
Apolipoprotein B and Cardiovascular Disease: Biomarker and Potential Therapeutic Target

 ,
, 
Abstract
:1. Introduction
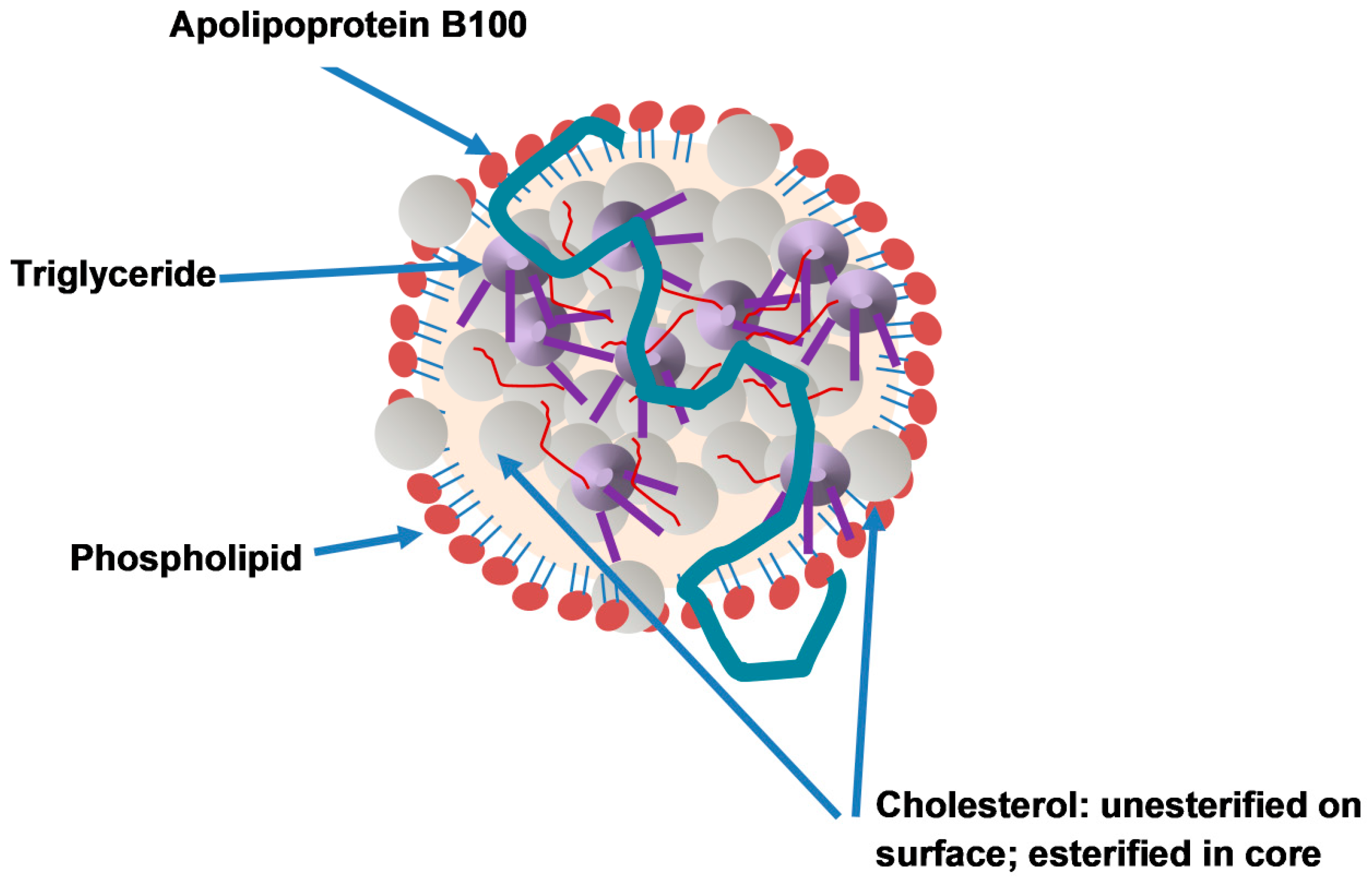
2. ApoB: Characteristics and Composition
3. Biomarkers for CVD: LDL-C and ApoB
3.1. The Lipid Profile and LDL-C as a Biomarker
3.2. Non-HDL-C as a Biomarker
3.3. ApoB as a Biomarker
3.4. ApoB to ApoA1 Ratio as a Biomarker
3.5. When Does ApoB Show an Advantage over HDL-C?
4. How Do Pro-Atherosclerotic Risk Factors Affect ApoB Levels?
4.1. ApoB, CVD and Demographics
4.2. Specific Risk Factors: Body Weight, Hypertension, Diabetes
4.3. The Clinical Significance of Small Dense LDL
5. ApoB as a Target of CVD Treatment
6. Future Perspectives
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Roth, G.A.; Forouzanfar, M.H.; Moran, A.E.; Barber, R.; Nguyen, G.; Feigin, V.L.; Naghavi, M.; Mensah, G.A.; Murray, C.J. Demographic and epidemiologic drivers of global cardiovascular mortality. N. Engl. J. Med. 2015, 372, 1333–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuster, V.; Voûte, J. MDGs: Chronic diseases are not on the agenda. Lancet 2005, 366, 1512–1514. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart disease and stroke statistics—2019 update: A report from the American heart association. Circulation 2019, 139, e56–e528. [Google Scholar] [PubMed]
- Gazzola, K.; Reeskamp, L.; van den Born, B.J. Ethnicity, lipids and cardiovascular disease. Curr. Opin. Lipidol. 2017, 28, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Szuszkiewicz-Garcia, M.M.; Davidson, J.A. Cardiovascular disease in diabetes mellitus, risk factors and medical therapy. Endocrinol. Metab. Clin. N. Am. 2014, 43, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Silverio, A.; Cavallo, P.; De Rosa, R.; Galasso, G. Big Health Data and Cardiovascular Diseases: A Challenge for Research, an Opportunity for Clinical Care. Front. Med. 2019, 6, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkhalil, M. Mechanistic Insights to Target Atherosclerosis Residual Risk. Curr. Probl. Cardiol. 2019, 46, 100432. [Google Scholar] [CrossRef]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J. Am. Coll. Cardiol. 2019, 74, 1376–1414. [Google Scholar] [CrossRef]
- Fruchart, J.C.; Sacks, F.; Hermans, M.P.; Assmann, G.; Brown, W.V.; Ceska, R.; Chapman, M.J.; Dodson, P.M.; Fioretto, P.; Ginsberg, H.N.; et al. The Residual Risk Reduction Initiative: A call to action to reduce residual vascular risk in patients with dyslipidemia. Am. J. Cardiol. 2008, 102, 1K–34K. [Google Scholar] [CrossRef]
- Trompet, S.; Packard, C.J.; Jukema, J.W. Plasma apolipoprotein-B is an important risk factor for CVD, and its assessment should be routine clinical practice. Curr. Opin. Lipidol. 2018, 29, 51–52. [Google Scholar] [CrossRef]
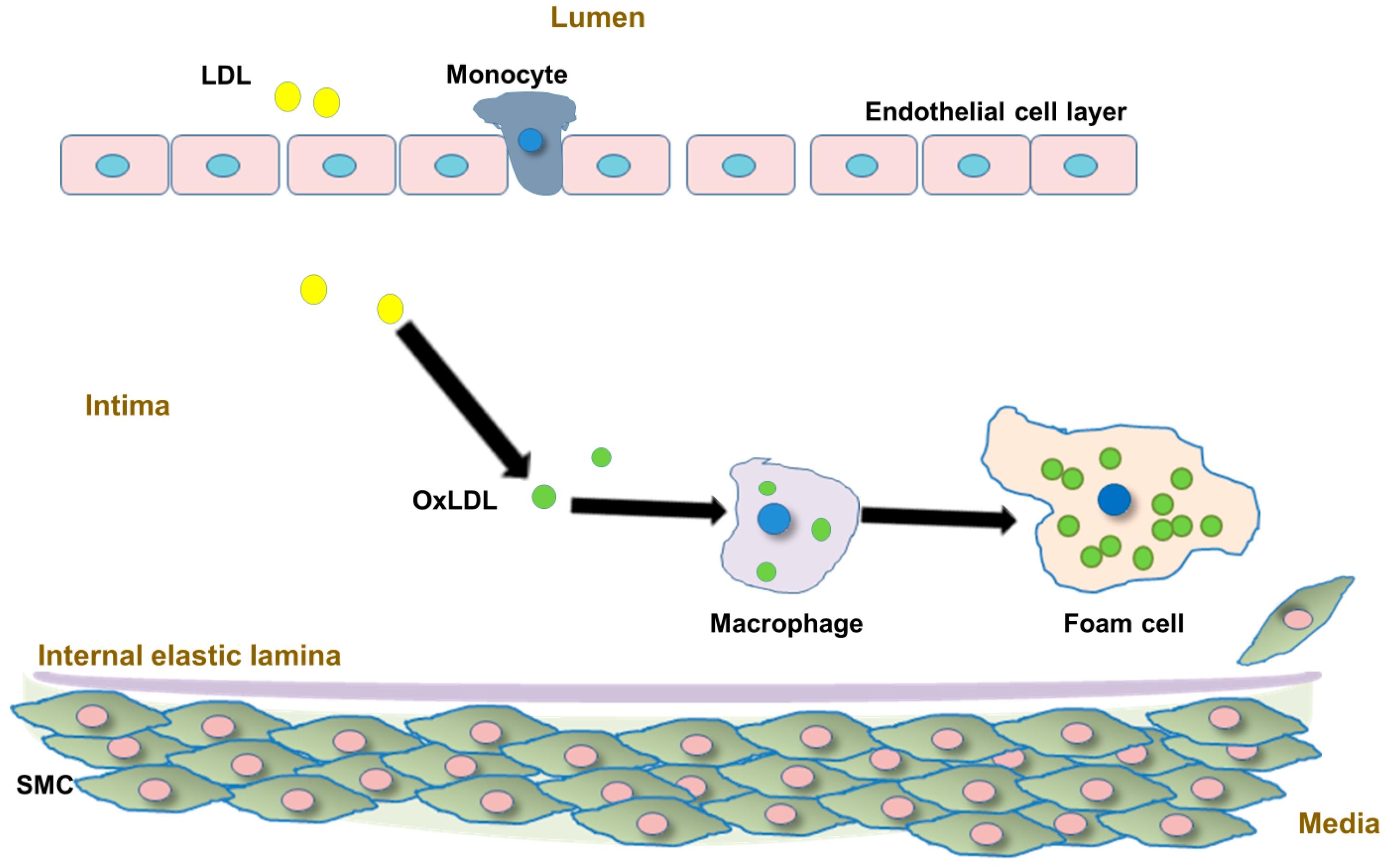
- Libby, P.; Tabas, I.; Fredman, G.; Fisher, E.A. Inflammation and its resolution as determinants of acute coronary syndromes. Circ. Res. 2014, 114, 1867–1879. [Google Scholar] [CrossRef] [Green Version]
- Maguire, E.M.; Pearce, S.W.A.; Xiao, Q. Foam Cell Formation: A New Target for Fighting Atherosclerosis and Cardiovascular Disease. Vascul. Pharmacol. 2019, 112, 54–71. [Google Scholar] [CrossRef] [PubMed]
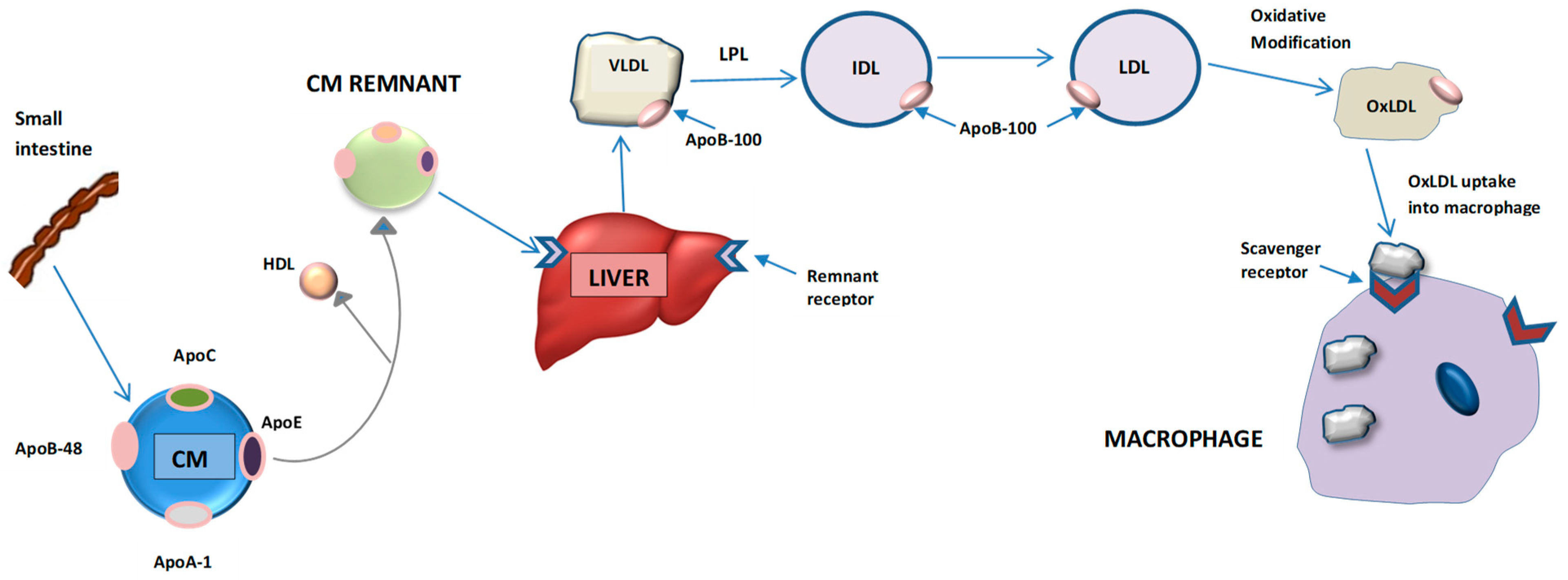
- Ginsberg, H.N. Lipoprotein Physiology. Endocrinol. Metab. Clin. North Am. 1998, 27, 503–519. [Google Scholar] [CrossRef]
- Olofsson, S.O.; Borèn, J. Apolipoprotein B: A clinically important apolipoprotein which assembles atherogenic lipoproteins and promotes the development of atherosclerosis. J. Intern. Med. 2005, 258, 395–410. [Google Scholar] [CrossRef] [PubMed]
- Von Zychlinski, A.; Williams, M.; McCormick, S.; Kleffmann, T. Absolute quantification of apolipoproteins and associated proteins on human plasma lipoproteins. J. Proteom. 2014, 106, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.Y. Metabolism and Modification of Apolipoprotein B-Containing Lipoproteins Involved in Dyslipidemia and Atherosclerosis. Biol. Pharm. Bull. 2016, 39, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Herscovitz, H.; Derksen, A.; Walsh, M.T.; McKnight, C.J.; Gantz, D.L.; Hadzopoulou Cladaras, M.; Zannis, V.; Curry, C.; Small, D.M. The N-terminal 17% of apoB binds tightly and irreversibly to emulsions modeling nascent very low density lipoproteins. J. Lipid Res. 2001, 42, 51–59. [Google Scholar] [CrossRef]
- Pulai, J.I.; Neuman, R.J.; Groenewegen, A.W.; Wu, J.; Schonfeld, G. Genetic heterogeneity in familial hypobetalipoproteinemia: Linkage and non-linkage to the apoB gene in Caucasian families. Am. J. Med. Genet. 1998, 76, 79–86. [Google Scholar] [CrossRef]
- Law, S.W.; Lackner, K.J.; Hospattankar, A.V.; Anchors, J.M.; Sakaguchi, A.Y.; Naylor, S.L.; Brewer, H., Jr. B. Human apolipoprotein B-100: Cloning, analysis of liver mRNA, and assignment of the gene to chromosome 2. Proc. Natl. Acad. Sci. USA 1985, 82, 8340–8344. [Google Scholar]
- Fisher, E.A.; Ginsberg, H.N. Complexity in the secretory pathway: The assembly and secretion of apolipoprotein B containing lipoproteins. J. Biol. Chem. 2002, 277, 17377–17380. [Google Scholar]
- Chan, L. Apolipoprotein B, the Major Protein Component of Triglyceride-rich and Low Density Lipoproteins. J. Biol. Chem. 1992, 267, 25621–25624. [Google Scholar] [CrossRef]
- Nakajima, K.; Nakano, T.; Tokita, Y.; Nagamine, T.; Inazu, A.; Kobayashi, J. Postprandial lipoprotein metabolism; VLDL vs chylomicrons. Clin. Chim. Acta 2011, 412, 1306–1318. [Google Scholar] [CrossRef] [Green Version]
- Powell, L.M.; Wallis, S.C.; Pease, R.J.; Edwards, Y.H.; Knott, T.J.; Scott, J. A novel form of specific RNA processing produces apolipoprotein B-48 in intestine. Cell 1987, 50, 831–840. [Google Scholar] [CrossRef]
- Davidson, N.O.; Shelness, G.S. Apolipoprotein B: mRNA editing, lipoprotein assembly, and presecretory degradation. Annu. Rev. Nutr. 2000, 20, 169–193. [Google Scholar] [CrossRef] [PubMed]
- Avramoglu, R.K.; Adeli, K. Hepatic regulation of apolipoprotein B. Rev. Endocr. Metab. Disord. 2004, 5, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Doonan, L.M.; Fisher, E.A.; Brodsky, J.L. Can modulators of apolipoproteinB biogenesis serve as an alternate target for cholesterol-lowering drugs? Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2018, 1863, 762–771. [Google Scholar] [CrossRef] [PubMed]
- Rustaeus, S.; Lindberg, K.; Stillemark, P.; Claesson, C.; Asp, L.; Larsson, T.; Borén, J.; Olofsson, S.O. Assembly of very low density lipoprotein: A two-step process of apolipoprotein B core lipidation. J. Nutr. 1999, 129, 463S–466S. [Google Scholar] [CrossRef] [PubMed]
- Kane, J.P. Apolipoprotein B: Structural and metabolic heterogeneity. Annu. Rev. Physiol. 1983, 45, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, A.J.; Barrett, P.H.; van Bockxmeer, F.M.; Burnett, J.R. Lipid disorders and mutations in the APOB gene. Clin. Chem. 2004, 50, 1725–1732. [Google Scholar] [CrossRef] [Green Version]
- Burnett, J.R.; Barrett, P.H.R. Apolipoprotein B metabolism: Tracer kinetics, models, and metabolic studies. Crit. Rev. Clin. Lab. Sci. 2002, 39, 89–137. [Google Scholar] [CrossRef]
- Segrest, J.P.; Jones, M.K.; Mishra, V.K.; Anantharamaiah, G.M.; Garber, D.W. ApoB-100 has a pentapartite structure composed of three amphipathic alpha-helical domains alternating with two amphipathic beta-strand domains. Detection by the computer program LOCATE. Arterioscler. Thromb. 1994, 14, 1674–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasiole, D.A.; Davis, R.A.; Attie, A.D. The physiological and molecular regulation of lipoprotein assembly and secretion. Mol. Biosyst. 2007, 3, 608–619. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.J.; Fisher, E.A. Apolipoprotein B: The crucial protein of atherogenic lipoproteins. In Atherosclerosis: Risks, Mechanisms, and Therapies; Wang, H., Patterson, C., Eds.; Wiley Blackwell: Hoboken, NJ, USA, 2015; pp. 291–305. [Google Scholar]
- Jiang, Z.G.; Liu, Y.; Hussain, M.M.; Atkinson, D.; McKnight, C.J. Reconstituting initial events during the assembly of apolipoprotein B-containing lipoproteins in a cell-free system. J. Mol. Biol. 2008, 383, 1181–1194. [Google Scholar] [CrossRef] [Green Version]
- Manchekar, M.; Kapil, R.; Sun, Z.; Segrest, J.P.; Dashti, N. Relationship between Amphipathic β Structures in the β1 Domain of Apolipoprotein B and the Properties of the Secreted Lipoprotein Particles in McA-RH7777 Cells. Biochemistry 2017, 56, 4084–4094. [Google Scholar] [CrossRef]
- Biterova, E.I.; Isupov, M.N.; Keegan, R.M.; Lebedev, A.A.; Sohail, A.A.; Liaqat, I.; Alanen, H.I.; Ruddock, L.W. The crystal structure of human microsomal triglyceride transfer protein. Proc. Natl. Acad. Sci. USA 2019, 116, 17251–17260. [Google Scholar] [CrossRef] [Green Version]
- Segrest, J.P.; Jones, M.K.; De Loof, H.; Dashti, N. Structure of apolipoprotein B100 in low density lipoproteins. J. Lipid Res. 2001, 42, 1346–1367. [Google Scholar] [CrossRef]
- Dixon, J.L.; Ginsberg, H.N. Regulation of hepatic secretion of apolipoprotein B-containing lipoproteins: Information obtained from cultured liver cells. J. Lipid Res. 1993, 34, 167–179. [Google Scholar] [CrossRef]
- Shelness, G.S.; Ingram, M.F.; Huang, X.F.; DeLozier, J.A. Apolipoprotein B in the Rough Endoplasmic Reticulum: Translation, Translocation and the Initiation of Lipoprotein Assembly. J. Nutr. 1999, 129, 456S–462S. [Google Scholar]
- Zhou, M.; Fisher, E.A.; Ginsberg, H.N. Regulated Co-translational ubiquitination of apolipoprotein B100. A new paradigm for proteasomal degradation of a secretory protein. J. Biol. Chem. 1998, 273, 24649–24653. [Google Scholar] [CrossRef] [Green Version]
- Rutledge, A.C.; Qiu, W.; Zhang, R.; Kohen-Avramoglu, R.; Nemat-Gorgani, N.; Adeli, K. Mechanisms targeting apolipoprotein B100 to proteasomal degradation: Evidence that degradation is initiated by BiP binding at the N terminus and the formation of a p97 complex at the C terminus. Arterioscler. Thromb. Vasc. Biol. 2009, 229, 579–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koerner, C.M.; Roberts, B.S.; Neher, S.B. Endoplasmic reticulum quality control in lipoprotein metabolism. Mol. Cell. Endocrinol. 2019, 498, 110547. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.A.; Zhou, M.; Mitchell, D.M.; Wu, X.; Omura, S.; Wang, H.; Goldberg, A.L.; Ginsberg, H.N. The degradation of apolipoprotein B100 is mediated by the ubiquitin-proteasome pathway and involves heat shock protein 70. J. Biol. Chem. 1997, 272, 20427–20434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, E.A.; Pan, M.; Chen, X.; Wu, X.; Wang, H.; Jamil, H.; Sparks, J.D.; Williams, K.J. The triple threat to nascent apolipoprotein B. Evidence for multiple, distinct degradative pathways. J. Biol. Chem. 2001, 276, 27855–27863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulinski, A.; Rustaeus, S.; Vance, J.E. Microsomal triacylglycerol transfer protein is required for lumenal accretion of triacylglycerol not associated with ApoB, as well as for ApoB lipidation. J. Biol. Chem. 2002, 277, 31516–31525. [Google Scholar] [CrossRef] [Green Version]
- Wetterau, J.R.; Combs, K.A.; Spinner, S.N.; Joiner, B.J. Protein disulfide isomerase is a component of the microsomal triglyceride transfer protein complex. J. Biol. Chem. 1990, 265, 9800–9807. [Google Scholar] [CrossRef]
- Innerarity, T.L.; Borén, J.; Yamanaka, S.; Olofsson, S.O. Biosynthesis of apolipoprotein B48-containing lipoproteins. Regulation by novel post-transcriptional mechanisms. J. Biol. Chem. 1996, 271, 2353–2356. [Google Scholar] [CrossRef] [Green Version]
- Brodsky, J.L.; Gusarova, V.; Fisher, E.A. Vesicular trafficking of hepatic apolipoprotein B100 and its maturation to very low-density lipoprotein particles; studies from cells and cellfree systems. Trends Cardiovasc. Med. 2004, 14, 127–132. [Google Scholar] [CrossRef]
- Sundaram, M.; Yao, Z. Recent progress in understanding protein and lipid factors affecting hepatic VLDL assembly and secretion. Nutr. Metab. 2010, 7, 35. [Google Scholar] [CrossRef] [Green Version]
- Olofsson, S.O.; Wiklund, O.; Borén, J. Apolipoproteins A-I and B: Biosynthesis, role in the development of atherosclerosis and targets for intervention against cardiovascular disease. Vasc. Health Risk Manag. 2007, 3, 491–502. [Google Scholar]
- Tiwari, S.; Siddiqi, S.A. Intracellular trafficking and secretion of VLDL. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1079–1086. [Google Scholar] [CrossRef] [Green Version]
- Aneni, E.C.; Osondu, C.U.; De La Cruz, J.; Martin, S.S.; Blaha, M.J.; Younus, A.; Feldman, T.; Agatston, A.S.; Veledar, E.; Nasir, K. Lipoprotein Sub-Fractions by Ion-Mobility Analysis and Its Association with Subclinical Coronary Atherosclerosis in High-Risk Individuals. J. Atheroscler. Thromb. 2019, 26, 50–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittendorfer, B.; Yoshino, M.; Patterson, B.W.; Klein, S. VLDL Triglyceride Kinetics in Lean, Overweight, and Obese Men and Women. J. Clin. Endocrinol. Metab. 2016, 101, 4151–4160. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.A.; Sharma, A.; Abramson, J.L.; Vaccarino, V.; Gillespie, C.; Vos, M.B. Caloric sweetener consumption and dyslipidemia among US adults. J. Am. Med. Assoc. 2010, 303, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Wojczynski, M.K.; Glasser, S.P.; Oberman, A.; Kabagambe, E.K.; Hopkins, P.N.; Tsai, M.Y.; Straka, R.J.; Ordovas, J.M.; Arnett, D.K. High-fat meal effect on LDL, HDL, and VLDL particle size and number in the Genetics of Lipid-Lowering drugs and diet network (GOLDN): An interventional study. Lipids Health Dis. 2011, 10, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adiels, M.; Mardinoglu, A.; Taskinen, M.R.; Borén, J. Kinetic Studies to Elucidate Impaired Metabolism of Triglyceride-rich Lipoproteins in Humans. Front. Physiol. 2015, 6, 342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, A.; Scott, J. A cross-species comparison of the apolipoprotein B domain that binds to the LDL receptor. J. Lipid Res. 1990, 31, 1109–1120. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Regulation of the activity of the low density lipoprotein receptor in human fibroblasts. Cell 1975, 6, 307–316. [Google Scholar] [CrossRef]
- Brunelli, R.; Greco, G.; Barteri, M.; Krasnowska, E.K.; Mei, G.; Natella, F.; Pala, A.; Rotella, S.; Ursini, F.; Zichella, L.; et al. One site on the apoB-100 specifically binds 17-beta-estradiol and regulates the overall structure of LDL. FASEB J. 2003, 17, 2127–2129. [Google Scholar] [CrossRef]
- Hevonoja, T.; Pentikäinen, M.O.; Hyvönen, M.T.; Kovanen, P.T.; Ala-Korpela, M. Structure of low density lipoprotein (LDL) particles: Basis for understanding molecular changes in modified LDL. Biochim. Biophys. Acta 2000, 1488, 189–210. [Google Scholar] [CrossRef]
- Bruikman, C.S.; Hovingh, G.K.; Kastelein, J.J. Molecular basis of familial hypercholesterolemia. Curr. Opin. Cardiol. 2017, 32, 262–266. [Google Scholar] [CrossRef]
- Reiss, A.B.; Shah, N.; Muhieddine, D.; Zhen, J.; Yudkevich, J.; Kasselman, L.J.; DeLeon, J. PCSK9 in cholesterol metabolism: From bench to bedside. Clin. Sci. 2018, 132, 1135–1153. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein-Szanto, A.J.P.; Bassi, D.E. Keep recycling going: New approaches to reduce LDL-C. Biochem. Pharmacol. 2019, 164, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Langer, T.; Strober, W.; Levy, R.I. The Metabolism of Low Density Lipoprotein in Familial Type II Hyperlipoproteinemia. J. Clin. Investig. 1972, 51, 1528–1536. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Olin, K.; Lee, I.; Chait, A.; Wight, T.N.; Innerarity, T.L. Identification of the principal proteoglycan-binding site in LDL. A single-point mutation in apo-B100 severely affects proteoglycan interaction without affecting LDL receptor binding. J. Clin. Investig. 1998, 101, 2658–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustafsson, M.; Borén, J. Mechanism of lipoprotein retention by the extracellular matrix. Curr. Opin. Lipidol. 2004, 15, 505–514. [Google Scholar] [CrossRef]
- Olsson, U.; Camejo, G.; Hurt-Camej, E. Possible functional interactions of apolipoprotein B-100 segments that associate with cell proteoglycans and the apoB/E receptor. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 149–155. [Google Scholar] [CrossRef]
- Hurt-Camejo, E.; Camejo, G. ApoB-100 Lipoprotein Complex Formation with Intima Proteoglycans as a Cause of Atherosclerosis and Its Possible Ex Vivo Evaluation as a Disease Biomarker. J. Cardiovasc. Dev. Dis. 2018, 5, 36. [Google Scholar] [CrossRef] [Green Version]
- Borèn, J.; Lee, I.; Zhu, W. Identification of the low density lipoprotein receptor-binding site in apolipoprotein B100 and the modulation of its binding activity by the carboxyl terminus in familial defective apoB100. J. Clin. Investig. 1998, 101, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Oörni, K.; Pentikäinen, M.O.; Ala-Korpela, M.; Kovanen, P.T. Aggregation, fusion, and vesicle formation of modified low density lipoprotein particles: Molecular mechanisms and effects on matrix interactions. J. Lipid Res. 2000, 41, 1703–1714. [Google Scholar] [CrossRef]
- Skålén, K.; Gustafsson, M.; Rydberg, E.K.; Hultén, L.M.; Wiklund, O.; Innerarity, T.L.; Borén, J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature 2002, 417, 750–754. [Google Scholar] [CrossRef]
- Lu, M.; Gursky, O. Aggregation and fusion of low-density lipoproteins in vivo and in vitro. Biomol. Concepts 2013, 4, 501–518. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.D. Hepatic uptake of chylomicron remnants. J. Lipid Res. 1997, 38, 2173–2192. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Wagner, W.D.; Pang, L.; Paka, L.; Curtiss, L.K.; DeLozier, J.A.; Shelness, G.S.; Young, C.S.; Pillarisetti, S. The NH2-terminal Region of Apolipoprotein B Is Sufficient for Lipoprotein Association with Glycosaminoglycans. J. Biol. Chem. 1998, 273, 35355–35361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebara, T.; Conde, K.; Kako, Y.; Liu, Y.; Xu, Y.; Ramakrishnan, R.; Goldberg, I.J.; Shachter, N.S. Delayed catabolism of apoB-48 lipoproteins due to decreased heparan sulfate proteoglycan production in diabetic mice. J. Clin. Investig. 2000, 105, 1807–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinagre, C.G.; Freitas, F.R.; de Mesquita, C.H.; Vinagre, J.C.; Mariani, A.C.; Kalil-Filho, R.; Maranhão, R.C. Removal of Chylomicron Remnants from the Bloodstream is Delayed in Aged Subjects. Aging Dis. 2018, 9, 748–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; de Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 139, e1082–e1143. [Google Scholar] [PubMed]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Bairey Merz, C.N.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; et al. American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2014, 63, 2889–2934. [Google Scholar] [PubMed] [Green Version]
- Friedewald, W.T.; Levy, R.I.; Fredrickson, D.S. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin. Chem. 1972, 18, 499–502. [Google Scholar] [CrossRef] [PubMed]
- Rifai, N.; Warnick, G.R.; McNamara, J.R. Measurement of low-density-lipoprotein cholesterol in serum: A status report. Clin. Chem. 1992, 38, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Hirany, S.; Li, D.; Jialal, I. A more valid measurement of low-density lipoprotein cholesterol in diabetic patients. Am. J. Med. 1997, 102, 48–53. [Google Scholar] [CrossRef]
- Scharnagl, H.; Nauck, M.; Wieland, H.; März, W. The friedewald formula underestimates LDL cholesterol at low concentrations. Clin. Chem. Lab. Med. 2001, 39, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Sawle, A.; Higgins, M.K.; Olivant, M.P.; Higgins, J.A. A rapid single-step centrifugation method for determination of HDL, LDL, and VLDL cholesterol, and TG, and identification of predominant LDL subclass. J. Lipid Res. 2002, 43, 335–343. [Google Scholar] [CrossRef]
- Rahman, F.; Blumenthal, R.S.; Jones, S.R. Fasting or Non-fasting Lipids for Atherosclerotic Cardiovascular Disease Risk Assessment and Treatment? Curr. Atheroscler. Rep. 2018, 20, 14. [Google Scholar] [CrossRef]
- Ridker, P.M. LDL cholesterol: Controversies and future therapeutic directions. Lancet 2014, 384, 607–617. [Google Scholar] [CrossRef]
- Silverman, M.G.; Ference, B.A.; Im, K.; Wiviott, S.D.; Giugliano, R.P.; Grundy, S.M.; Braunwald, E.; Sabatine, M.S. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: A systematic review and meta- analysis. J. Am. Med. Assoc. 2016, 316, 1289–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boekholdt, S.M.; Hovingh, G.K.; Mora, S.; Arsenault, B.J.; Amarenco, P.; Pedersen, T.R.; LaRosa, J.C.; Waters, D.D.; DeMicco, D.A.; Simes, R.J.; et al. Very low levels of atherogenic lipoproteins and the risk for cardiovascular events: A meta-analysis of statin trials. J. Am. Coll. Cardiol. 2014, 64, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Cholesterol Treatment Trialists’ (CTT) Collaboration; Fulcher, J.; O’Connell, R.; Voysey, M.; Emberson, J.; Blackwell, L.; Mihaylova, B.; Simes, J.; Collins, R.; Kirby, A.; et al. Efficacy and safety of LDL-lowering therapy among men and women: Meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet 2015, 385, 1397–1405. [Google Scholar]
- Kilgore, M.; Muntner, P.; Woolley, J.M.; Sharma, P.; Bittner, V.; Rosenson, R.S. Discordance between high non-HDL cholesterol and high LDL-cholesterol among US adults. J. Clin. Lipidol. 2014, 8, 86–93. [Google Scholar] [CrossRef] [Green Version]
- Diffenderfer, M.R.; Schaefer, E.J. The composition and metabolism of large and small LDL. Curr. Opin. Lipidol. 2014, 25, 221–226. [Google Scholar] [CrossRef]
- Contois, J.H.; McConnell, J.P.; Sethi, A.A.; Csako, G.; Devaraj, S.; Hoefner, D.M.; Warnick, G.R.; AACC Lipoproteins and Vascular Diseases Division Working Group on Best Practices. Apolipoprotein B and cardiovascular disease risk: Position statement from the AACC Lipoproteins and Vascular Diseases Division Working Group on Best Practices. Clin. Chem. 2009, 55, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Otvos, J.D. Measurement of triglyceride-rich lipoproteins by nuclear magnetic resonance spectroscopy. Clin. Cardiol. 1999, 22, II21–II27. [Google Scholar] [CrossRef]
- Goff, D.C., Jr.; Lloyd-Jones, D.M.; Bennett, G. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014, 129, S49–S73. [Google Scholar] [CrossRef] [Green Version]
- Preiss, D.; Kristensen, S.L. The New Pooled Cohort Equations Risk Calculator. Can. J. Cardiol. 2015, 31, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.C.; Ahn, H.Y.; Han, K.H.; Park, S.W.; Park, C.Y. Prediction of future cardiovascular disease with an equation to estimate apolipoprotein B in patients with high cardiovascular risk: An analysis from the TNT and IDEAL study. Lipids Health Dis. 2017, 16, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weitgasser, R.; Ratzinger, M.; Hemetsberger, M.; Siostrzonek, P. LDL-cholesterol and cardiovascular events: The lower the better? Wiener Medizinische Wochenschrift 2018, 168, 108. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, E.A.; Myasoedova, V.A.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. Small Dense Low-Density Lipoprotein as Biomarker for Atherosclerotic Diseases. Oxid. Med. Cell. Longev. 2017, 2017, e1273042. [Google Scholar] [CrossRef]
- Averna, M.; Stroes, E. Lipid alterations beyond LDL expert working group. How to assess and manage cardiovascular risk associated with lipid alterations beyond LDL. Atheroscler. Suppl. 2017, 26, 16–24. [Google Scholar] [CrossRef]
- Virani, S.S. Non-HDL cholesterol as a metric of good quality of care: Opportunities and challenges. Tex. Heart Inst. J. 2011, 38, 160–162. [Google Scholar]
- Cannon, C.P.; Braunwald, E.; McCabe, C.H.; Rader, D.J.; Rouleau, J.L.; Belder, R.; Joyal, S.V.; Hill, K.A.; Pfeffer, M.A.; Skene, A.M. Pravastatin or Atorvastatin Evaluation and Infection Therapy-Thrombolysis in Myocardial Infarction 22 Investigators. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N. Engl. J. Med. 2004, 350, 1495–1504. [Google Scholar] [CrossRef]
- Bachorik, P.S.; Ross, J.W. National Cholesterol Education Program recommendations for measurement of low-density lipoprotein cholesterol: Executive summary. The National Cholesterol Education Program Working Group on Lipoprotein Measurement. Clin. Chem. 1995, 41, 1414–1420. [Google Scholar] [CrossRef]
- Davidson, M.H.; Maki, K.C.; Pearson, T.A.; Pasternak, R.C.; Deedwania, P.C.; McKenney, J.M.; Fonarow, G.C.; Maron, D.J.; Ansell, B.J.; Clark, L.T.; et al. Results of the National Cholesterol Education (NCEP) Program Evaluation Project Utilizing Novel E-Technology (NEPTUNE) II survey and implications for treatment under the recent NCEP Writing Group recommendations. Am. J. Cardiol. 2005, 96, 556–563. [Google Scholar] [CrossRef]
- Khanji, M.Y.; Bicalho, V.V.; van Waardhuizen, C.N.; Ferket, B.S.; Petersen, S.E.; Hunink, M.G. Cardiovascular risk assessment: A systematic review of guidelines. Ann. Intern. Med. 2016, 165, 713–722. [Google Scholar] [CrossRef]
- Soran, H.; Ho, J.H.; Adam, S.; Durrington, P.N. Non-HDL cholesterol should not generally replace LDL cholesterol in the management of hyperlipidaemia. Curr. Opin. Lipidol. 2019, 30, 263–272. [Google Scholar] [CrossRef]
- Hermans, M.P.; Ahn, S.A.; Rousseau, M.F. Novel unbiased equations to calculate triglyceride rich lipoprotein cholesterol from routine non-fasting lipids. Cardiovasc. Diabetol. 2014, 13, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundy, S.M.; Stone, N.J. Elevated apolipoprotein B as a risk-enhancing factor in 2018 cholesterol guidelines. J. Clin. Lipidol. 2019, 13, 356–359. [Google Scholar] [CrossRef] [PubMed]
- Sniderman, A.D.; Williams, K.; Contois, J.H.; Monroe, H.M.; McQueen, M.J.; de Graaf, J.; Furberg, C.D. A meta-analysis of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circ. Cardiovasc. Qual. Outcomes 2011, 4, 337–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhu, P.K.; Musaad, S.M.; Remaley, A.T.; Buehler, S.S.; Strider, S.; Derzon, J.H.; Vesper, H.W.; Ranne, A.; Shaw, C.S.; Christenson, R.H. Lipoprotein Biomarkers and Risk of Cardiovascular Disease: A Laboratory Medicine Best Practices (LMBP) Systematic Review. J. Appl. Lab. Med. 2016, 1, 214–229. [Google Scholar] [CrossRef] [Green Version]
- Pedro-Botet, J.; Mantilla-Morató, T.; Díaz-Rodríguez, Á.; Brea-Hernando, Á.; González-Santos, P.; Hernández-Mijares, A.; Pintó, X.; Millán Núñez-Cortés, J. El papel de la dislipemia aterogénica en las guías de práctica clínica [The role of atherogenic dyslipidaemia in clinical practice guidelines]. Clin. Investig. Arterioscler. 2016, 28, 65–70. [Google Scholar]
- de Nijs, T.; Sniderman, A.; de Graaf, J. Apo B versus non-HDL-cholesterol: Diagnosis and cardiovascular risk assessment. Crit. Rev. Clin. Laboratory Sci. 2013, 50, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. ESC Scientific Document Group. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Barter, P.J.; Ballantyne, C.M.; Carmena, R.; Castro Cabezas, M.; Chapman, M.J.; Couture, P.; de Graaf, J.; Durrington, P.N.; Faergeman, O.; Frohlich, J.; et al. Apo B versus cholesterol in estimating cardiovascular risk and in guiding therapy: Report of the thirty-person/ten-country panel. J. Int. Med. 2006, 259, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Holewijn, S.; den Heijer, M.; van Tits, L.J.; Swinkels, D.W.; Stalenhoef, A.F.; de Graaf, J. Apolipoprotein B, non-HDL cholesterol and LDL cholesterol for identifying individuals at increased cardiovascular risk. J. Intern. Med. 2010, 268, 567–577. [Google Scholar] [CrossRef]
- Carr, S.S.; Hooper, A.J.; Sullivan, D.R.; Burnett, J.R. Non-HDL-cholesterol and apolipoprotein B compared with LDL-cholesterol in atherosclerotic cardiovascular disease risk assessment. Pathology 2019, 51, 148–154. [Google Scholar] [CrossRef]
- Langlois, M.R.; Sniderman, A.D. Non-HDL Cholesterol or apoB: Which to Prefer as a Target for the Prevention of Atherosclerotic Cardiovascular Disease? Curr. Cardiol. Rep. 2020, 22, 67. [Google Scholar] [CrossRef] [PubMed]
- Meeusen, J.W.; Donato, L.J.; Jaffe, A.S. Should apolipoprotein B replace LDL cholesterol as therapeutic targets are lowered? Curr. Opin. Lipidol. 2016, 27, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Sampson, U.K.; Fazio, S.; Linton, M.F. Residual cardiovascular risk despite optimal LDL cholesterol reduction with statins: The evidence, etiology, and therapeutic challenges. Curr. Atheroscler. Rep. 2014, 14, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Jones, D.; Adams, R.J.; Brown, T.M.; Carnethon, M.; Dai, S.; De Simone, G.; Ferguson, T.B.; Ford, E.; Furie, K.; Gillespie, C.; et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Executive summary: Heart disease and stroke statistics--2010 update: A report from the American Heart Association. Circulation 2010, 121, 948–954. [Google Scholar]
- Sniderman, A.D.; Pencina, M.; Thanassoulis, G. ApoB: The power of physiology to transform the prevention of cardiovascular disease. Circ. Res. 2019, 124, 1425–1427. [Google Scholar] [CrossRef]
- Cantey, E.P.; Wilkins, J.T. Discordance between lipoprotein particle number and cholesterol content: An update. Curr. Opin. Endocrinol. Diabetes Obes. 2018, 25, 130–136. [Google Scholar] [CrossRef]
- Sniderman, A.D.; Vu, H.; Cianflone, K. The effect of moderate hypertriglyceridemia on the relation of plasma total and LDL apoB levels. Atherosclerosis 1991, 89, 109–116. [Google Scholar] [CrossRef]
- Durrington, P.N.; Bolton, C.N.; Hartog, H. Serum and lipoprotein apolipoprotein B levels in normal subjects and patients with hyperlipoproteinemia. Clin. Chim. Acta 1978, 82, 151–160. [Google Scholar] [CrossRef]
- Benn, M.; Nordestgaard, B.G.; Jensen, G.B.; Tybjaerg-Hansen, A. Improving prediction of ischemic cardiovascular disease in the general population using apolipoprotein B: The Copenhagen City Heart Study. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 661–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pischon, T.; Girman, C.J.; Sacks, F.M.; Rifai, N.; Stampfer, M.J.; Rimm, E.B. Non-high-density lipoprotein cholesterol and apolipoprotein B in the prediction of coronary heart disease in men. Circulation 2005, 112, 3375–3383. [Google Scholar] [CrossRef] [Green Version]
- Walldius, G.; Jungner, I.; Holme, I.; Aastveit, A.H.; Kolar, W.; Steiner, E. High apolipoprotein B, low apolipoprotein A-I, and improvement in the prediction of fatal myocardial infarction (AMORIS study): A prospective study. Lancet 2001, 358, 2026–2033. [Google Scholar] [CrossRef]
- Ohwada, T.; Sakamoto, T.; Kanno, Y.; Yokokawa, S.; Amami, K.; Nakazato, K.; Takeishi, Y.; Watanabe, K. Apolipoprotein B correlates with intra-plaque necrotic core volume in stable coronary artery disease. PLoS ONE 2019, 14, e0212539. [Google Scholar] [CrossRef] [Green Version]
- Krauss, R.M. Atherogenicity of triglyceride-rich lipoproteins. Am. J. Cardiol. 1998, 81, 13B–17B. [Google Scholar] [CrossRef]
- Graziani, M.S.; Zanolla, L.; Righetti, G.; Marchetti, C.; Mocarelli, P.; Marcovina, S.M. Plasma apolipoproteins A-I and B in survivors of myocardial infarction and in a control group. Clin. Chem. 1998, 44, 134–140. [Google Scholar] [CrossRef]
- Westerveld, H.T.; van Lennep, J.E.; van Lennep, H.W.; Liem, A.H.; de Boo, J.A.; van der Schouw, Y.T.; Erkelens, D.W. Apolipoprotein B and coronary artery disease in women: A cross-sectional study in women undergoing their first coronary angiography. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1101–1107. [Google Scholar] [CrossRef] [Green Version]
- Sniderman, A.D. Counterpoint: To (measure apo) B or not to (measure apo) B: A critique of modern medical decision-making. Clin. Chem. 1997, 43, 1310–1314. [Google Scholar] [CrossRef]
- Sacks, F.M.; Alaupovic, P.; Moye, L.A.; Cole, T.G.; Sussex, B.; Stampfer, M.J.; Pfeffer, M.A.; Braunwald, E. VLDL, apolipoproteins B, CIII, and E, and risk of recurrent coronary events in the Cholesterol and Recurrent Events (CARE) trial. Circulation 2000, 102, 1886–1892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sniderman, A.D.; de Graaf, J.; Thanassoulis, G.; Tremblay, A.J.; Martin, S.S.; Couture, P. The spectrum of type III hyperlipoproteinemia. J. Clin. Lipidol. 2018, 12, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Steffen, B.T.; Guan, W.; Remaley, A.T.; McConnell, J.P.; Palamalai, V.; Tsai, M.Y. A comparison of three apolipoprotein B methods and their associations with incident coronary heart disease risk over a 12-year follow-up period: The Multi-Ethnic Study of Atherosclerosis. J. Clin. Lipidol. 2018, 12, 300–304. [Google Scholar] [CrossRef]
- Hermans, M.P.; Sacks, F.M.; Ahn, S.A.; Rousseau, M.F. Non-HDL-cholesterol as valid surrogate to apolipoprotein B100 measurement in diabetes: Discriminant Ratio and unbiased equivalence. Cardiovasc. Diabetol. 2011, 10, 20. [Google Scholar] [CrossRef] [Green Version]
- Hwang, Y.C.; Ahn, H.Y.; Lee, W.J.; Park, C.Y.; Park, S.W. An equation to estimate the concentration of serum apolipoprotein B. PLoS ONE 2012, 7, e51607. [Google Scholar] [CrossRef]
- de Vries, M.A.; van Santen, S.S.; Klop, B.; van der Meulen, N.; van Vliet, M.; van de Geijn, G.M.; van der Zwan-van Beek, E.M.; Birnie, E.; Liem, A.H.; de Herder, W.W.; et al. Erythrocyte-bound apolipoprotein B in atherosclerosis and mortality. Eur. J. Clin. Investig. 2017, 47, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Walldius, G.; Jungner, I. The apoB/apoA-I ratio: A strong, new risk factor for cardiovascular disease and a target for lipid-lowering therapy–a review of the evidence. J. Intern. Med. 2006, 259, 493–519. [Google Scholar] [CrossRef]
- Bachorik, P.S.; Lovejoy, K.L.; Carroll, M.D.; Johnson, C.L. Apolipoprotein B and AI distributions in the United States, 1988-1991: Results of the National Health and Nutrition Examination Survey III (NHANES III). Clin. Chem. 1997, 43, 2364–2378. [Google Scholar] [CrossRef] [Green Version]
- Barter, P.J.; Rye, K.A. The rationale for using apoA-I as a clinical marker of cardiovascular risk. J. Intern. Med. 2006, 239, 447–454. [Google Scholar] [CrossRef]
- Walldius, G.; Jungner, I. Is there a better marker of cardiovascular risk than LDL cholesterol? Apolipoproteins B and A-I--new risk factors and targets for therapy. Nutr. Metab. Cardiovasc. Dis. 2007, 17, 565–571. [Google Scholar] [CrossRef]
- Holme, I.; Aastveit, A.H.; Jungner, I.; Walldius, G. Relationships between lipoprotein components and risk of myocardial infarction: Age, gender and short versus longer follow-up periods in the Apolipoprotein Mortality Risk Study (AMORIS). J. Intern. Med. 2008, 264, 30–38. [Google Scholar] [CrossRef]
- Yusuf, S.; Hawken, S.; Ounpuu, S.; Dans, T.; Avezum, A.; Lanas, F.; McQueen, M.; Budaj, A.; Pais, P.; Varigos, J. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): Case-control study. Lancet 2004, 264, 937–952. [Google Scholar] [CrossRef]
- Tian, M.; Li, R.; Shan, Z.; Wang, D.W.; Jiang, J.; Cui, G. Comparison of apolipoprotein B/A1 ratio, Framingham risk score and TC/HDL-c for predicting clinical outcomes in patients undergoing percutaneous coronary intervention. Lipids Health Dis. 2019, 18, 202. [Google Scholar] [CrossRef] [Green Version]
- Ivert, T.; Malmström, H.; Hammar, N.; Carlsson, A.C.; Wändell, P.E.; Holzmann, M.J.; Jungner, I.; Ärnlöv, J.; Walldius, G. Cardiovascular events in patients under age fifty with early findings of elevated lipid and glucose levels-The AMORIS study. PLoS ONE 2018, 13, e0201972. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Johnson, J.; Fisher, R.M.; Romero-Corral, A.; Somers, V.K.; Lopez-Jimenez, F.; Ohrvik, J.; Walldius, G.; Hellenius, M.L.; Hamsten, A. Concentration of apolipoprotein B is comparable with the apolipoprotein B/apolipoprotein A-I ratio and better than routine clinical lipid measurements in predicting coronary heart disease mortality: Findings from a multi-ethnic US population. Eur. Heart J. 2009, 30, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Sierra-Johnson, J.; Romero-Corral, A.; Somers, V.K.; Lopez-Jimenez, F.; Walldius, G.; Hamsten, A.; Hellenius, M.L.; Fisher, R.M. ApoB/apoA-I ratio: An independent predictor of insulin resistance in US non-diabetic subjects. Eur. Heart J. 2007, 28, 2637–2643. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.S.; Qasim, A.N.; Mehta, N.N.; Wolfe, M.; Terembula, K.; Schwartz, S.; Iqbal, N.; Schutta, M.; Bagheri, R.; Reilly, M.P. Apolipoprotein B but not LDL cholesterol is associated with coronary artery calcification in type 2 diabetic whites. Diabetes 2009, 58, 1887–1892. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Shang, D.; Shao, J.; Dai, S.; Ge, X.; Hao, C.; Zhu, T. Prognostic significance of carotid plaque presence in peritoneal dialysis patients and its association with the apolipoprotein B/apolipoprotein A1 ratio. Nephrology 2020, 25, 919–928. [Google Scholar] [CrossRef]
- Karasek, D.; Vaverkova, H.; Cibickova, L.; Gajdova, J.; Kubickova, V. Apolipoprotein B vs non-high-density lipoprotein cholesterol: Association with endothelial hemostatic markers and carotid intima-media thickness. J. Clin. Lipidol. 2017, 11, 442–449. [Google Scholar] [CrossRef] [PubMed]
- McClelland, R.L.; Jorgensen, N.W.; Budoff, M.; Blaha, M.J.; Post, W.S.; Kronmal, R.A.; Bild, D.E.; Shea, S.; Liu, K.; Watson, K.E.; et al. 10-year coronary heart disease risk prediction using coronary artery calcium and traditional risk factors: Derivation in the multi-ethnic study of atherosclerosis with validation in the Heinz Nixdorf recall study and the Dallas Heart Study. J. Am. Coll. Cardiol. 2015, 66, 1643–1653. [Google Scholar] [CrossRef] [Green Version]
- Sequí-Domínguez, I.; Cavero-Redondo, I.; Álvarez-Bueno, C.; Pozuelo-Carrascosa, D.P.; Nuñez de Arenas-Arroyo, S.; Martínez-Vizcaíno, V. Accuracy of Pulse Wave Velocity Predicting Cardiovascular and All-Cause Mortality. A Systematic Review and Meta-Analysis. J. Clin. Med. 2020, 9, 2080. [Google Scholar] [CrossRef]
- Jagannathan, R.; Patel, S.A.; Ali, M.K.; Narayan, K. Global updates on cardiovascular disease mortality trends and attribution of traditional risk factors. Curr. Diab. Rep. 2019, 19, 44. [Google Scholar] [CrossRef]
- Dieter, B.P.; Tuttle, K.R. Dietary strategies for cardiovascular health. Trends Cardiovasc. Med. 2017, 27, 295–313. [Google Scholar] [CrossRef] [PubMed]
- Isakadze, N.; Mehta, P.K.; Law, K.; Dolan, M.; Lundberg, G.P. Addressing the gap in physician preparedness to assess cardiovascular risk in women: A comprehensive approach to cardiovascular risk assessment in women. Curr. Treat. Options Cardiovasc. Med. 2019, 21, 47. [Google Scholar] [CrossRef]
- Cannon, C.P. Cardiovascular disease and modifiable cardiometabolic risk factors. Clin. Cornerstone 2007, 8, 11–28. [Google Scholar] [CrossRef]
- D’Agostino, R.B., Sr.; Vasan, R.S.; Pencina, M.J.; Wolf, P.A.; Cobain, M.; Massaro, J.M.; Kannel, W.B. General cardiovascular risk profile for use in primary care: The Framingham Heart Study. Circulation 2008, 117, 743–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, P.W.; D’Agostino, R.B.; Levy, D.; Belanger, A.M.; Silbershatz, H.; Kannel, W.B. Prediction of coronary heart disease using risk factor categories. Circulation 1998, 97, 1837–1847. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.; Sniderman, A.D.; Sattar, N.; D’Agostino, R., Jr.; Wagenknecht, L.E.; Haffner, S.M. Comparison of the associations of apolipoprotein B and low-density lipoprotein cholesterol with other cardiovascular risk factors in the Insulin Resistance Atherosclerosis Study (IRAS). Circulation 2003, 108, 2312–2316. [Google Scholar] [CrossRef] [Green Version]
- Rosenblit, P.D. Extreme Atherosclerotic Cardiovascular Disease (ASCVD) risk recognition. Curr. Diab. Rep. 2019, 19, 61. [Google Scholar] [CrossRef] [PubMed]
- Enkhmaa, B.; Anuurad, E.; Zhang, W.; Berglund, L. Significant associations between lipoprotein(a) and corrected apolipoprotein B-100 levels in African-Americans. Atherosclerosis 2014, 235, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Tsimikas, S.; Clopton, P.; Brilakis, E.S.; Marcovina, S.M.; Khera, A.; Miller, E.R.; de Lemos, J.A.; Witztum, J.L. Relationship of oxidized phospholipids on apolipoprotein B-100 particles to race/ethnicity, apolipoprotein(a) isoform size, and cardiovascular risk factors: Results from the Dallas Heart Study. Circulation 2009, 119, 1711–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthan, N.R.; Jalbert, S.M.; Barrett, P.H.; Dolnikowski, G.G.; Schaefer, E.J.; Lichtenstein, A.H. Gender-specific differences in the kinetics of nonfasting TRL, IDL, and LDL apolipoprotein B-100 in men and premenopausal women. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1838–1843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungner, I.; Marcovina, S.M.; Walldius, G.; Holme, I.; Kolar, W.; Steiner, E. Apolipoprotein B and A-I values in 147 576 Swedish males and females, standardized according to the World Health Organization–International Federation of Clinical Chemistry First International Reference Materials. Clin. Chem. 1998, 44, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Matthan, N.R.; Jalbert, S.M.; Lamon-Fava, S.; Dolnikowski, G.G.; Welty, F.K.; Barrett, H.R.; Schaefer, E.J.; Lichtenstein, A.H. TRL, IDL, and LDL apolipoprotein B-100 and HDL apolipoprotein A-I kinetics as a function of age and menopausal status. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1691–1696. [Google Scholar] [CrossRef] [PubMed]
- Dansinger, M.L.; Williams, P.T.; Superko, H.R.; Schaefer, E.J. Effects of weight change on apolipoprotein B-containing emerging atherosclerotic cardiovascular disease (ASCVD) risk factors. Lipids Health Dis. 2019, 18, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundfør, T.M.; Svendsen, M.; Heggen, E.; Dushanov, S.; Klemsdal, T.O.; Tonstad, S. BMI modifies the effect of dietary fat on atherogenic lipids: A randomized clinical trial. Am. J. Clin. Nutr. 2019, 110, 832–841. [Google Scholar] [CrossRef]
- Lamantia, V.; Sniderman, A.; Faraj, M. Nutritional management of hyperapoB. Nutr. Res. Rev. 2016, 29, 202–233. [Google Scholar] [CrossRef]
- Wilkins, J.T.; Li, R.C.; Sniderman, A.; Chan, C.; Lloyd-Jones, D.M. Discordance between apolipoprotein B and LDL-cholesterol in young adults predicts coronary artery calcification. J. Am. Coll. Cardiol. 2016, 67, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Sniderman, A.D.; Islam, S.; McQueen, M.; Pencina, M.; Furberg, C.D.; Thanassoulis, G.; Yusuf, S. Age and cardiovascular risk attributable to apolipoprotein B, low-density lipoprotein cholesterol or non-high-density lipoprotein cholesterol. J. Am. Heart Assoc. 2016, 5, e003665. [Google Scholar] [CrossRef] [PubMed]
- Varvel, S.A.; Dayspring, T.D.; Edmonds, Y.; Thiselton, D.L.; Ghaedi, L.; Voros, S.; McConnell, J.P.; Sasinowski, M.; Dall, T.; Warnick, G.R. Discordance between apolipoprotein B and low-density lipoprotein particle number is associated with insulin resistance in clinical practice. J. Clin. Lipidol. 2015, 9, 247–255. [Google Scholar] [CrossRef] [Green Version]
- Paredes, S.; Fonseca, L.; Ribeiro, L.; Ramos, H.; Oliveira, J.C.; Palma, I. Novel and traditional lipid profiles in metabolic syndrome reveal a high atherogenicity. Sci. Rep. 2019, 9, 11792. [Google Scholar] [CrossRef] [Green Version]
- Faraj, M.; Messier, L.; Bastard, J.P.; Tardif, A.; Godbout, A.; Prud’homme, D.; Rabasa-Lhoret, R. Apolipoprotein B: A predictor of inflammatory status in postmenopausal overweight and obese women. Diabetologia 2006, 49, 1637–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, Y.C.; Ahn, H.Y.; Park, S.W.; Park, C.Y. Apolipoprotein B and non-HDL cholesterol are more powerful predictors for incident type 2 diabetes than fasting glucose or glycated hemoglobin in subjects with normal glucose tolerance: A 3.3-year retrospective longitudinal study. Acta Diabetol. 2014, 51, 941–946. [Google Scholar] [CrossRef]
- Cartier, L.J.; St-Coeur, S.; Robin, A.; Lagace, M.; Douville, P. Impact of the Martin/Hopkins modified equation for estimating LDL-C on lipid target attainment in a high risk patient population. Clin. Biochem. 2020, 76, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.; Ascaso, J.F. Control of the overall lipid profile. Clin. Investig. Arterioscler. 2019, 31, 34–41. [Google Scholar] [PubMed]
- Lamantia, V.; Bissonnette, S.; Wassef, H.; Cyr, Y.; Baass, A.; Dufour, R.; Rabasa-Lhoret, R.; Faraj, M. ApoB-lipoproteins and dysfunctional white adipose tissue: Relation to risk factors for type 2 diabetes in humans. J. Clin. Lipidol. 2017, 11, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Ley, S.H.; Harris, S.B.; Connelly, P.W.; Mamakeesick, M.; Gittelsohn, J.; Wolever, T.M.; Hegele, R.A.; Zinman, B.; Hanley, A.J. Association of apolipoprotein B with incident type 2 diabetes in an aboriginal Canadian population. Clin. Chem. 2010, 56, 666–670. [Google Scholar] [CrossRef] [Green Version]
- Salomaa, V.; Havulinna, A.; Saarela, O.; Zeller, T.; Jousilahti, P.; Jula, A.; Muenzel, T.; Aromaa, A.; Evans, A.; Kuulasmaa, K.; et al. Thirty-one novel biomarkers as predictors for clinically incident diabetes. PLoS ONE 2010, 5, e10100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onat, A.; Can, G.; Hergenç, G.; Yazici, M.; Karabulut, A.; Albayrak, S. Serum apolipoprotein B predicts dyslipidemia, metabolic syndrome and, in women, hypertension and diabetes, independent of markers of central obesity and inflammation. Int. J. Obes. 2007, 31, 1119–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhailidis, D.P.; Elisaf, M.; Rizzo, M.; Berneis, K.; Griffin, B.; Zambon, A.; Athyros, V.; de Graaf, J.; März, W.; Parhofer, K.G.; et al. “European panel on low density lipoprotein (LDL) subclasses”: A statement on the pathophysiology, atherogenicity and clinical significance of LDL subclasses: Executive summary. Curr. Vasc. Pharmacol. 2011, 9, 531–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Ren, F.G.; Guan, K.P. Predictive Value of Small Dense Low-Density Lipoprotein in Coronary Heart Disease in the Chinese Population. Clin. Lab. 2021, 67, 5. [Google Scholar] [CrossRef]
- Berneis, K.K.; Krauss, R.M. Metabolic origins and clinical significance of LDL heterogeneity. J. Lipid Res. 2002, 43, 1363–1379. [Google Scholar] [CrossRef] [Green Version]
- Packard, C.J.; Demant, T.; Stewart, J.P.; Bedford, D.; Caslake, M.J.; Schwertfeger, G.; Bedynek, A.; Shepherd, J.; Seidel, D. Apolipoprotein B metabolism and the distribution of VLDL and LDL subfractions. J. Lipid Res. 2000, 41, 305–318. [Google Scholar] [CrossRef]
- Hayashi, T.; Hirano, T.; Shiobara, T.; Suguro, T.; Adachi, M. Small dense LDL concentration is closely associated with serum apolipoprotein B, comparisons of non-LDL cholesterol or LDL cholesterol. Rinsho Byori 2006, 54, 569–575. [Google Scholar] [PubMed]
- Vekic, J.; Zeljkovic, A.; Jelic-Ivanovic, Z.; Spasojevic-Kalimanovska, V.; Bogavac-Stanojevic, N.; Memon, L.; Spasic, S. Small, dense LDL cholesterol and apolipoprotein B: Relationship with serum lipids and LDL size. Atherosclerosis 2009, 207, 496–501. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.; Spinas, G.A.; Cesur, M.; Ozbalkan, Z.; Rini, G.B.; Berneis, K. Atherogenic lipoprotein phenotype and LDL size and subclasses in drug-naïve patients with early rheumatoid arthritis. Atherosclerosis 2009, 207, 502–506. [Google Scholar] [CrossRef]
- Olusi, S.O.; George, S. Prevalence of LDL atherogenic phenotype in patients with systemic lupus erythematosus. Vasc. Health Risk Manag. 2011, 7, 75–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizvi, A.A.; Cuadra, S.; Nikolic, D.; Giglio, R.V.; Montalto, G.; Rizzo, M. Gestational diabetes and the metabolic syndrome: Can obesity and small, dense low density lipoproteins be key mediators of this association? Curr. Pharm. Biotechnol. 2014, 15, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.; Berneis, K.; Altinova, A.E.; Toruner, F.B.; Akturk, M.; Ayvaz, G.; Rini, G.B.; Spinas, G.A.; Arslan, M. Atherogenic lipoprotein phenotype and LDL size and subclasses in women with gestational diabetes. Diabet Med. 2008, 25, 1406–1411. [Google Scholar] [CrossRef]
- Hirano, T. Small Dense LDL Tied to Diabetic Retinopathy-Similarity to Atherosclerosis. J. Atheroscler. Thromb. 2021, in press. [Google Scholar] [CrossRef]
- Chapman, M.J.; Orsoni, A.; Tan, R.; Mellett, N.A.; Nguyen, A.; Robillard, P.; Giral, P.; Thérond, P.; Meikle, P.J. LDL subclass lipidomics in atherogenic dyslipidemia: Effect of statin therapy on bioactive lipids and dense LDL. Lipid Res. 2020, 61, 911–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viktorinova, A.; Malickova, D.; Svitekova, K.; Choudhury, S.; Krizko, M. Low-density lipoprotein cholesterol-to-apolipoprotein B ratio as a potential indicator of LDL particle size and plasma atherogenicity in type 2 diabetes. Diabetes Res. Clin. Pract. 2021, 176, 108858. [Google Scholar] [CrossRef] [PubMed]
- Hirano, T.; Kodera, R.; Hirashima, T.; Suzuki, N.; Aoki, E.; Hosoya, M.; Oshima, T.; Hayashi, T.; Koba, S.; Ohta, M.; et al. Metabolic Properties of Low density Lipoprotein (LDL) Triglycerides in Patients with Type 2 Diabetes, Comparison with Small Dense LDL-Cholesterol. J. Atheroscler. Thromb. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Khoo, C.; Furtado, J.; Sacks, F.M. Apolipoprotein C-III and the metabolic basis for hypertriglyceridemia and the dense low-density lipoprotein phenotype. Circulation 2010, 121, 1722–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, M.; Pernice, V.; Frasheri, A.; Berneis, K. Atherogenic lipoprotein phenotype and LDL size and subclasses in patients with peripheral arterial disease. Atherosclerosis 2008, 197, 237–241. [Google Scholar] [CrossRef]
- Rizzo, M.; Berneis, K. Who needs to care about small, dense low-density lipoproteins? Int. J. Clin. Pract. 2007, 61, 1949–1956. [Google Scholar] [CrossRef]
- Kou, M.; Ding, N.; Ballew, S.H.; Salameh, M.J.; Martin, S.S.; Selvin, E.; Heiss, G.; Ballantyne, C.M.; Matsushita, K.; Hoogeveen, R.C. Conventional and Novel Lipid Measures and Risk of Peripheral Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1229–1238. [Google Scholar] [CrossRef]
- Sjögren, P.; Fredrikson, G.N.; Samnegard, A.; Ericsson, C.G.; Ohrvik, J.; Fisher, R.M.; Nilsson, J.; Hamsten, A. High plasma concentrations of autoantibodies against native peptide 210 of apoB-100 are related to less coronary atherosclerosis and lower risk of myocardial infarction. Eur. Heart J. 2008, 29, 2218–2226. [Google Scholar] [CrossRef]
- Barter, P.J. CETP and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2029–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barter, P.J.; Brewer, H.B., Jr.; Chapman, M.J.; Hennekens, C.H.; Rader, D.J.; Tall, A.R. Cholesteryl ester transfer protein: A novel target for raising HDL and inhibiting atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R. Functions of cholesterol ester transfer protein and relationship to coronary artery disease risk. J. Clin. Lipidol. 2010, 4, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Ference, B.A.; Kastelein, J.; Ginsberg, H.N.; Chapman, M.J.; Nicholls, S.J.; Ray, K.K.; Packard, C.J.; Laufs, U.; Brook, R.D.; Oliver-Williams, C.; et al. Association of genetic variants related to CETP inhibitors and statins with lipoprotein levels and cardiovascular risk. J. Am. Med. Assoc. 2017, 318, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, S.J.; Tuzcu, E.M.; Brennan, D.M.; Tardif, J.C.; Nissen, S.E. Cholesteryl ester transfer protein inhibition, high-density lipoprotein raising, and progression of coronary atherosclerosis: Insights from ILLUSTRATE (Investigation of Lipid Level Management Using Coronary Ultrasound to Assess Reduction of Atherosclerosis by CETP Inhibition and HDL Elevation). Circulation 2008, 118, 2506–2514. [Google Scholar]
- Hegele, R.A. CETP inhibitors–a new inning? N. Engl. J. Med. 2017, 377, 1284–1285. [Google Scholar] [CrossRef]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Illuminate Investigators. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lincoff, A.M.; Nicholls, S.J.; Riesmeyer, J.S.; Barter, P.J.; Brewer, H.B.; Fox, K.A.A.; Gibson, C.M.; Granger, C.; Menon, V.; Montalescot, G.; et al. Evacetrapib and cardiovascular outcomes in high-risk vascular disease. N. Engl. J. Med. 2017, 376, 1933–1942. [Google Scholar] [CrossRef]
- Bowman, L.; Hopewell, J.C.; Chen, F.; Wallendszus, K.; Stevens, W.; Collins, R.; Wiviott, S.D.; Cannon, C.P.; Braunwald, E.; Sammons, E.; et al. Effects of Anacetrapib in Patients with Atherosclerotic Vascular Disease. N. Engl. J. Med. 2017, 377, 1217–1227. [Google Scholar] [CrossRef] [PubMed]
- Grabie, M.; Tai, C.H.; Frishman, W.H. Is Anacetrapib Better Than Its CETP Inhibitor Counterparts? Cardiol. Rev. 2019, 27, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Blauw, L.L.; Noordam, R.; Soidinsalo, S.; Blauw, C.A.; Li-Gao, R.; de Mutsert, R.; Berbée, J.F.P.; Wang, Y.; van Heemst, D.; Rosendaal, F.R.; et al. Mendelian randomization reveals unexpected effects of CETP on the lipoprotein profile. Eur. J. Hum. Genet. 2019, 27, 422–431. [Google Scholar] [CrossRef] [Green Version]
- Ference, B.A.; Kastelein, J.; Ray, K.K.; Ginsberg, H.N.; Chapman, M.J.; Packard, C.J.; Laufs, U.; Oliver-Williams, C.; Wood, A.M.; Butterworth, A.S.; et al. Association of Triglyceride-Lowering LPL Variants and LDL-C-Lowering LDLR Variants With Risk of Coronary Heart Disease. J. Am. Med. Assoc. 2019, 321, 364–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganji, S.H.; Kamanna, V.S.; Kashyap, M.L. Niacin and cholesterol: Role in cardiovascular disease (review). J. Nutr. Biochem. 2003, 14, 298–305. [Google Scholar] [CrossRef]
- Bays, H.E.; Shah, A.; Lin, J.; Sisk, C.M.; Dong, Q.; Maccubbin, D. Consistency of extended-release niacin/laropiprant effects on Lp(a), ApoB, non-HDL-C, Apo A1, and ApoB/ApoA1 ratio across patient subgroups. Am. J. Cardiovasc. Drugs 2012, 12, 197–206. [Google Scholar] [CrossRef]
- Birjmohun, R.S.; Hutten, B.A.; Kastelein, J.J.; Stroes, E.S. Increasing HDL cholesterol with extended-release nicotinic acid: From promise to practice. Neth. J. Med. 2004, 62, 229–234. [Google Scholar] [PubMed]
- Cheng, K.; Wu, T.J.; Wu, K.K.; Sturino, C.; Metters, K.; Gottesdiener, K.; Wright, S.D.; Wang, Z.; O’Neill, G.; Lai, E.; et al. Antagonism of the prostaglandin D2 receptor 1 suppresses nicotinic acid-induced vasodilation in mice and humans. Proc. Natl. Acad. Sci. USA 2006, 103, 6682–6687. [Google Scholar] [CrossRef] [Green Version]
- Illingworth, D.R.; Stein, E.A.; Mitchel, Y.B.; Dujovne, C.A.; Frost, P.H.; Knopp, R.H.; Tun, P.; Zupkis, R.V.; Greguski, R.A. Comparative effects of lovastatin and niacin in primary hypercholesterolemia: A prospective trial. Arch. Intern. Med. 1994, 154, 1586–1595. [Google Scholar] [CrossRef]
- Knopp, R.H.; Ginsberg, J.; Albers, J.J.; Hoff, C.; Ogilvie, J.T.; Warnick, G.R.; Burrows, E.; Retzlaff, B.; Poole, M. Contrasting effects of unmodified and time-release forms of niacin on lipoproteins in hyperlipidemic subjects: Clues to mechanism of action of niacin. Metabolism 1985, 34, 642–650. [Google Scholar] [CrossRef]
- Mills, E.; Prousky, J.; Raskin, G.; Gagnier, J.; Rachlis, B.; Montori, V.M.; Juurlink, D. The safety of over-the-counter niacin: A randomized placebo-controlled trial [ISRCTN18054903]. BMC Clin. Pharmacol. 2003, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- Paolini, J.F.; Mitchel, Y.B.; Reyes, R.; Kher, U.; Lai, E.; Watson, D.J.; Norquist, J.M.; Meehan, A.G.; Bays, H.E.; Davidson, M.; et al. Effects of laropiprant on nicotinic acid-induced flushing in patients with dyslipidemia. Am. J. Cardiol. 2008, 101, 625–630. [Google Scholar] [CrossRef]
- Sturino, C.F.; O’Neill, G.; Lachance, N.; Boyd, M.; Berthelette, C.; Labelle, M.; Li, L.; Roy, B.; Scheigetz, J.; Tsou, N.; et al. Discovery of a potent and selective prostaglandin D2 receptor antagonist, [(3R)-4-(4-chloro-benzyl)-7-fluoro-5-(methylsulfonyl)-1,2,3,4-tetrahydrocy clopenta[b]indol-3-yl]-acetic acid (MK-0524). J. Med. Chem. 2007, 50, 794–806. [Google Scholar] [CrossRef]
- Ballantyne, C.; Gleim, G.; Liu, N.; Sisk, C.M.; Johnson-Levonas, A.O.; Mitchel, Y. Efficacy and safety profile of coadministered ER niacin/laropiprant and simvastatin in dyslipidaemia. J. Clin. Lipidol. 2012, 6, 235–243. [Google Scholar] [CrossRef]
- Maccubbin, D.; Bays, H.E.; Olsson, A.G.; Elinoff, V.; Elis, A.; Mitchel, Y.; Sirah, W.; Betteridge, A.; Reyes, R.; Yu, Q.; et al. Lipid-modifying efficacy and tolerability of extended-release niacin/laropiprant in patients with primary hypercholesterolaemia or mixed dyslipidaemia. Int. J. Clin. Pract. 2008, 62, 1959–1970. [Google Scholar] [CrossRef]
- Haynes, R.; Valdes-Marquez, E.; Hopewell, J.C.; Chen, F.; Li, J.; Parish, S.; Landray, M.J.; Armitage, J.; Baigent, C.; HPS2-THRIVE Collaborative Group, HPS2-THRIVE Writing Committee members; et al. Serious Adverse Effects of Extended-release Niacin/Laropiprant: Results from the Heart Protection Study 2-Treatment of HDL to Reduce the Incidence of Vascular Events (HPS2-THRIVE) Trial. Clin. Ther. 2019, 41, 1767–1777. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Liu, Y.; Kwok, S.; Hama, S.; France, M.; Eatough, R.; Pemberton, P.; Schofield, J.; Siahmansur, T.J.; Malik, R.; et al. Effect of Extended-Release Niacin on High-Density Lipoprotein (HDL) Functionality, Lipoprotein Metabolism, and Mediators of Vascular Inflammation in Statin-Treated Patients. J. Am. Heart. Assoc. 2015, 4, e001508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomlinson, B.; Chan, P.; Zhang, Y.; Liu, Z.; Lam, C.W.K. Pharmacokinetics of current and emerging treatments for hypercholesterolemia. Expert Opin. Drug Metab. Toxicol. 2020, 16, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Colhoun, H.M.; Leiter, L.A.; Müller-Wieland, D.; Cariou, B.; Ray, K.K.; Tinahones, F.J.; Domenger, C.; Letierce, A.; Israel, M.; Samuel, R.; et al. Effect of alirocumab on individuals with type 2 diabetes, high triglycerides, and low high-density lipoprotein cholesterol. Cardiovasc. Diabetol. 2020, 19, 14. [Google Scholar] [CrossRef] [Green Version]
- Ying, Q.; Chan, D.C.; Watts, G.F. New Insights into the Regulation of Lipoprotein Metabolism by PCSK9: Lessons From Stable Isotope Tracer Studies in Human Subjects. Front. Physiol. 2021, 12, 603910. [Google Scholar] [CrossRef]
- Feng, X.; Berklein, F.; Rane, P.B.; Habib, M.; Lin, P.J. Patient Characteristics and Treatment Patterns among Medicare Beneficiaries Initiating PCSK9 Inhibitor Therapy. Cardiovasc. Drugs Ther. 2021, 35, 965–973. [Google Scholar] [CrossRef]
- Tokgozoglu, L.; Kayikcioglu, M. Familial Hypercholesterolemia: Global Burden and Approaches. Curr. Cardiol. Rep. 2021, 23, 151. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Li, Q.; Tian, X.Q.; Qian, H.Y.; Yang, Y.J. Mipomersen is a promising therapy in the management of hypercholesterolemia: A meta-analysis of randomized controlled trials. Am. J. Cardiovasc. Drugs 2014, 14, 367–376. [Google Scholar] [CrossRef]
- Mullick, A.E.; Fu, W.; Graham, M.J.; Lee, R.G.; Witchell, D.; Bell, T.A.; Whipple, C.P.; Crooke, R.M. Antisense oligonucleotide reduction of apoB-ameliorated atherosclerosis in LDL receptor-deficient mice. J. Lipid Res. 2011, 52, 885–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Zhang, Y.; Xu, C.B. Apolipoprotein B, the villain in the drama? Eur. J. Pharmacol. 2015, 748, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Fogacci, F.; Ferri, N.; Toth, P.P.; Ruscica, M.; Corsini, A.; Cicero, A. Efficacy and Safety of Mipomersen: A Systematic Review and Meta-Analysis of Randomized Clinical Trials. Drugs 2019, 79, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.drugs.com/history/kynamro.html (accessed on 4 August 2021).
- Sirwi, A.; Hussain, M.M. Lipid transfer proteins in the assembly of apoB-containing lipoproteins. J. Lipid Res. 2018, 59, 1094–1102. [Google Scholar] [CrossRef] [Green Version]
- Sahebkar, A.; Watts, G.F. New therapies targeting apoB metabolism for high-risk patients with inherited dyslipidaemias: What can the clinician expect? Cardiovasc. Drugs Ther. 2013, 27, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.A. Recent advances in elucidating the role of the microsomal triglyceride transfer protein in apolipoprotein B lipoprotein assembly. Curr. Opin. Lipidol. 1997, 8, 131–137. [Google Scholar] [CrossRef]
- Alonso, R.; Cuevas, A.; Mata, P. Lomitapide: A review of its clinical use, efficacy, and tolerability. Core Evid. 2019, 14, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Kolovou, G.; Diakoumakou, O.; Kolovou, V.; Fountas, E.; Stratakis, S.; Zacharis, E.; Liberopoulos, E.N.; Matsouka, F.; Tsoutsinos, A.; Mastorakou, I.; et al. Microsomal triglyceride transfer protein inhibitor (lomitapide) efficacy in the treatment of patients with homozygous familial hypercholesterolaemia. Eur. J. Prev. Cardiol. 2019, 27, 157–165. [Google Scholar] [CrossRef]
- Spinler, S. The pharmacology and therapeutic use of dabigatran etexilate. J. Clin. Pharmacol. 2013, 53, 1–13. [Google Scholar]
- Cate, H.T. Dabigatran and apolipoprotein B. Heart 2016, 102, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Joseph, P.; Pare, G.; Wallentin, L.; Connolly, S.; Yusuf, S.; Wang, J.; Ezekowitz, M.; Eikelboom, J.; Siegbahn, A.; Reilly, P.; et al. Dabigatran etexilate and reduction in serum apolipoprotein B. Heart 2016, 102, 57–62. [Google Scholar] [CrossRef]
- Zhao, X.; Ma, X.; Luo, X.; Shi, Z.; Deng, Z.; Jin, Y.; Xiao, Z.; Tan, L.; Liu, P.; Jiang, S.; et al. Efficacy and safety of bempedoic acid alone or combining with other lipid-lowering therapies in hypercholesterolemic patients: A meta-analysis of randomized controlled trials. BMC Pharmacol. Toxicol. 2020, 21, 86. [Google Scholar] [CrossRef]
- Ray, K.K.; Bays, H.E.; Catapano, A.L.; Lalwani, N.D.; Bloedon, L.T.; Sterling, L.R.; Robinson, P.L.; Ballantyne, C.M.; CLEAR Harmony Trial. Safety and Efficacy of Bempedoic Acid to Reduce LDL Cholesterol. N. Engl. J. Med. 2019, 380, 1022–1032. [Google Scholar] [CrossRef]
- Ruscica, M.; Zimetti, F.; Adorni, M.P.; Sirtori, C.R.; Lupo, M.G.; Ferri, N. Pharmacological aspects of ANGPTL3 and ANGPTL4 inhibitors: New therapeutic approaches for the treatment of atherogenic dyslipidemia. Pharmacol. Res. 2020, 153, 104653. [Google Scholar] [CrossRef]
- Pu, X.; Sale, M.; Yang, F.; Zhang, Y.; Davis, J.D.; Al-Huniti, N. Population pharmacokinetics and exposure-response modeling for evinacumab in homozygous familial hypercholesterolemia. CPT Pharmacomet. Syst. Pharmacol. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Meng, F.; Yang, W.; Liang, L.; Wang, H.; Fu, Z. Efficacy and Safety of Evinacumab for the Treatment of Hypercholesterolemia: A Meta-Analysis. J. Cardiovasc. Pharmacol. 2021, 78, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Warden, B.A.; Duell, P.B. Evinacumab for treatment of familial hypercholesterolemia. Expert Rev. Cardiovasc. Ther. 2021, 19, 739–751. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Underberg, J.A. Systematic Review: Evaluating the Effect of Lipid-Lowering Therapy on Lipoprotein and Lipid Values. Cardiovasc. Drugs Ther. 2013, 27, 465–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elam, M.; Lovato, L.C.; Ginsberg, H. Role of fibrates in cardiovascular disease prevention, the ACCORD-Lipid perspective. Curr. Opin. Lipidol. 2011, 22, 55–61. [Google Scholar] [CrossRef]
- Bays, H.E.; McKenney, J.M.; Dujovne, C.A.; Schrott, H.G.; Zema, M.J.; Nyberg, J.; MacDougall, D.E. Effectiveness and tolerability of a new lipid-altering agent, gemcabene, in patients with low levels of high-density lipoprotein cholesterol. Am. J. Cardiol. 2003, 92, 538–543. [Google Scholar] [CrossRef]
- Warden, B.A.; Duell, P.B. Inclisiran: A Novel Agent for Lowering Apolipoprotein B-containing Lipoproteins. J. Cardiovasc. Pharmacol. 2021, 78, e157–e174. [Google Scholar] [CrossRef]
- Soška, V.; Kyselák, O. Some causes of poor adherence to long-term statin therapy and their solution. Vnitřní Lékařství 2018, 64, 923–927. [Google Scholar] [CrossRef]
- De Vera, M.A.; Bhole, V.; Burns, L.C.; Lacaille, D. Impact of statin adherence on cardiovascular disease and mortality outcomes: A systematic review. Br. J. Clin. Pharmacol. 2014, 78, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Kobiyama, K.; Saigusa, R.; Ley, K. Vaccination against atherosclerosis. Curr. Opin. Immunol. 2019, 59, 15–24. [Google Scholar] [CrossRef]
- Amirfakhryan, H. Vaccination against atherosclerosis: An overview. Hell. J. Cardiol. 2020, 61, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.; Su, B.; Meng, X. Recent Advances in the Development of Vaccines for Diabetes, Hypertension, and Atherosclerosis. J. Diabetes Res. 2018, 24, 1638462. [Google Scholar] [CrossRef] [PubMed]
- Gisterå, A.; Klement, M.L.; Polyzos, K.A.; Mailer, R.; Duhlin, A.; Karlsson, M.; Ketelhuth, D.; Hansson, G.K. Low-Density Lipoprotein-Reactive T Cells Regulate Plasma Cholesterol Levels and Development of Atherosclerosis in Humanized Hypercholesterolemic Mice. Circulation 2018, 138, 2513–2526. [Google Scholar] [CrossRef] [PubMed]
- Klingenberg, R.; Lebens, M.; Hermansson, A.; Fredrikson, G.N.; Strodthoff, D.; Rudling, M.; Ketelhuth, D.F.; Gerdes, N.; Holmgren, J.; Nilsson, J.; et al. Intranasal immunization with an apolipoprotein B-100 fusion protein induces antigen-specific regulatory T cells and reduces atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 946–952. [Google Scholar] [CrossRef] [Green Version]
- Chyu, K.Y.; Shah, P.K. In Pursuit of an Atherosclerosis Vaccine. Circ. Res. 2018, 123, 1121–1123. [Google Scholar] [CrossRef]
- Nghiem, N.; Knight, J.; Mizdrak, A.; Blakely, T.; Wilson, N. Preventive Pharmacotherapy for Cardiovascular Disease: A Modelling Study Considering Health Gain, Costs, and Cost-Effectiveness when Stratifying by Absolute Risk. Sci. Rep. 2019, 9, 19562. [Google Scholar] [CrossRef]
- Sidney, S.; Sorel, M.E.; Quesenberry, C.P.; Jaffe, M.G.; Solomon, M.D.; Nguyen-Huynh, M.N.; Go, A.S.; Rana, J.S. Comparative Trends in Heart Disease, Stroke, and All-Cause Mortality in the United States and a Large Integrated Healthcare Delivery System. Am. J. Med. 2018, 131, 829–836. [Google Scholar] [CrossRef]
- Sniderman, A.D.; Thanassoulis, G.; Williams, K.; Pencina, M. Risk of premature cardiovascular disease vs the number of premature cardiovascular events. JAMA Cardiol. 2016, 1, 492–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sidney, S.; Quesenberry, C.P., Jr.; Jaffe, M.G.; Sorel, M.; Nguyen-Huynh, M.N.; Kushi, L.H.; Go, A.S.; Rana, J.S. Recent Trends in Cardiovascular Mortality in the United States and Public Health Goals. JAMA Cardiol. 2016, 1, 594–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunbar, S.B.; Khavjou, O.A.; Bakas, T.; Hunt, G.; Kirch, R.A.; Leib, A.R.; Morrison, R.S.; Poehler, D.C.; Roger, V.L.; Whitsel, L.P.; et al. Projected Costs of Informal Caregiving for Cardiovascular Disease: 2015 to 2035: A Policy Statement from the American Heart Association. Circulation 2018, 137, e558–e577. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.G.; Huijgen, R.; Ray, K.; Persons, J.; Kastelein, J.J.; Pencina, M.J. Determining When to Add Nonstatin Therapy: A Quantitative Approach. J. Am. Coll. Cardiol. 2016, 68, 2412–2421. [Google Scholar] [CrossRef] [PubMed]
- Rosei, E.A.; Salvetti, M. Management of Hypercholesterolemia, Appropriateness of Therapeutic Approaches and New Drugs in Patients with High Cardiovascular Risk. High Blood Press. Cardiovasc. Prev. 2016, 23, 217–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langlois, M.R.; Nordestgaard, B.G.; Langsted, A.; Chapman, M.J.; Aakre, K.M.; Baum, H.; Borén, J.; Bruckert, E.; Catapano, A.; Cobbaert, C.; et al. European Atherosclerosis Society (EAS) and the European Federation of Clinical Chemistry and Laboratory Medicine (EFLM) Joint Consensus Initiative. Quantifying atherogenic lipoproteins for lipid-lowering strategies: Consensus-based recommendations from EAS and EFLM. Clin. Chem. Lab. Med. 2020, 58, 496–517. [Google Scholar] [PubMed] [Green Version]
- Solati, Z.; Ravandi, A. Lipidomics of Bioactive Lipids in Acute Coronary Syndromes. Int. J. Mol. Sci. 2019, 20, 1051. [Google Scholar] [CrossRef] [Green Version]
- Cobbaert, C.M.; Althaus, H.; Begcevic Brkovic, I.; Ceglarek, U.; Coassin, S.; Delatour, V.; Deprez, L.; Dikaios, I.; Dittrich, J.; Hoofnagle, A.N.; et al. Towards an SI-Traceable Reference Measurement System for Seven Serum Apolipoproteins Using Bottom-Up Quantitative Proteomics: Conceptual Approach Enabled by Cross-Disciplinary/Cross-Sector Collaboration. Clin. Chem. 2021, 67, 478–489. [Google Scholar] [CrossRef]
- Bodde, M.C.; Hermans, M.; Jukema, J.W.; Schalij, M.J.; Lijfering, W.M.; Rosendaal, F.R.; Romijn, F.; Ruhaak, L.R.; van der Laarse, A.; Cobbaert, C.M. Apolipoproteins A1, B, and apoB/apoA1 ratio are associated with first ST-segment elevation myocardial infarction but not with recurrent events during long-term follow-up. Clin. Res. Cardiol. 2019, 108, 520–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contois, J.H.; Delatour, V. Apolipoprotein B measurement: Need for standardization. J. Clin. Lipidol. 2018, 12, 264–265. [Google Scholar] [CrossRef]
- Delatour, V.; Clouet-Foraison, N.; Gaie-Levrel, F.; Marcovina, S.M.; Hoofnagle, A.N.; Kuklenyik, Z.; Caulfield, M.P.; Otvos, J.D.; Krauss, R.M.; Kulkarni, K.R.; et al. Comparability of Lipoprotein Particle Number Concentrations Across ES-DMA, NMR, LC-MS/MS, Immunonephelometry, and VAP: In Search of a Candidate Reference Measurement Procedure for apoB and non-HDL-P Standardization. Clin. Chem. 2018, 64, 1485–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, T.Y.; Chen, J.D. Low-density lipoprotein cholesterol/apolipoprotein B ratio is superior to apolipoprotein B alone in the diagnosis of coronary artery calcification. Coron. Artery Dis. 2021, 32, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Drexel, H.; Larcher, B.; Mader, A.; Vonbank, A.; Heinzle, C.F.; Moser, B.; Zanolin-Purin, D.; Saely, C.H. The LDL-C/ApoB ratio predicts major cardiovascular events in patients with established atherosclerotic cardiovascular disease. Atherosclerosis 2021, 329, 44–49. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Therapy | Type of Compound | Mechanism of Effect on ApoB |
|---|---|---|
| statins | Competitive inhibitors of HMG-CoA reductase | Lower apoB concentration by decreasing entry of apoB-containing lipoproteins LDL and VLDL into plasma |
| anacetrapib (development discontinued) | Small molecule oxazolidinone | Potent selective CETP inhibitor. Reduces apoB-containing lipoprotein particles |
| niacin | Nicotinic acid (vitamin B3) | Modulates liver synthesis of triglycerides, limiting VLDL assembly, resulting in intrahepatic apo B degradation |
| evolocumab and alirocumab | Fully human anti-PCSK9 monoclonal antibodies | PCSK9 inhibitors increase hepatic LDL receptors, which remove apoB-containing LDL particles from the circulation. Lp(a) also decreased, mechanism not understood |
| mipomersen (development discontinued) | Synthetic phosphorothioate antisense oligonucleotide apoB inhibitor | Prevents translation of the apoB mRNA into protein, leading to decreased VLDL and LDL |
| lomitapide | Small molecule that binds directly to and inhibits MTP | Inhibition of MTP in hepatocytes and enterocytes by lomitapide reduces plasma levels of all ApoB-containing lipoproteins. |
| dabigatran | Novel, synthetic, specific, non-peptide thrombin inhibitor | Antithrombotic effect due to binding competitively to the active site on human thrombin. ApoB lowering is a pleiotropic effect, mechanism unclear. |
| bempedoic acid | 8-hydroxy-2,2,14,14-tetramethylpentadecaned-ioic acid | Inhibits ATP-citrate lyase in the liver, which decreases liver cholesterol synthesis and reduces serum LDL levels by upregulating LDL receptors. |
| evinacumab | Fully human monoclonal antibody directed against ANGPTL3 | Antagonizes ANGPTL3-mediated inhibition of lipoprotein lipase and endothelial lipase, increasing clearance of apoB-containing lipoproteins. |
| fibrates | Amphipathic carboxylic acids that act as peroxisome proliferator receptor α agonists | Reduce plasma triglycerides by inhibiting their hepatic synthesis and increasing their catabolism. Lower LDL-C, non-HDL-C and apoB. |
| inclisiran | siRNA conjugated to triantennary N-acetylgalactosamine carbohydrates | Inhibits PCSK9, thereby reducing levels of apoB-containing lipoproteins. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behbodikhah, J.; Ahmed, S.; Elyasi, A.; Kasselman, L.J.; De Leon, J.; Glass, A.D.; Reiss, A.B. Apolipoprotein B and Cardiovascular Disease: Biomarker and Potential Therapeutic Target. Metabolites 2021, 11, 690. https://doi.org/10.3390/metabo11100690
Behbodikhah J, Ahmed S, Elyasi A, Kasselman LJ, De Leon J, Glass AD, Reiss AB. Apolipoprotein B and Cardiovascular Disease: Biomarker and Potential Therapeutic Target. Metabolites. 2021; 11(10):690. https://doi.org/10.3390/metabo11100690
Chicago/Turabian StyleBehbodikhah, Jennifer, Saba Ahmed, Ailin Elyasi, Lora J. Kasselman, Joshua De Leon, Amy D. Glass, and Allison B. Reiss. 2021. "Apolipoprotein B and Cardiovascular Disease: Biomarker and Potential Therapeutic Target" Metabolites 11, no. 10: 690. https://doi.org/10.3390/metabo11100690