
Effects of Hyperhomocysteinemia on the Platelet-Driven Contraction of Blood Clots

 , and
, and
Abstract
:
1. Introduction
2. Results
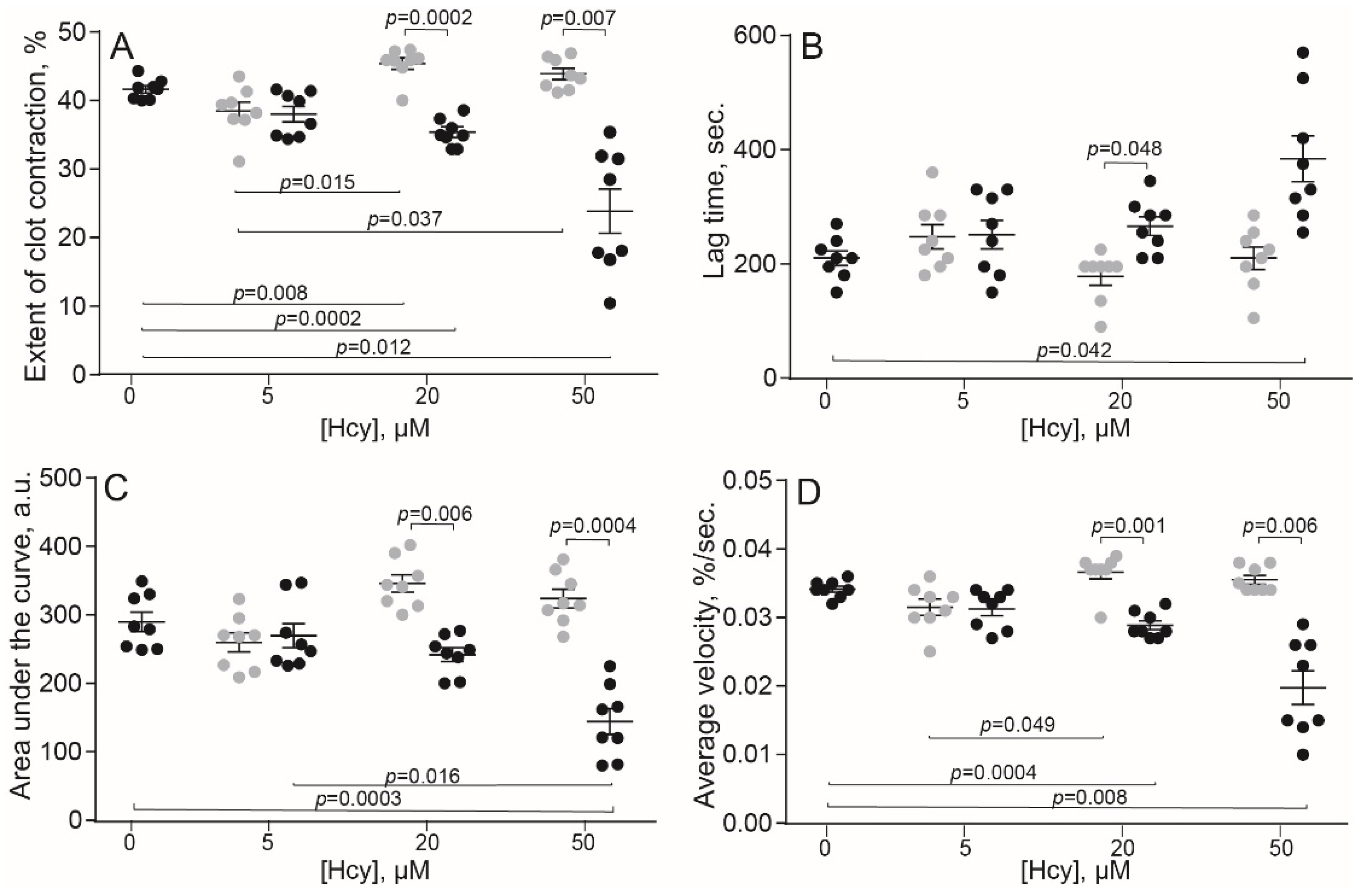
2.1. Hcy Modulates Clot Contraction In Vitro via Affecting Platelet Functionality
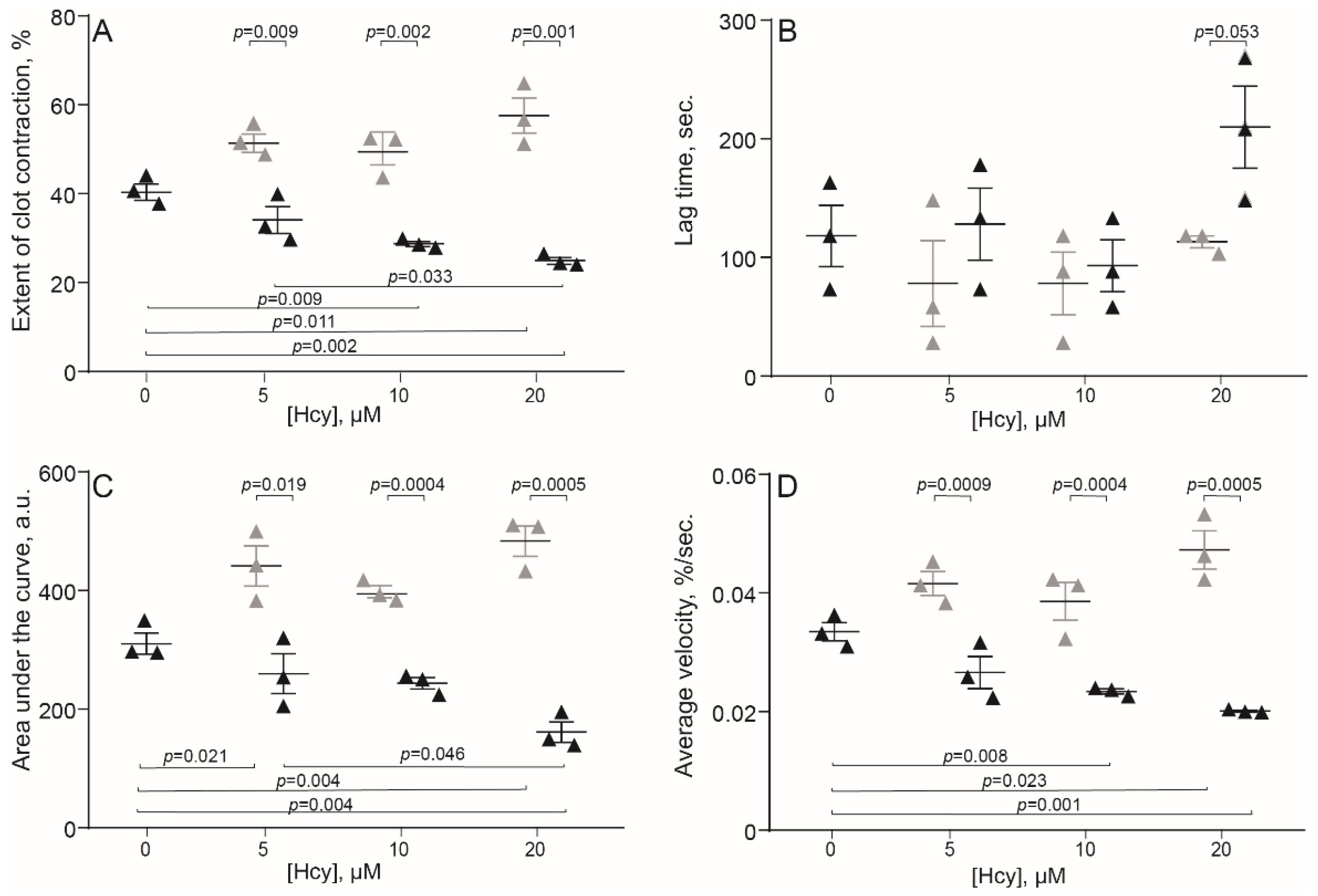
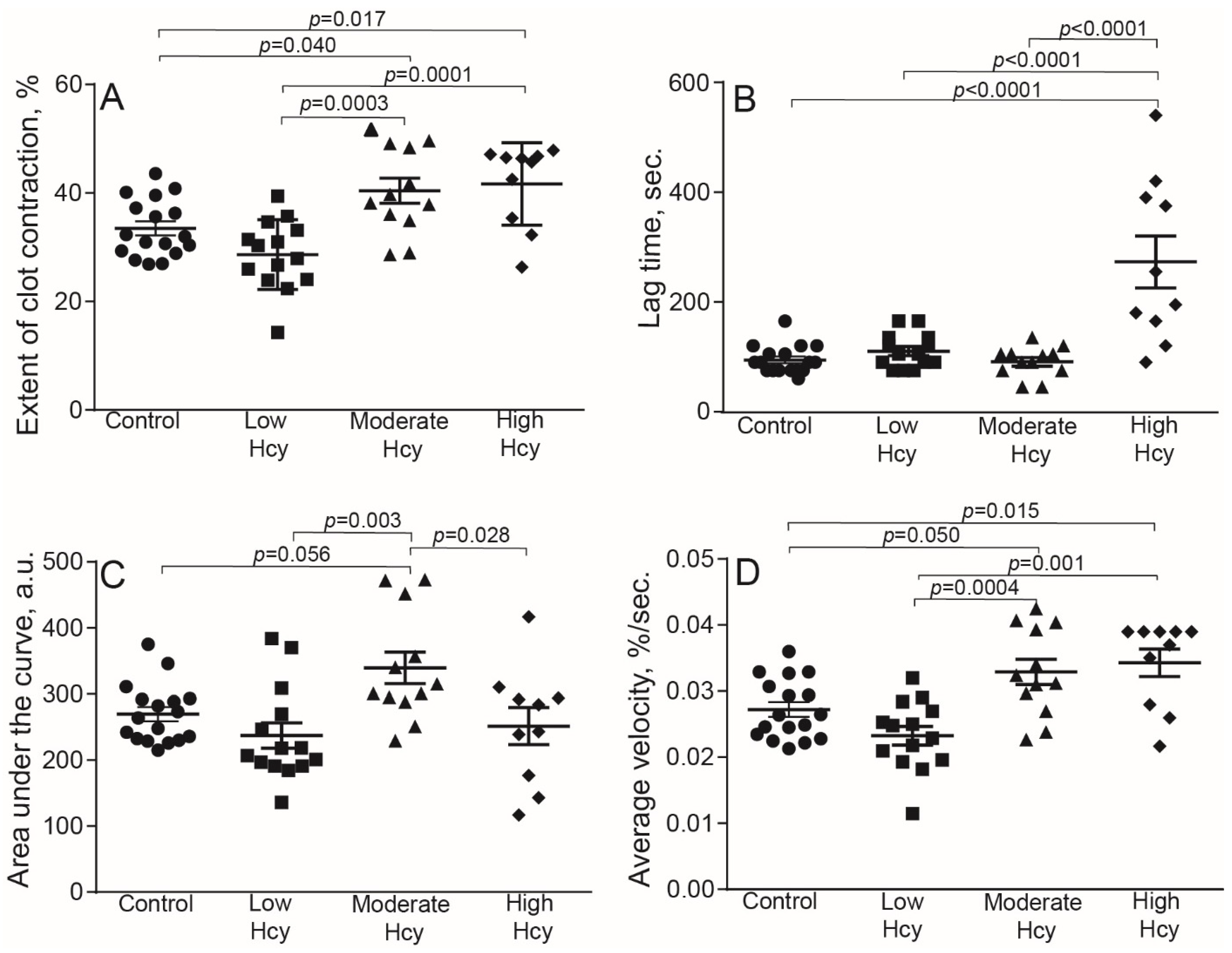
2.2. HHcy Modulates Clot Contraction In Vivo
2.3. HHcy-Induced Changes in the Blood Composition That Can Affect Clot Contraction
3. Discussion
4. Materials and Methods
4.1. Human Blood Samples
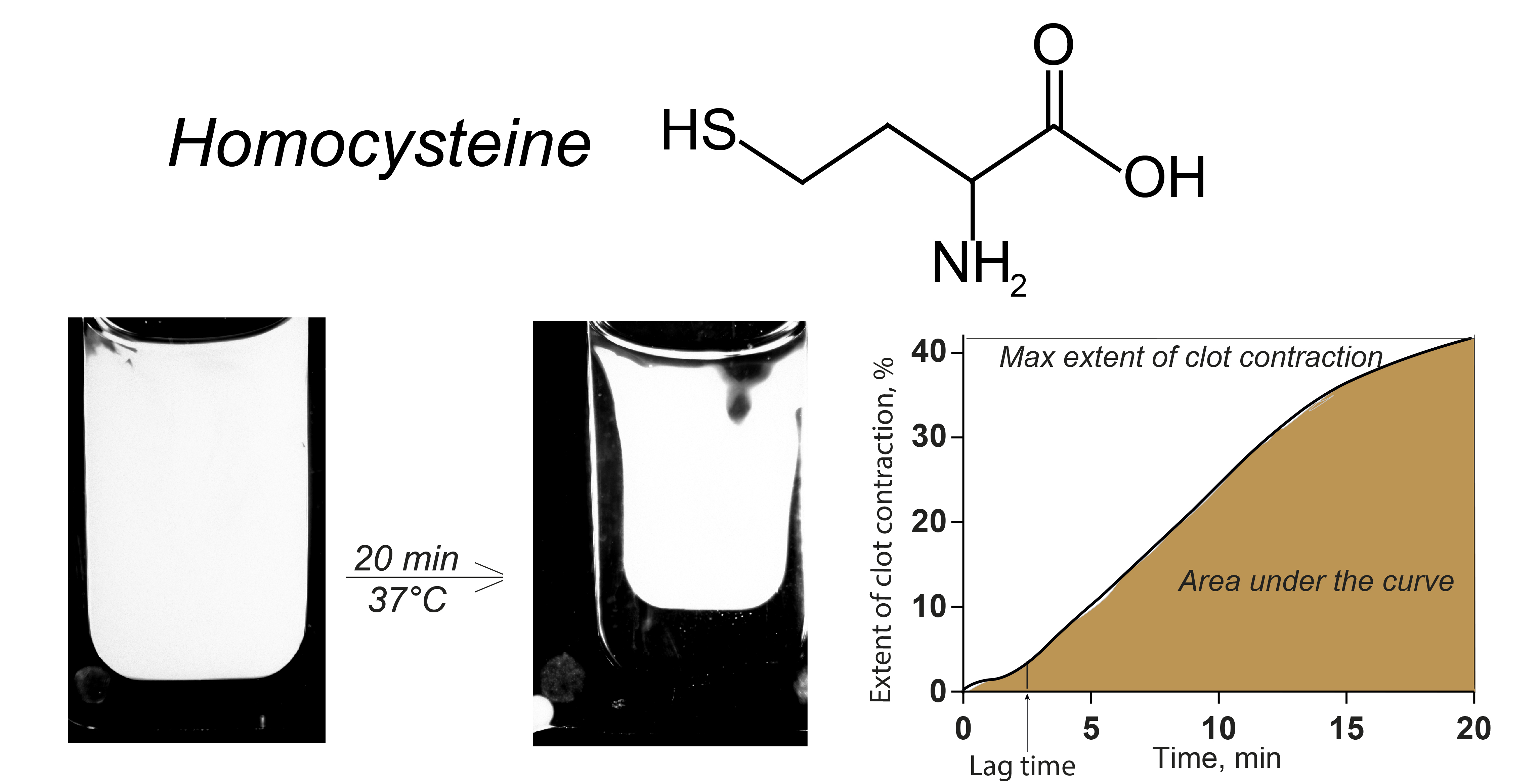
4.2. Blood Clot Contraction Assay
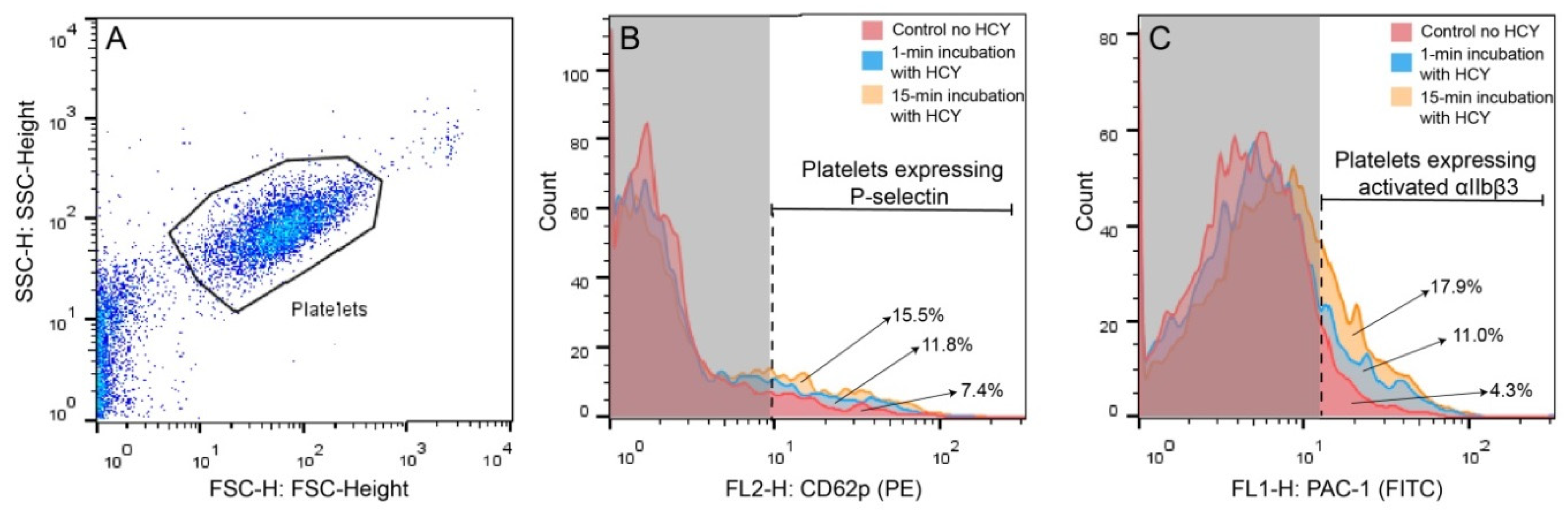
4.3. Flow Cytometry of Human Platelets
4.4. Rat Model of Hyperhomocysteinemia (HHcy)
4.5. Determination of Fibrinogen Concentration in Blood Plasma
4.6. Determination of Hcy Levels in Blood Plasma
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rehman, T.; Shabbir, M.A.; Inam-Ur-Raheem, M.; Manzoor, M.F.; Ahmad, N.; Liu, Z.; Ahmad, M.H.; Siddeeg, A.; Abid, M.; Aadil, R.M. Cysteine and homocysteine as biomarker of various diseases. Food Sci. Nutr. 2020, 8, 4696–4707. [Google Scholar] [CrossRef]
- Rozen, R. Genetic modulation of homocysteinemia. Semin. Thromb. Hemost. 2000, 26, 255–262. [Google Scholar] [CrossRef]
- Lentz, S.R.; Haynes, W.G. Homocysteine: Is it a clinically important cardiovascular risk factor? Clevel. Clin. J. Med. 2004, 71, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, D.W. Hyperhomocysteinemia and oxidative stress: Time for a reality check? Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1182–1184. [Google Scholar] [CrossRef] [Green Version]
- Kostić, S.; Mićovic, Ž.; Andrejević, L.; Cvetković, S.; Stamenković, A.; Stanković, S.; Obrenović, R.; Labudović-Borović, M.; Hrnčić, D.; Jakovljević, V.; et al. The effects of l-cysteine and N-acetyl-l-cysteine on homocysteine metabolism and haemostatic markers, and on cardiac and aortic histology in subchronically methionine-treated Wistar male rats. Mol. Cell. Biochem. 2019, 451, 43–54. [Google Scholar] [CrossRef]
- Herrmann, W.; Herrmann, M.; Obeid, R. Hyperhomocysteinaemia: A critical review of old and new aspects. Curr. Drug Metab. 2007, 8, 17–31. [Google Scholar] [CrossRef]
- Zaric, B.L.; Obradovic, M.; Bajic, V.; Haidara, M.A.; Jovanovic, M.; Isenovic, E.R. Homocysteine and hyperhomocysteinaemia. Curr. Med. Chem. 2018, 26, 2948–2961. [Google Scholar] [CrossRef] [PubMed]
- McCully, K.S. Homocysteine and vascular disease. Nat. Med. 1996, 2, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Den Heijer, M.; Rosendaal, F.R.; Blom, H.J.; Gerrits, W.B.J.; Bos, G.M.J. Hyperhomocysteinemia and venous thrombosis: A meta-analysis. Thromb. Haemost. 1998, 80, 874–877. [Google Scholar] [CrossRef] [PubMed]
- Boers, G.H.J.; Smals, A.G.H.; Trijbels, F.J.M.; Fowler, B.; Bakkeren, J.A.J.M.; Schoonderwaldt, H.C.; Kleijer, W.J.; Kloppenborg, P.W.C. Heterozygosity for homocystinuria in premature peripheral and cerebral occlusive arterial disease. N. Engl. J. Med. 1985, 313, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Riba, R.; Nicolaou, A.; Troxler, M.; Homer-Vaniasinkam, S.; Naseem, K.M. Altered platelet reactivity in peripheral vascular disease complicated with elevated plasma homocysteine levels. Atherosclerosis 2004, 175, 69–75. [Google Scholar] [CrossRef]
- Wald, D.S.; Law, M.; Morris, J.K. Homocysteine and cardiovascular disease: Evidence on causality from a meta-analysis. Br. Med. J. 2002, 325, 1202–1206. [Google Scholar] [CrossRef] [Green Version]
- Den Heijer, M.; Lewington, S.; Clarke, R. Homocysteine, MTHFR and risk of venous thrombosis: A meta-analysis of published epidemiological studies. J. Thromb. Haemost. 2005, 3, 292–299. [Google Scholar] [CrossRef]
- Van Guldener, C.; Stehouwer, C.D.A. Hyperhomocysteinemia, vascular pathology, and endothelial dysfunction. Semin. Thromb. Hemost. 2000, 26, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Welch, G.N.; Loscalzo, J. Homocysteine and atherothrombosis. N. Engl. J. Med. 1998, 338, 1042–1050. [Google Scholar] [CrossRef]
- Spencer, C.G.C.; Martin, S.C.; Felmeden, D.C.; Blann, A.D.; Beevers, G.D.; Lip, G.Y.H. Relationship of homocysteine to markers of platelet and endothelial activation in ‘high risk’ hypertensives: A substudy of the anglo-scandinavian cardiac outcomes trial. Int. J. Cardiol. 2004, 94, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Lawrence de Koning, A.B.; Werstuck, G.H.; Zhou, J.; Austin, R.C. Hyperhomocysteinemia and its role in the development of atherosclerosis. Clin. Biochem. 2003, 36, 431–441. [Google Scholar] [CrossRef]
- Cook, J.W.; Taylor, L.M.; Orloff, S.L.; Landry, G.J.; Moneta, G.L.; Porter, J.M. Homocysteine and arterial disease. Vascul. Pharmacol. 2002, 38, 293–300. [Google Scholar] [CrossRef]
- Coppola, A.; Davi, G.; De Stefano, V.; Mancini, F.P.; Cerbone, A.M.; Di Minno, G. Homocysteine, coagulation, platelet function, and thrombosis. Semin. Thromb. Hemost. 2000, 26, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, G.M.; Kane, W.H. Activation of endogenous factor V by a homocysteine-induced vascular endothelial cell activator. J. Clin. Investig. 1986, 77, 1909–1916. [Google Scholar] [CrossRef] [Green Version]
- Lentz, S.R.; Sadler, J.E. Inhibition of thrombomodulin surface expression and protein C activation by the thrombogenic agent homocysteine. J. Clin. Investig. 1991, 88, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Lentz, S.R. Mechanisms of homocysteine-induced atherothrombosis. J. Thromb. Haemost. 2005, 3, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Austin, R.C.; Lentz, S.R.; Werstuck, G.H. Role of hyperhomocysteinemia in endothelial dysfunction and atherothrombotic disease. Cell Death Differ. 2004, 11, S56–S64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutus, B.; Rabini, R.A.; Staffolani, R.; Ricciotti, R.; Fumelli, P.; Moretti, N.; Martarelli, D.; Mazzanti, L. Homocysteine-induced inhibition of nitric oxide production in platelets: A study on healthy and diabetic subjects. Diabetologia 2001, 44, 979–982. [Google Scholar] [CrossRef] [Green Version]
- Leoncini, G.; Pascale, R.; Signorello, M.G. Effects of homocysteine on L-arginine transport and nitric oxide formation in human platelets. Eur. J. Clin. Investig. 2003, 33, 713–719. [Google Scholar] [CrossRef]
- Kim, O.V.; Litvinov, R.I.; Alber, M.S.; Weisel, J.W. Quantitative structural mechanobiology of platelet-driven blood clot contraction. Nat. Commun. 2017, 8, 1274. [Google Scholar] [CrossRef]
- Peshkova, A.; Malyasyov, D.; Bredikhin, R.; Le Minh, G.; Andrianova, I.; Tutwiler, V.; Nagaswami, C.; Weisel, J.; Litvinov, R. Reduced contraction of blood clots in venous thromboembolism is a potential thrombogenic and embologenic mechanism. TH Open 2018, 2, e104–e115. [Google Scholar] [CrossRef] [Green Version]
- Evtugina, N.G.; Peshkova, A.D.; Pichugin, A.A.; Weisel, J.W.; Litvinov, R.I. Impaired contraction of blood clots precedes and predicts postoperative venous thromboembolism. Sci. Rep. 2020, 10, 18261. [Google Scholar] [CrossRef]
- Martins, P.J.F.; Galdieri, L.C.; Souza, F.G.; Andersen, M.L.; Benedito-Silva, A.A.; Tufik, S.; D’Almeida, V. Physiological variation in plasma total homocysteine concentrations in rats. Life Sci. 2005, 76, 2621–2629. [Google Scholar] [CrossRef] [PubMed]
- Tutwiler, V.; Peshkova, A.D.; Andrianova, I.A.; Khasanova, D.R.; Weisel, J.W.; Litvinov, R.I. Contraction of blood clots is impaired in acute ischemic stroke. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Minh, G.; Peshkova, A.D.; Andrianova, I.A.; Sibgatullin, T.B.; Maksudova, A.N.; Weisel, J.W.; Litvinov, R.I. Impaired contraction of blood clots as a novel prothrombotic mechanism in systemic lupus erythematosus. Clin. Sci. 2018, 132, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, O.V.; Nevzorova, T.A.; Mordakhanova, E.R.; Ponomareva, A.A.; Andrianova, I.A.; Le Minh, G.; Daminova, A.G.; Peshkova, A.D.; Alber, M.S.; Vagin, O.; et al. Fatal dysfunction and disintegration of thrombin-stimulated platelets. Haematologica 2019, 104, 1866–1878. [Google Scholar] [CrossRef]
- De Vriese, A.S.; Blom, H.J.; Heil, S.G.; Mortier, S.; Kluijtmans, L.A.J.; Van De Voorde, J.; Lameire, N.H. Endothelium-derived hyperpolarizing factor-mediated renal vasodilatory response is impaired during acute and chronic hyperhomocysteinemia. Circulation 2004, 109, 2331–2336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bełtowski, J.; Wójcicka, G.; Wojtak, A. Effect of experimental hyperhomocysteinemia on plasma lipid profile, insulin sensitivity and paraoxonase 1 in the rat. Adipobiology 2012, 4, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Ma, S.; Wang, T.; Zhao, C.; Li, Y.; Yin, J.; Liu, C.; Gao, C.; Sun, L.; Yue, W.; et al. A novel rat model of heart failure induced by high methionine diet showing evidence of association between hyperhomocysteinemia and activation of NF-kappaB. Am. J. Transl. Res. 2016, 8, 117–124. [Google Scholar] [PubMed]
- Gerasimova, E.; Yakovleva, O.; Burkhanova, G.; Ziyatdinova, G.; Khaertdinov, N.; Sitdikova, G. Effects of maternal hyperhomocysteinemia on the early physical development and neurobehavioral maturation of rat offspring. Bionanoscience 2017, 7, 155–158. [Google Scholar] [CrossRef]
- Yakovleva, O.; Bogatova, K.; Mukhtarova, R.; Yakovlev, A.; Shakhmatova, V.; Gerasimova, E.; Ziyatdinova, G.; Hermann, A.; Sitdikova, G. Hydrogen sulfide alleviates anxiety, motor, and cognitive dysfunctions in rats with maternal hyperhomocysteinemia via mitigation of oxidative stress. Biomolecules 2020, 10, 995. [Google Scholar] [CrossRef]
- Tutwiler, V.; Litvinov, R.I.; Lozhkin, A.P.; Peshkova, A.D.; Lebedeva, T.; Ataullakhanov, F.I.; Spiller, K.L.; Cines, D.B.; Weisel, J.W. Kinetics and mechanics of clot contraction are governed by the molecular and cellular composition of the blood. Blood 2016, 127, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Peshkova, A.D.; Safiullina, S.I.; Evtugina, N.G.; Baras, Y.S.; Ataullakhanov, F.I.; Weisel, J.W.; Litvinov, R.I. Premorbid hemostasis in women with a history of pregnancy loss. Thromb. Haemost. 2019, 119. [Google Scholar] [CrossRef]
- Peshkova, A.D.; Evdokimova, T.A.; Sibgatullin, T.B.; Ataullakhanov, F.I.; Litvinov, R.I.; Weisel, J.W. Accelerated spatial fibrin growth and impaired contraction of blood clots in patients with rheumatoid arthritis. Int. J. Mol. Sci. 2020, 21, 9434. [Google Scholar] [CrossRef]
- Litvinov, R.I.; Khismatullin, R.R.; Shakirova, A.Z.; Litvinov, T.R.; Nagaswami, C.; Peshkova, A.D.; Weisel, J.W. Morphological signs of intravital contraction (retraction) of pulmonary thrombotic emboli. Bionanoscience 2018, 8, 428–433. [Google Scholar] [CrossRef]
- Khismatullin, R.R.; Nagaswami, C.; Shakirova, A.Z.; Vrtková, A.; Procházka, V.; Gumulec, J.; Mačák, J.; Litvinov, R.I.; Weisel, J.W. Quantitative morphology of cerebral thrombi related to intravital contraction and clinical features of ischemic stroke. Stroke 2020, 51, 3640–3650. [Google Scholar] [CrossRef]
- Cines, D.B.; Lebedeva, T.; Nagaswami, C.; Hayes, V.; Massefski, W.; Litvinov, R.I.; Rauova, L.; Lowery, T.J.; Weisel, J.W. Clot contraction: Compression of erythrocytes into tightly packed polyhedra and redistribution of platelets and fibrin. Blood 2014, 123, 1596–1603. [Google Scholar] [CrossRef] [Green Version]
- Tutwiler, V.; Peshkova, A.D.; Le Minh, G.; Zaitsev, S.; Litvinov, R.I.; Cines, D.B.; Weisel, J.W. Blood clot contraction differentially modulates internal and external fibrinolysis. J. Thromb. Haemost. 2019, 17, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Lam, W.A.; Chaudhuri, O.; Crow, A.; Webster, K.D.; Li, T.-D.; Kita, A.; Huang, J.; Fletcher, D.A. Mechanics and contraction dynamics of single platelets and implications for clot stiffening. Nat. Mater. 2011, 10, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoncini, G.; Bruzzese, D.; Signorello, M.G. A role for PLCγ2 in platelet activation by homocysteine. J. Cell. Biochem. 2007, 100, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Luo, F.; Liu, X.; Wang, S.; Chen, H. Effect of homocysteine on platelet activation induced by collagen. Nutrition 2006, 22, 69–75. [Google Scholar] [CrossRef]
- Signorello, M.G.; Segantin, A.; Passalacqua, M.; Leoncini, G. Homocysteine decreases platelet NO level via protein kinase C activation. Nitric Oxide 2009, 20, 104–113. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Z.; Shi, S.; Gao, F.; Wu, J.; Dong, S.; Zhang, W.; Liu, Y.; Zhong, X. Calcium sensing receptor initiating cystathionine-gamma-lyase/hydrogen sulfide pathway to inhibit platelet activation in hyperhomocysteinemia rat. Exp. Cell Res. 2017, 358, 171–181. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, X.; Shi, S.; Li, H.; Gao, F.; Zhong, X.; Wang, Y. Exogenous hydrogen sulfide protects from endothelial cell damage, platelet activation, and neutrophils extracellular traps formation in hyperhomocysteinemia rats. Exp. Cell Res. 2018, 370, 434–443. [Google Scholar] [CrossRef]
- Sen, U.; Mishra, P.K.; Tyagi, N.; Tyagi, S.C. Homocysteine to hydrogen sulfide or hypertension. Cell Biochem. Biophys. 2010, 57, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Karolczak, K.; Olas, B. Mechanism of action of homocysteine and its thiolactone in hemostasis system. Physiol. Res. 2009, 58, 623–633. [Google Scholar] [CrossRef]
- Malinowska, J.; Olas, B. Homocysteine and its thiolactone-mediated modification of fibrinogen affect blood platelet adhesion. Platelets 2012, 23, 409–412. [Google Scholar] [CrossRef] [Green Version]
- Mohan, I.V.; Jagroop, I.A.; Mikhailidis, D.P.; Stansby, G.P. Homocysteine activates platelets in vitro. Clin. Appl. Thromb. 2008, 14, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Fuglsang, J.; Stender, M.; Zhou, J.; Møller, J.; Falk, E.; Ravn, H.B. Platelet activity and in vivo arterial thrombus formation in rats with mild hyperhomocysteinaemia. Blood Coagul. Fibrinolysis 2002, 13, 683–689. [Google Scholar] [CrossRef]
- Weisel, J.W.; Litvinov, R.I. Mechanisms of fibrin polymerization and clinical implications. Blood 2013, 121, 1712–1719. [Google Scholar] [CrossRef] [Green Version]
- Ueland, P.M. Homocysteine species as components of plasma redox thiol status. Clin. Chem. 1995, 41, 340–342. [Google Scholar] [CrossRef] [Green Version]
- Sauls, D.L.; Lockhart, E.; Warren, M.E.; Lenkowski, A.; Wilhelm, S.E.; Hoffman, M. Modification of fibrinogen by homocysteine thiolactone increases resistance to fibrinolysis: A potential mechanism of the thrombotic tendency in hyperhomocysteinemia. Biochemistry 2006, 45, 2480–2487. [Google Scholar] [CrossRef]
- Genoud, V.; Lauricella, A.M.; Kordich, L.C.; Quintana, I. Impact of homocysteine-thiolactone on plasma fibrin networks. J. Thromb. Thrombolysis 2014, 38, 540–545. [Google Scholar] [CrossRef]
- Nienaber-Rousseau, C.; De Lange, Z.; Pieters, M. Homocysteine influences blood clot properties alone and in combination with total fibrinogen but not with fibrinogen γ′ in Africans. Blood Coagul. Fibrinolysis 2015, 26, 389–395. [Google Scholar] [CrossRef]
- Marchi, R.; Carvajal, Z.; Weisel, J.W. Comparison of the effect of different homocysteine concentrations on clot formation using human plasma and purified fibrinogen. Thromb. Haemost. 2008, 99, 451–452. [Google Scholar] [CrossRef] [Green Version]
- Sauls, D.L.; Arnold, E.K.; Bell, C.W.; Allen, J.C.; Hoffman, M. Pro-thrombotic and pro-oxidant effects of diet-induced hyperhomocysteinemia. Thromb. Res. 2007, 120, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Lauricella, A.M.; Quintana, I.L.; Kordich, L.C. Effects of homocysteine thiol group on fibrin networks: Another possible mechanism of harm. Thromb. Res. 2002, 107, 75–79. [Google Scholar] [CrossRef]
- Lauricella, A.M.; Quintana, I.; Castañon, M.; Sassetti, B.; Kordich, L. Influence of homocysteine on fibrin network lysis. Blood Coagul. Fibrinolysis 2006, 17, 181–186. [Google Scholar] [CrossRef]
- Undas, A.; Brozek, J.; Jankowski, M.; Siudak, Z.; Szczeklik, A.; Jakubowski, H. Plasma homocysteine affects fibrin clot permeability and resistance to lysis in human subjects. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1397–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sauls, D.L.; Wolberg, A.S.; Hoffman, M. Elevated plasma homocysteine leads to alterations in fibrin clot structure and stability: Implications for the mechanism of thrombosis in hyperhomocysteinemia. J. Thromb. Haemost. 2003, 1, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Quintana, I.L.; Oberholzer, M.V.; Kordich, L.; Lauricella, A.M. Impaired fibrin gel permeability by high homocysteine levels. Thromb. Res. 2011, 127, 35–38. [Google Scholar] [CrossRef]
- Rojas, A.M.; Kordich, L.; Lauricella, A.M. Homocysteine modifies fibrin clot deformability: Another possible explanation of harm. Biorheology 2009, 46, 379–387. [Google Scholar] [CrossRef]
- Ziyatdinova, G.; Grigor’eva, L.; Morozov, M.; Gilmutdinov, A.; Budnikov, H. Electrochemical oxidation of sulfur-containing amino acids on an electrode modified with multi-walled carbon nanotubes. Microchim. Acta 2009, 165, 353–359. [Google Scholar] [CrossRef]
- Lee, P.T.; Lowinsohn, D.; Compton, R.G. Simultaneous detection of homocysteine and cysteine in the presence of ascorbic acid and glutathione using a nanocarbon modified electrode. Electroanalysis 2014, 26, 1488–1496. [Google Scholar] [CrossRef] [Green Version]
- Gaifullina, A.S.; Lazniewska, J.; Gerasimova, E.V.; Burkhanova, G.F.; Rzhepetskyy, Y.; Tomin, A.; Rivas-Ramirez, P.; Huang, J.; Cmarko, L.; Zamponi, G.W.; et al. A potential role for T-type calcium channels in homocysteinemia-induced peripheral neuropathy. Pain 2019, 160, 2798–2810. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Markers | Untreated Platelets (n = 4) | Hcy-Treated Platelets (n = 4) | |
|---|---|---|---|
| 1-min Incubation | 15-min Incubation | ||
| P-selectin | 6.9 (5.3–8.6) | 8.4 (5.4–11.3) † | 12.9 (11.5–15.8) * |
| Activated integrin αIIbβ3 | 2.8 (2.0–3.0) | 9.7 (3.5–13.4) † | 15.4 (11.7–18.0) * |
| Parameters | Hcy Levels in Blood | |||
|---|---|---|---|---|
| Control 3.6–7.8 μM (n = 17) | Low HHcy 3.6–9.9 μM (n = 14) | Moderate HHcy 10–50 μM (n = 12) | High HHcy 51–122 μM (n = 10) | |
| Extent of clot contraction, % | 33.5 ± 1.3 § | 28.6 ± 1.7 § | 40.4 ± 2.3 *,† | 41.7 ± 2.4 *,† |
| Lag time, s | 94 ± 6 § | 110 ± 8 § | 91 ± 9 § | 273 ± 47 *,† |
| Area under curve, a.u. | 270 ± 11 | 237 ± 19 | 340 ± 24 †,§ | 252 ± 28 |
| Parameters | Hcy Levels in Blood | |||
|---|---|---|---|---|
| Control 3.6–7.8 μM (n = 17) | Low HHcy 3.6–9.9 μM (n = 14) | Moderate HHcy 10–50 μM (n = 12) | High HHcy 51–122 μM (n = 10) | |
| Fibrinogen, g/L | 3.5 ± 0.2 § | 3.1 ± 0.4 § | 2.4 ± 0.4 | 1.3 ± 0.2 *,† |
| Platelets, ×1012/L | 1.2 ± 0.1 § | 1.3 ± 0.2 § | 1.0 ± 0.3 § | 13.2 ± 3.1 *,† |
| Red blood cells, ×1012/L | 5.3 ± 0.4 | 6.6 ± 0.7 § | 3.6 ± 0.3 *,† | 3.9 ± 0.4 † |
| Parameters of Blood Composition | Parameters of Blood Clot Contraction Kinetics | |||
|---|---|---|---|---|
| Extent of Clot Contraction | Lag Time | Area under Curve | Average Velocity | |
| Hcy blood levels in vivo | 0.56 *** | 0.43 ** | - | 0.56 *** |
| Fibrinogen levels | −0.37 *** | - | - | −0.38 ** |
| Platelet counts | - | 0.29 * | - | - |
| RBCs counts | −0.42 ** | - | −0.29 * | −0.42 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Litvinov, R.I.; Peshkova, A.D.; Le Minh, G.; Khaertdinov, N.N.; Evtugina, N.G.; Sitdikova, G.F.; Weisel, J.W. Effects of Hyperhomocysteinemia on the Platelet-Driven Contraction of Blood Clots. Metabolites 2021, 11, 354. https://doi.org/10.3390/metabo11060354
Litvinov RI, Peshkova AD, Le Minh G, Khaertdinov NN, Evtugina NG, Sitdikova GF, Weisel JW. Effects of Hyperhomocysteinemia on the Platelet-Driven Contraction of Blood Clots. Metabolites. 2021; 11(6):354. https://doi.org/10.3390/metabo11060354
Chicago/Turabian StyleLitvinov, Rustem I., Alina D. Peshkova, Giang Le Minh, Nail N. Khaertdinov, Natalia G. Evtugina, Guzel F. Sitdikova, and John W. Weisel. 2021. "Effects of Hyperhomocysteinemia on the Platelet-Driven Contraction of Blood Clots" Metabolites 11, no. 6: 354. https://doi.org/10.3390/metabo11060354