |
| rs569421885 | 3 × 10−37 | Total testosterone levels | [103] | |
| rs2816177 | 7 × 10−9 | Type 2 diabetes | [104] | |
| rs67563284 | 2 × 10−8 | White matter microstructure | [105] | |
| SOAT2 | rs11170417 | 7 × 10−6 | Age-related macular degeneration progression | [106] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hai, Q.; Smith, J.D. Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its Discovery to Clinical Trials and the Genomics Era. Metabolites 2021, 11, 543. https://doi.org/10.3390/metabo11080543
Hai Q, Smith JD. Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its Discovery to Clinical Trials and the Genomics Era. Metabolites. 2021; 11(8):543. https://doi.org/10.3390/metabo11080543
Chicago/Turabian StyleHai, Qimin, and Jonathan D. Smith. 2021. "Acyl-Coenzyme A: Cholesterol Acyltransferase (ACAT) in Cholesterol Metabolism: From Its Discovery to Clinical Trials and the Genomics Era" Metabolites 11, no. 8: 543. https://doi.org/10.3390/metabo11080543