The Relationship of the Mechanisms of the Pathogenesis of Multiple Sclerosis and the Expression of Endogenous Retroviruses

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Endogenous and Exogenous Factors Affecting the Development of MS
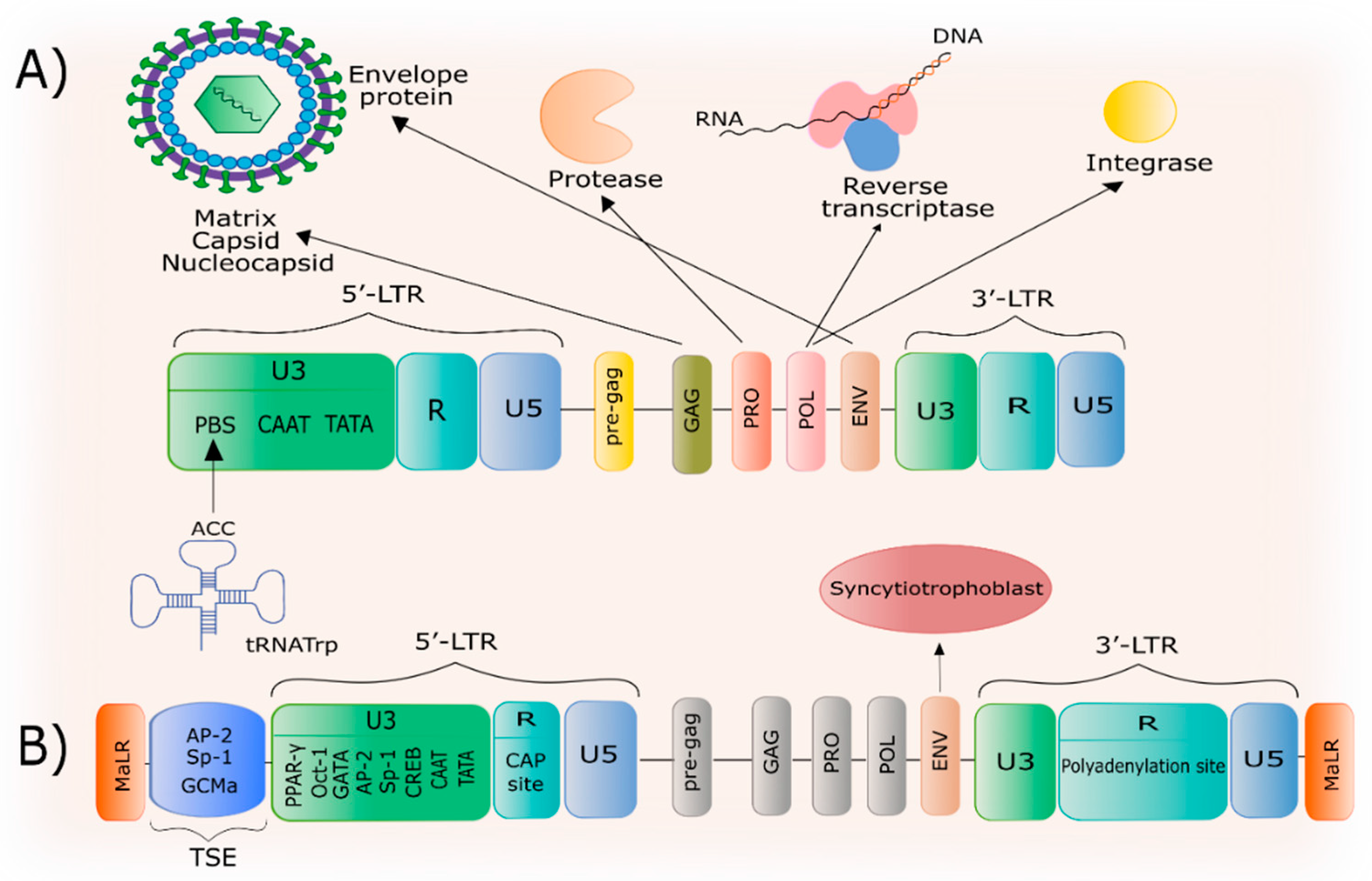
2. General Characteristics of HERV-W
3. The Structure of the HERV-W Family and the ERVWE1 Gene
4. Epigenetic Mechanisms of Regulation of HERV-W Expression
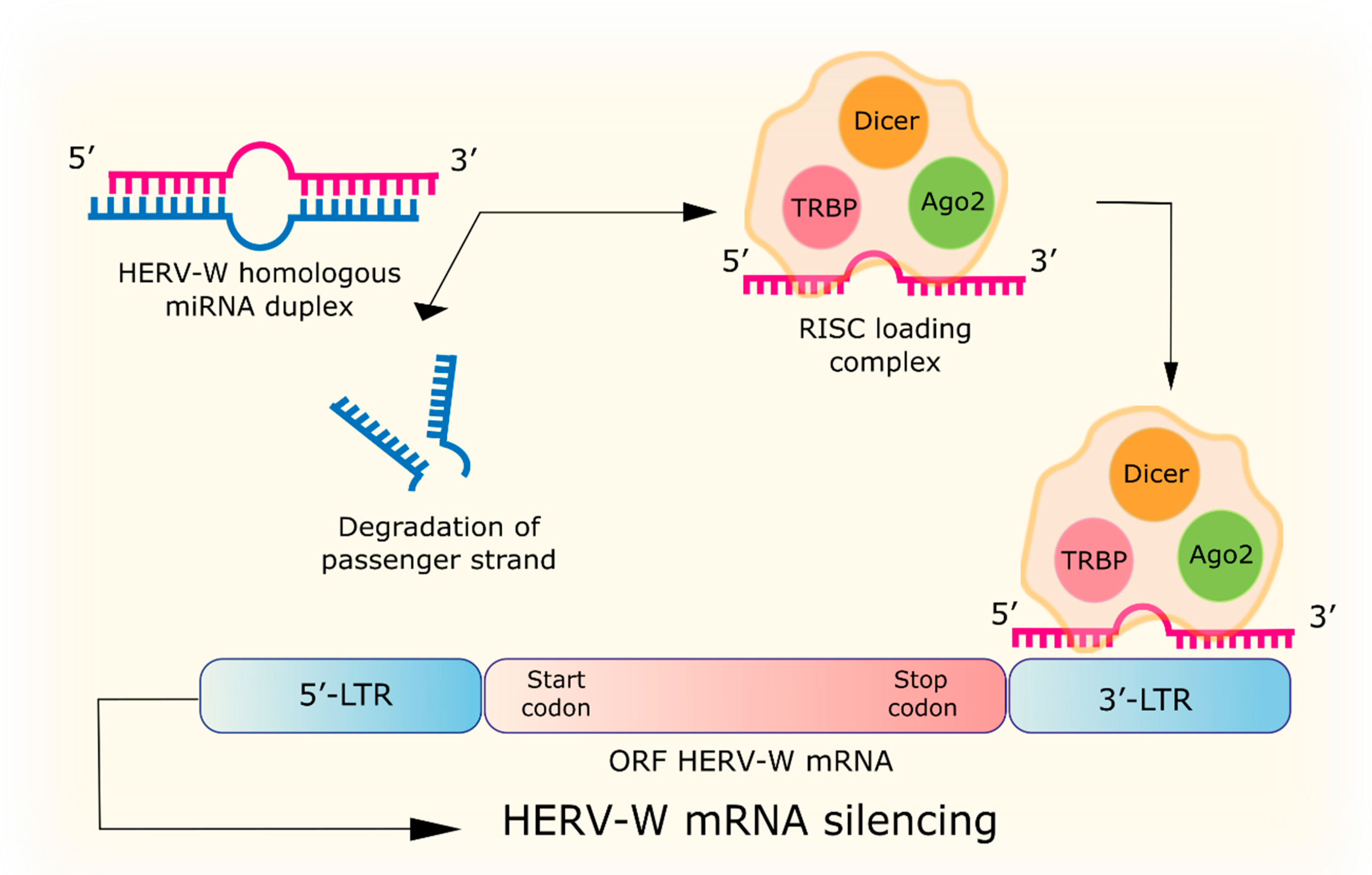
4.1. RNA Interference
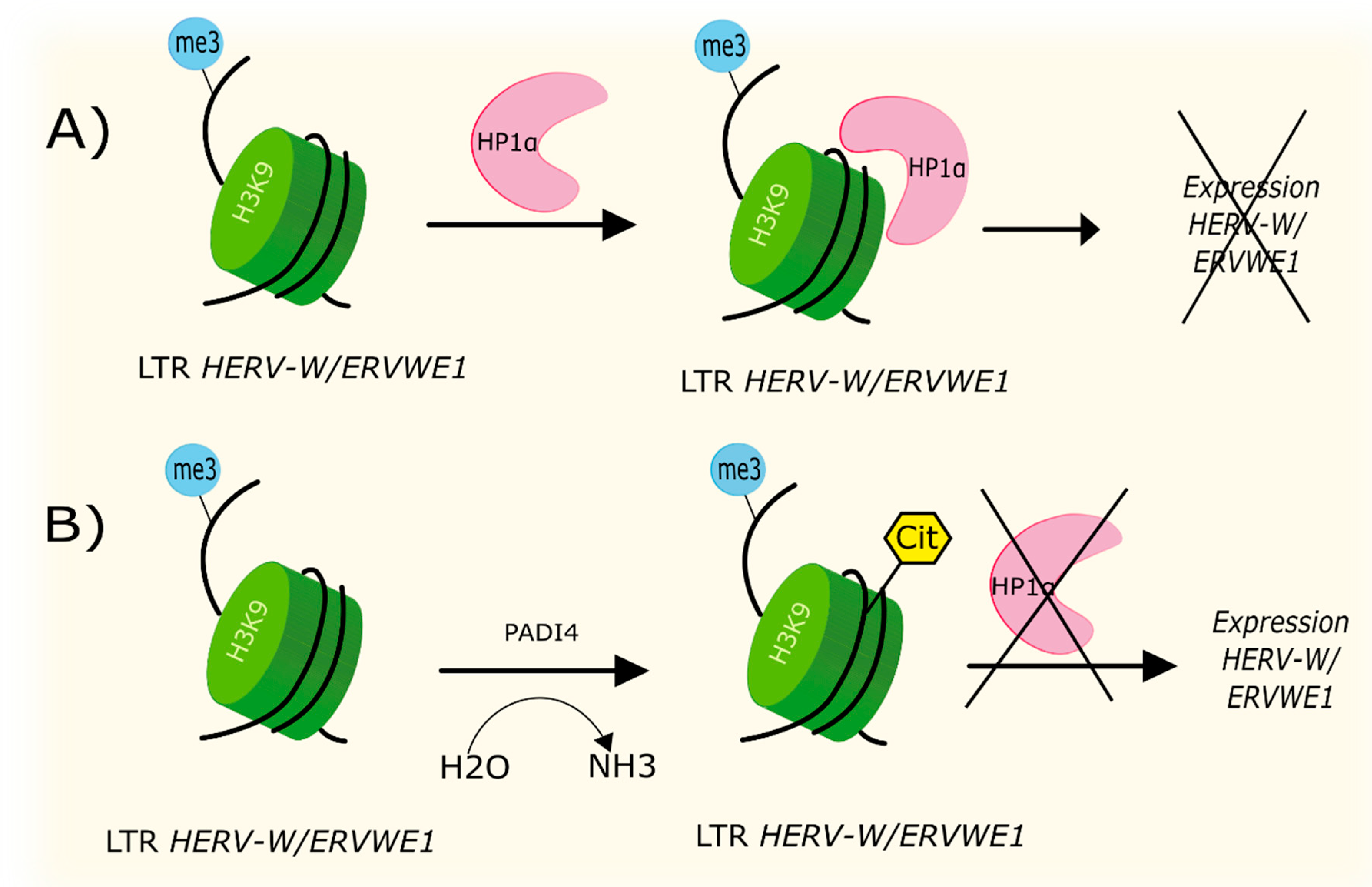
4.2. Citrullination
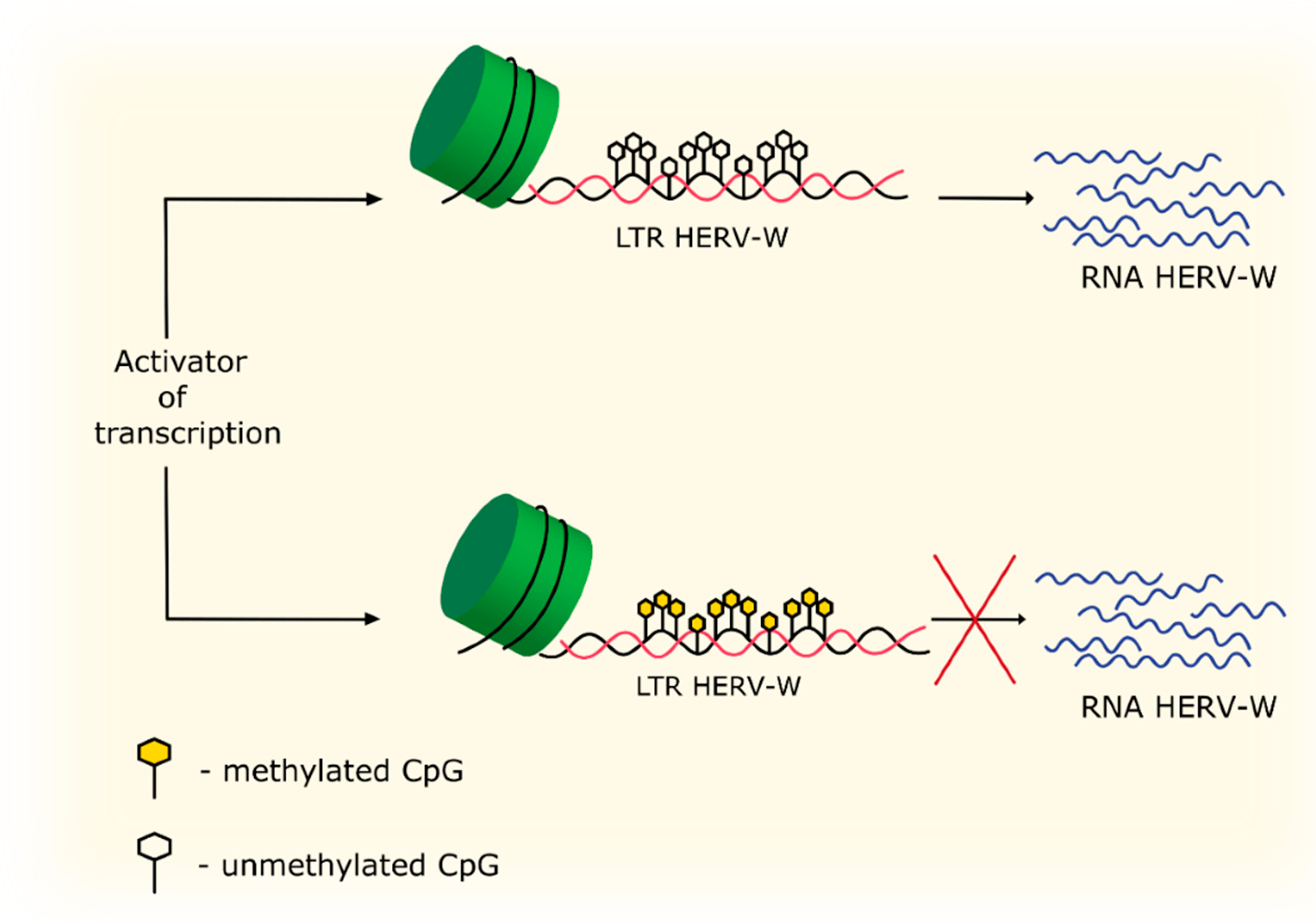
4.3. Methylation and Acetylation
5. Changes in LTR Regions and the Effects of Transcription Factors
6. The Role of the Immune System in Activation of HERV-W Transcription
7. Activation of HERV-W Transcription upon Exposure to Exogenous Viruses
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wallin, M.T.; Culpepper, W.J.; Nichols, E.; Bhutta, Z.A.; Gebrehiwot, T.T.; Hay, S.I.; Khalil, I.A.; Krohn, K.J.; Liang, X.; Naghavi, M.; et al. Global, regional, and national burden of multiple sclerosis 1990–2016: A systematic analysis for the Global Burden of Disease Study. Lancet Neurol. 2019, 18, 269–285. [Google Scholar] [CrossRef] [Green Version]
- Rosati, G.; Pinna, L.; Granieri, E.; Aiello, I.; De Bastiani, P.; Tola, R.; Agnetti, V.; Pirisi, A. The distribution of multiple sclerosis in Sardinia. Rivista di Patologia Nervosa e Mentale 1977, 98, 46–64. [Google Scholar]
- Houzen, H.; Niino, M.; Hirotani, M.; Fukazawa, T.; Kikuchi, S.; Tanaka, K.; Sasaki, H. Increased prevalence, incidence, and female predominance of multiple sclerosis in northern Japan. J. Neurol. Sci. 2012, 323, 117–122. [Google Scholar] [CrossRef] [PubMed]
- McCombe, P.A.; Greer, J.M. Female reproductive issues in multiple sclerosis. Mult. Scler. J. 2013, 19, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.-L.; Vodehnalova, K.; Kalincik, T.; Signori, A.; Havrdova, E.K.; Lechner-Scott, J.; Skibina, O.G.; Eastaugh, A.; Taylor, L.; Baker, J.; et al. Association of Pregnancy with the Onset of Clinically Isolated Syndrome. JAMA Neurol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Vukusic, S.; Hutchinson, M.; Hours, M.; Moreau, T.; Cortinovis-Tourniaire, P.; Adeleine, P.; Confavreux, C. Pregnancy and multiple sclerosis (the PRIMS study): Clinical predictors of post-partum relapse. Brain 2004, 127, 1353–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Smolders, J.; Menheere, P.; Kessels, A.; Damoiseaux, J.; Hupperts, R. Association of vitamin D metabolite levels with relapse rate and disability in multiple sclerosis. Mult. Scler. J. 2008, 14, 1220–1224. [Google Scholar] [CrossRef]
- Becker, J.; Callegaro, D.; Lana-Peixoto, M.A.; Talim, N.; Vidaletti, T.; Correa, M.D.P.; Gomes, I. Hypovitaminosis D association with disease activity in relapsing remitting multiple sclerosis in Brazil. J. Neurol. Sci. 2016, 363, 236–239. [Google Scholar] [CrossRef]
- Simpson, S., Jr.; Van Der Mei, I.; Lucas, R.M.; Ponsonby, A.-L.; Broadley, S.; Blizzard, L.; Taylor, B.; Dear, K.; Dwyer, T.; Taylor, B.V. Sun exposure across the life course significantly modulates early multiple sclerosis clinical course. Front. Neurol. 2018, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Tremlett, H.; Zhu, F.; Ascherio, A.; Munger, K.L. Sun exposure over the life course and associations with multiple sclerosis. Neurology 2018, 90, e1191–e1199. [Google Scholar] [CrossRef] [PubMed]
- Ramagopalan, S.V.; Heger, A.; Berlanga, A.J.; Maugeri, N.J.; Lincoln, M.R.; Burrell, A.; Handunnetthi, L.; Handel, A.E.; Disanto, G.; Orton, S.-M. A ChIP-seq defined genome-wide map of vitamin D receptor binding: Associations with disease and evolution. Genome Res. 2010, 20, 1352–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peeters, L.M.; Vanheusden, M.; Somers, V.; Van Wijmeersch, B.; Stinissen, P.; Broux, B.; Hellings, N. Cytotoxic CD4+ T Cells Drive Multiple Sclerosis Progression. Front. Immunol. 2017, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saul, L.; Mair, I.; Ivens, A.; Brown, P.; Samuel, K.; Campbell, J.D.M.; Soong, D.Y.; Kamenjarin, N.; Mellanby, R.J. 1,25-Dihydroxyvitamin D3 Restrains CD4+ T Cell Priming Ability of CD11c+ Dendritic Cells by Upregulating Expression of CD31. Front. Immunol. 2019, 10, 600. [Google Scholar] [CrossRef]
- Cao, Y.; Goods, B.A.; Raddassi, K.; Nepom, G.T.; Kwok, W.W.; Love, J.C.; Hafler, D.A. Functional inflammatory profiles distinguish myelin-reactive T cells from patients with multiple sclerosis. Sci. Transl. Med. 2015, 7, 287ra74. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Hildeman, D.; Tripathi, P.; Velu, C.S.; Grimes, H.L.; Zheng, Y. Coordination of IL-7 receptor and T-cell receptor signaling by cell-division cycle 42 in T-cell homeostasis. Proc. Natl. Acad. Sci. USA 2010, 107, 18505–18510. [Google Scholar] [CrossRef] [Green Version]
- Berge, T.; Leikfoss, I.S.; Brorson, I.S.; Bos, S.D.; Page, C.M.; Gustavsen, M.W.; Bjolgerud, A.; Holmoy, T.; Celius, E.G.; Damoiseaux, J.; et al. The multiple sclerosis susceptibility genes TAGAP and IL2RA are regulated by vitamin D in CD4+ T cells. Genes Immun. 2016, 17, 118–127. [Google Scholar] [CrossRef] [Green Version]
- Alcina, A.; Vandenbroeck, K.; Otaegui, D.; Saiz, A.; Gonzalez, J.R.; Fernandez, O.; Cavanillas, M.L.; Cenit, M.C.; Arroyo, R.; Alloza, I.; et al. The autoimmune disease-associated KIF5A, CD226 and SH2B3 gene variants confer susceptibility for multiple sclerosis. Genes Immun. 2010, 11, 439–445. [Google Scholar] [CrossRef]
- Gross, C.C.; Schulte-Mecklenbeck, A.; Rünzi, A.; Kuhlmann, T.; Posevitz-Fejfár, A.; Schwab, N.; Schneider-Hohendorf, T.; Herich, S.; Held, K.; Konjević, M. Impaired NK-mediated regulation of T-cell activity in multiple sclerosis is reconstituted by IL-2 receptor modulation. Proc. Natl. Acad. Sci. USA 2016, 113, E2973–E2982. [Google Scholar] [CrossRef] [Green Version]
- Vandenbroeck, K.; Alvarez, J.; Swaminathan, B.; Alloza, I.; Comabella, M.; Urcelay, E.; Comabella, M.; Alcina, A.; Fedetz, M.; Ortiz, A.M.; et al. A cytokine gene screen uncovers SOCS1 as genetic risk factor for multiple sclerosis. Genes Immun. 2011, 13, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Toghi, M.; Taheri, M.; Arsang-Jang, S.; Ohadi, M.; Mirfakhraie, R.; Mazdeh, M.; Sayad, A. SOCS gene family expression profile in the blood of multiple sclerosis patients. J. Neurol. Sci. 2017, 375, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Lünemann, J.D.; Jelčić, I.; Roberts, S.; Lutterotti, A.; Tackenberg, B.; Martin, R.; Münz, C. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-γ and IL-2. J. Exp. Med. 2008, 205, 1763–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, D.F.; Serafini, B.; Piras, E.; Severa, M.; Coccia, E.M.; Rosicarelli, B.; Ruggieri, S.; Gasperini, C.; Buttari, F.; Centonze, D.; et al. Increased CD8+ T Cell Response to Epstein-Barr Virus Lytic Antigens in the Active Phase of Multiple Sclerosis. PLOS Pathog. 2013, 9, e1003220. [Google Scholar] [CrossRef] [PubMed]
- Tejada-Simon, M.V.; Zang, Y.C.Q.; Hong, J.; Rivera, V.M.; Zhang, J. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann. Neurol. 2003, 53, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Vanheusden, M.; Broux, B.; Welten, S.P.M.; Peeters, L.M.; Panagioti, E.; Van Wijmeersch, B.; Somers, V.; Stinissen, P.; Arens, R.; Hellings, N. Cytomegalovirus infection exacerbates autoimmune mediated neuroinflammation. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Oldstone, M.B. Molecular Mimicry: Its Evolution from Concept to Mechanism as a Cause of Autoimmune Diseases. Monoclon. Antibodies Immunodiagn. Immunother. 2014, 33, 158–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cossu, D.; Cocco, E.; Paccagnini, D.; Masala, S.; Ahmed, N.; Frau, J.; Marrosu, M.G.; Sechi, L.A. Association of Mycobacterium avium subsp. paratuberculosis with Multiple Sclerosis in Sardinian Patients. PLOS ONE 2011, 6, e18482. [Google Scholar] [CrossRef]
- Ivanova, M.V.; Kolkova, N.I.; Morgunova, E.Y.; Pashko, Y.P.; Zigangirova, N.A.; Zakharova, M.N. Role of Chlamydia in Multiple Sclerosis. Bull. Exp. Biol. Med. 2015, 159, 646–648. [Google Scholar] [CrossRef]
- Kazemi, S.; Aghaee, B.L.; Soltanian, A.R.; Mazdeh, M.; Taheri, M.; Alikhani, M.Y. Investigation of Chlamydia pneumoniae Infection in Patients with Multiple Sclerosis: A Case-Control Study. Avicenna J. Clin. Microb. Infect. 2020, 7, 36–39. [Google Scholar] [CrossRef]
- Berer, K.; Gerdes, L.A.; Cekanaviciute, E.; Jia, X.; Xiao, L.; Xia, Z.; Liu, C.; Klotz, L.; Stauffer, U.; Baranzini, S.E. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10719–10724. [Google Scholar] [CrossRef] [Green Version]
- Mameli, G.; Poddighe, L.; Astone, V.; Delogu, G.; Arru, G.; Sotgiu, S.; Serra, C.; Dolei, A. Novel reliable real-time PCR for differential detection of MSRVenv and syncytin-1 in RNA and DNA from patients with multiple sclerosis. J. Virol. Methods 2009, 161, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Küry, P.; Nath, A.; Créange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.-P.; Perron, H. Human Endogenous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Li, J.; Wang, F.; Oliver, M.T.; Tipton, T.; Gao, Y.; Jiang, S.-W. (Albert) Syncytin-1 modulates placental trophoblast cell proliferation by promoting G1/S transition. Cell. Signal. 2013, 25, 1027–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, S.; Echeverría, N.; Moratorio, G.; Landoni, A.I.; Dighiero, G.; Cristina, J.; Oppezzo, P.; Moreno, P. Human endogenous retrovirus np9 gene is over expressed in chronic lymphocytic leukemia patients. Leuk. Res. Rep. 2014, 3, 70–72. [Google Scholar] [CrossRef] [Green Version]
- Van Horssen, J.; Van Der Pol, S.; Nijland, P.; Amor, S.; Perron, H. Human endogenous retrovirus W in brain lesions: Rationale for targeted therapy in multiple sclerosis. Mult. Scler. Relat. Disord. 2016, 8, 11–18. [Google Scholar] [CrossRef] [Green Version]
- Perron, H.; Germi, R.; Bernard, C.; Garcia-Montojo, M.; Deluen, C.; Farinelli, L.; Faucard, R.; Veas, F.; Stefas, I.; O Fabriek, B.; et al. Human endogenous retrovirus type W envelope expression in blood and brain cells provides new insights into multiple sclerosis disease. Mult. Scler. J. 2012, 18, 1721–1736. [Google Scholar] [CrossRef]
- Kremer, D.; Gruchot, J.; Weyers, V.; Oldemeier, L.; Göttle, P.; Healy, L.; Jang, J.H.; Xu, Y.K.T.; Volsko, C.; Dutta, R. pHERV-W envelope protein fuels microglial cell-dependent damage of myelinated axons in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 15216–15225. [Google Scholar] [CrossRef] [Green Version]
- Brudek, T.; Christensen, T.; Aagaard, L.; Petersen, T.; Hansen, H.J.; Møller-Larsen, A. B cells and monocytes from patients with active multiple sclerosis exhibit increased surface expression of both HERV-H Env and HERV-W Env, accompanied by increased seroreactivity. Retrovirology 2009, 6, 104. [Google Scholar] [CrossRef] [Green Version]
- Arru, G.; Mameli, G.; Astone, V.; Serra, C.; Huang, Y.-M.; Link, H.; Fainardi, E.; Castellazzi, M.; Granieri, E.; Fernandez, M.; et al. Multiple Sclerosis and HERV-W/MSRV: A Multicentric Study. Int. J. Biomed. Sci. 2007, 3, 292–297. [Google Scholar]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The Sequence of the Human Genome. Sci. Transl. Med. 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [Green Version]
- Villesen, P.; Aagaard, L.; Wiuf, C.; Pedersen, F.S. Identification of endogenous retroviral reading frames in the human genome. Retrovirology 2004, 1, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oja, M.; Sperber, G.O.; Blomberg, J.; Kaski, S. Self-Organizing Map-Based Discovery and Visualization of Human Endogenous Retroviral Sequence Groups. Int. J. Neural Syst. 2005, 15, 163–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandi, N.; Tramontano, E. Human Endogenous Retroviruses Are Ancient Acquired Elements Still Shaping Innate Immune Responses. Front. Immunol. 2018, 9, 2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoni, F.; Guerra, J.; Luban, J. HERV-H RNA is abundant in human embryonic stem cells and a precise marker for pluripotency. Retrovirology 2012, 9, 111. [Google Scholar] [CrossRef] [Green Version]
- Bonnaud, B.; Beliaeff, J.; Bouton, O.; Oriol, G.; Duret, L.; Mallet, F. Natural history of the ERVWE1 endogenous retroviral locus. Retrovirology 2005, 2, 57. [Google Scholar] [CrossRef] [Green Version]
- Boeke, J.; Stoye, J. Retrotransposons, Endogenous Retroviruses, and the Evolution of Retroelements; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1997. [Google Scholar]
- Vargiu, L.; Rodriguez-Tomé, P.; Sperber, G.O.; Cadeddu, M.; Grandi, N.; Blikstad, V.; Tramontano, E.; Blomberg, J. Classification and characterization of human endogenous retroviruses; mosaic forms are common. Retrovirology 2016, 13, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, D.J. Endogenous retroviruses in the human genome sequence. Genome Biol. 2001, 2, 1017. [Google Scholar] [CrossRef]
- Nelson, P.N.; Carnegie, P.; Martin, J.; Ejtehadi, H.D.; Hooley, P.; Roden, D.; Rowland-Jones, S.; Warren, P.; Astley, J.; Murray, P.G. Demystified…Human endogenous retroviruses. Mol. Pathol. 2003, 56, 11. [Google Scholar] [CrossRef] [Green Version]
- Rolland, A.; Jouvin-Marche, E.; Viret, C.; Faure, M.; Perron, H.; Marche, P.N. The Envelope Protein of a Human Endogenous Retrovirus-W Family Activates Innate Immunity through CD14/TLR4 and Promotes Th1-Like Responses. J. Immunol. 2006, 176, 7636–7644. [Google Scholar] [CrossRef]
- Kremer, D.; Schichel, T.; Förster, M.; Tzekova, N.; Bernard, C.; Van Der Valk, P.; Van Horssen, J.; Hartung, H.-P.; Perron, H.; Küry, P. Human endogenous retrovirus type W envelope protein inhibits oligodendroglial precursor cell differentiation. Ann. Neurol. 2013, 74, 721–732. [Google Scholar] [CrossRef]
- Duperray, A.; Barbe, D.; Raguenez, G.; Weksler, B.B.; Romero, I.A.; Couraud, P.-O.; Perron, H.; Marche, P.N. Inflammatory response of endothelial cells to a human endogenous retrovirus associated with multiple sclerosis is mediated by TLR4. Int. Immunol. 2015, 27, 545–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlíček, A.; Pačes, J.; Elleder, D.; Hejnar, J. Processed pseudogenes of human endogenous retroviruses generated by LINEs: Their integration, stability, and distribution. Genome Res. 2002, 12, 391–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grandi, N.; Cadeddu, M.; Blomberg, J.; Tramontano, E. Contribution of type W human endogenous retroviruses to the human genome: Characterization of HERV-W proviral insertions and processed pseudogenes. Retrovirology 2016, 13, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laufer, G.; Mayer, J.; Mueller, B.F.; Mueller-Lantzsch, N.; Ruprecht, K. Analysis of transcribed human endogenous retrovirus W env loci clarifies the origin of multiple sclerosis-associated retrovirus envsequences. Retrovirology 2009, 6, 37. [Google Scholar] [CrossRef] [Green Version]
- Voisset, C.; Bouton, O.; Bedin, F.; Duret, L.; Mandrand, B.; Mallet, F.; Paranhos-Baccalà, G. Chromosomal Distribution and Coding Capacity of the Human Endogenous Retrovirus HERV-W Family. AIDS Res. Hum. Retroviruses 2000, 16, 731–740. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Nellåker, C.; Yolken, R.; Karlsson, H. A systematic evaluation of expression of HERV-W elements; influence of genomic context, viral structure and orientation. BMC Genom. 2011, 12, 22. [Google Scholar] [CrossRef] [Green Version]
- Blond, J.-L.; Besème, F.; Duret, L.; Bouton, O.; Bedin, F.; Perron, H.; Mandrand, B.; Mallet, F. Molecular Characterization and Placental Expression of HERV-W, a New Human Endogenous Retrovirus Family. J. Virol. 1999, 73, 1175–1185. [Google Scholar] [CrossRef] [Green Version]
- Prudhomme, S.; Oriol, G.; Mallet, F. A Retroviral Promoter and a Cellular Enhancer Define a Bipartite Element Which Controls env ERVWE1 Placental Expression. J. Virol. 2004, 78, 12157–12168. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, K.; Richter, C.; Backes, C.; Meese, E.; Ruprecht, K.; Mayer, J. Comprehensive Analysis of Human Endogenous Retrovirus Group HERV-W Locus Transcription in Multiple Sclerosis Brain Lesions by High-Throughput Amplicon Sequencing. J. Virol. 2013, 87, 13837–13852. [Google Scholar] [CrossRef] [Green Version]
- Place, R.F.; Li, L.-C.; Pookot, D.; Noonan, E.J.; Dahiya, R. MicroRNA-373 induces expression of genes with complementary promoter sequences. Proc. Natl. Acad. Sci. USA 2008, 105, 1608–1613. [Google Scholar] [CrossRef] [Green Version]
- Bagga, S.; Bracht, J.; Hunter, S.; Massirer, K.; Holtz, J.; Eachus, R.; Pasquinelli, A.E. Regulation by let-7 and lin-4 miRNAs Results in Target mRNA Degradation. Cell 2005, 122, 553–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Li, J.; Li, J.; Yang, Y.; Kang, X.; Li, Y.; Wu, X.; Zhu, Q.; Zhou, Y.; Hu, Y. Up-regulation of microRNA-203 in influenza A virus infection inhibits viral replication by targeting DR1. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Cox, M.B.; Cairns, M.J.; Gandhi, K.S.; Carroll, A.P.; Moscovis, S.; Stewart, G.J.; Broadley, S.; Scott, R.J.; Booth, D.R.; Lechner-Scott, J.; et al. MicroRNAs miR-17 and miR-20a Inhibit T Cell Activation Genes and Are Under-Expressed in MS Whole Blood. PLOS ONE 2010, 5, e12132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meira, M.; Sievers, C.; Hoffmann, F.; Rasenack, M.; Kuhle, J.; Derfuss, T.; Kappos, L.; Lindberg, R.L.P. Unraveling Natalizumab Effects on Deregulated miR-17 Expression in CD4+T Cells of Patients with Relapsing-Remitting Multiple Sclerosis. J. Immunol. Res. 2014, 2014, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arru, G.; Leoni, S.; Pugliatti, M.; Mei, A.; Serra, C.; Delogu, L.G.; Manetti, R.; Dolei, A.; Sotgiu, S.; Mameli, G. Natalizumab inhibits the expression of human endogenous retroviruses of the W family in multiple sclerosis patients: A longitudinal cohort study. Mult. Scler. J. 2013, 20, 174–182. [Google Scholar] [CrossRef]
- Hakim, S.T.; Alsayari, M.; McLean, D.C.; Saleem, S.; Addanki, K.C.; Aggarwal, M.; Mahalingam, K.; Bagasra, O. A large number of the human microRNAs target lentiviruses, retroviruses, and endogenous retroviruses. Biochem. Biophys. Res. Commun. 2008, 369, 357–362. [Google Scholar] [CrossRef]
- Mameli, G.; Astone, V.; Khalili, K.; Serra, C.; Sawaya, B.E.; Dolei, A. Regulation of the syncytin-1 promoter in human astrocytes by multiple sclerosis-related cytokines. Virology 2007, 362, 120–130. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Azebi, S.; England, P.; Christensen, T.; Møller-Larsen, A.; Petersen, T.; Batsche, E.; Muchardt, C. Citrullination of Histone H3 Interferes with HP1-Mediated Transcriptional Repression. PLoS Genet. 2012, 8, e1002934. [Google Scholar] [CrossRef] [Green Version]
- Warren, K.G.; Catz, I. Autoantibodies to myelin basic protein within multiple sclerosis central nervous system tissue. J. Neurol. Sci. 1993, 115, 169–176. [Google Scholar] [CrossRef]
- Libich, D.S.; Hill, C.M.; Bates, I.R.; Hallett, F.R.; Armstrong, S.; Siemiarczuk, A.; Harauz, G. Interaction of the 18.5-kD isoform of myelin basic protein with Ca2+-calmodulin: Effects of deimination assessed by intrinsic Trp fluorescence spectroscopy, dynamic light scattering, and circular dichroism. Protein Sci. 2003, 12, 1507–1521. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Jo, S.; Park, C.-S.; Song, H.-J.; Park, Z.-Y. Myelin basic protein as a binding partner and calmodulin adaptor for the BKCa channel. Proteomics 2007, 7, 2591–2602. [Google Scholar] [CrossRef] [PubMed]
- Moscarello, M.A.; Wood, D.D.; Ackerley, C.; Boulias, C. Myelin in multiple sclerosis is developmentally immature. J. Clin. Investig. 1994, 94, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.D.; Moscarello, M.A.; Bilbao, J.M.; O’Connors, P. Acute multiple sclerosis (marburg type) is associated with developmentally immature myelin basic protein. Ann. Neurol. 1996, 40, 18–24. [Google Scholar] [CrossRef]
- Wood, D.D.; A Ackerley, C.; Brand, B.V.D.; Zhang, L.; Raijmakers, R.; Mastronardi, F.G.; A Moscarello, M. Myelin localization of peptidylarginine deiminases 2 and 4: Comparison of PAD2 and PAD4 activities. Lab. Investig. 2008, 88, 354–364. [Google Scholar] [CrossRef] [Green Version]
- Faigle, W.; Cruciani, C.; Wolski, W.; Roschitzki, B.; Puthenparampil, M.; Tomas-Ojer, P.; Sellés-Moreno, C.; Zeis, T.; Jelcic, I.; Schaeren-Wiemers, N.; et al. Brain Citrullination Patterns and T Cell Reactivity of Cerebrospinal Fluid-Derived CD4+ T Cells in Multiple Sclerosis. Front. Immunol. 2019, 10, 540. [Google Scholar] [CrossRef] [Green Version]
- Jasse, L.; Vukusic, S.; Durand-Dubief, F.; Vartin, C.; Piras, C.; Bernard, M.; Pélisson, D.; Confavreux, C.; Vighetto, A.; Tilikete, C. Persistent visual impairment in multiple sclerosis: Prevalence, mechanisms and resulting disability. Mult. Scler. J. 2013, 19, 1618–1626. [Google Scholar] [CrossRef]
- Bazelier, M.T.; Mueller-Schotte, S.; Leufkens, H.G.; Uitdehaag, B.M.; Van Staa, T.P.; De Vries, F. Risk of cataract and glaucoma in patients with multiple sclerosis. Mult. Scler. J. 2011, 18, 628–638. [Google Scholar] [CrossRef]
- Bhattacharya, S.K.; Bhat, M.B.; Takahara, H. Modulation of Peptidyl Arginine Deiminase 2 and Implication for Neurodegeneration. Curr. Eye Res. 2006, 31, 1063–1071. [Google Scholar] [CrossRef]
- Bhattacharya, S.K.; Sinicrope, B.; Rayborn, M.E.; Hollyfield, J.G.; Bonilha, V.L. Age-related reduction in retinal deimination levels in the F344BN rat. Aging Cell 2008, 7, 441–444. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene Body Methylation Can Alter Gene Expression and Is a Therapeutic Target in Cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.S.; Teaberry, V.S.; Bland, A.E.; Huang, Z.; Whitaker, R.S.; Baba, T.; Fujii, S.; Secord, A.A.; Berchuck, A.; Murphy, S.K. Elevated MAL expression is accompanied by promoter hypomethylation and platinum resistance in epithelial ovarian cancer. Int. J. Cancer 2010, 126, 1378–1389. [Google Scholar] [PubMed]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Deng, F.-Y.; Lü, X.; Lei, S.-F. Susceptibility Genes for Multiple Sclerosis Identified in a Gene-Based Genome-Wide Association Study. J. Clin. Neurol. 2015, 11, 311–318. [Google Scholar] [CrossRef] [Green Version]
- Douvaras, P.; Rusielewicz, T.; Kim, K.H.; Haines, J.D.; Casaccia, P.; Fossati, V. Epigenetic Modulation of Human Induced Pluripotent Stem Cell Differentiation to Oligodendrocytes. Int. J. Mol. Sci. 2016, 17, 614. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.-L.; Kuhlmann, T.; Miron, V.E.; Leong, S.Y.; Fang, J.; Gris, P.; Kennedy, T.E.; Almazan, G.; Antel, J. Oligodendrocyte Progenitor Cell Susceptibility to Injury in Multiple Sclerosis. Am. J. Pathol. 2013, 183, 516–525. [Google Scholar] [CrossRef]
- Szpakowski, S.; Sun, X.; Lage, J.M.; Dyer, A.; Rubinstein, J.; Kowalski, D.; Sasaki, C.; Costa, J.; Lizardi, P.M. Loss of epigenetic silencing in tumors preferentially affects primate-specific retroelements. Gene 2009, 448, 151–167. [Google Scholar] [CrossRef] [Green Version]
- Matoušková, M.; Blažková, J.; Pajer, P.; Pavlíček, A.; Hejnar, J. CpG methylation suppresses transcriptional activity of human syncytin-1 in non-placental tissues. Exp. Cell Res. 2006, 312, 1011–1020. [Google Scholar] [CrossRef]
- Weber, S.; Hofmann, A.; Herms, S.; Hoffmann, P.; Doerfler, W. Destabilization of the human epigenome: Consequences of foreign DNA insertions. Epigenomics 2015, 7, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Weber, S.; Jung, S.; Doerfler, W. DNA methylation and transcription in HERV (K, W, E) and LINE sequences remain unchanged upon foreign DNA insertions. Epigenomics 2016, 8, 157–165. [Google Scholar] [CrossRef]
- Chomyk, A.M.; Volsko, C.; Tripathi, A.; Deckard, S.A.; Trapp, B.D.; Fox, R.J.; Dutta, R. DNA methylation in demyelinated multiple sclerosis hippocampus. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Nierop, G.P.; Van Luijn, M.M.; Michels, S.; Samira, S.; Melief, M.-J.; Janssen, M.; Langerak, A.W.; Ouwendijk, W.J.D.; Hintzen, R.Q.; Verjans, G.M. Phenotypic and functional characterization of T cells in white matter lesions of multiple sclerosis patients. Acta Neuropathol. 2017, 134, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Bos, S.D.; Page, C.M.; Andreassen, B.K.; Elboudwarej, E.; Gustavsen, M.W.; Briggs, F.B.; Quach, H.; Leikfoss, I.S.; Bjølgerud, A.; Berge, T.; et al. Genome-Wide DNA Methylation Profiles Indicate CD8+ T Cell Hypermethylation in Multiple Sclerosis. PLoS ONE 2015, 10, e0117403. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Hurst, T.; Pace, M.J.; Katzourakis, A.; Phillips, R.; Klenerman, P.; Frater, J.; Magiorkinis, G. Human endogenous retrovirus (HERV) expression is not induced by treatment with the histone deacetylase (HDAC) inhibitors in cellular models of HIV-1 latency. Retrovirology 2016, 13, 10. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Liu, T.; Zhao, Z.; Chen, Y.; Zeng, J.; Liu, S.; Zhu, F. Mutations in 3′-long terminal repeat of HERV-W family in chromosome 7 upregulate syncytin-1 expression in urothelial cell carcinoma of the bladder through interacting with c-Myb. Oncogene 2013, 33, 3947–3958. [Google Scholar] [CrossRef]
- Yu, J.S.; Hayashi, T.; Seboun, E.; Sklar, R.M.; Doolittle, T.H.; Hauser, S.L. Fos RNA accumulation in multiple sclerosis white matter tissue. J. Neurol. Sci. 1991, 103, 209–215. [Google Scholar] [CrossRef]
- Gimenez, J.; Montgiraud, C.; Pichon, J.-P.; Bonnaud, B.; Arsac, M.; Ruel, K.; Bouton, O.; Mallet, F. Custom human endogenous retroviruses dedicated microarray identifies self-induced HERV-W family elements reactivated in testicular cancer upon methylation control. Nucleic Acids Res. 2010, 38, 2229–2246. [Google Scholar] [CrossRef]
- Cramer, S.P.; Simonsen, H.; Frederiksen, J.; Rostrup, E.; Larsson, H.B.W. Abnormal blood–brain barrier permeability in normal appearing white matter in multiple sclerosis investigated by MRI. NeuroImage Clin. 2014, 4, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Schenk, G.J.; Dijkstra, S.; Hof, A.J.V.H.; Van Der Pol, S.M.A.; Drexhage, J.A.R.; Van Der Valk, P.; Reijerkerk, A.; Van Horssen, J.; De Vries, H.E. Roles for HB-EGF and CD9 in multiple sclerosis. Glia 2013, 61, 1890–1905. [Google Scholar] [CrossRef]
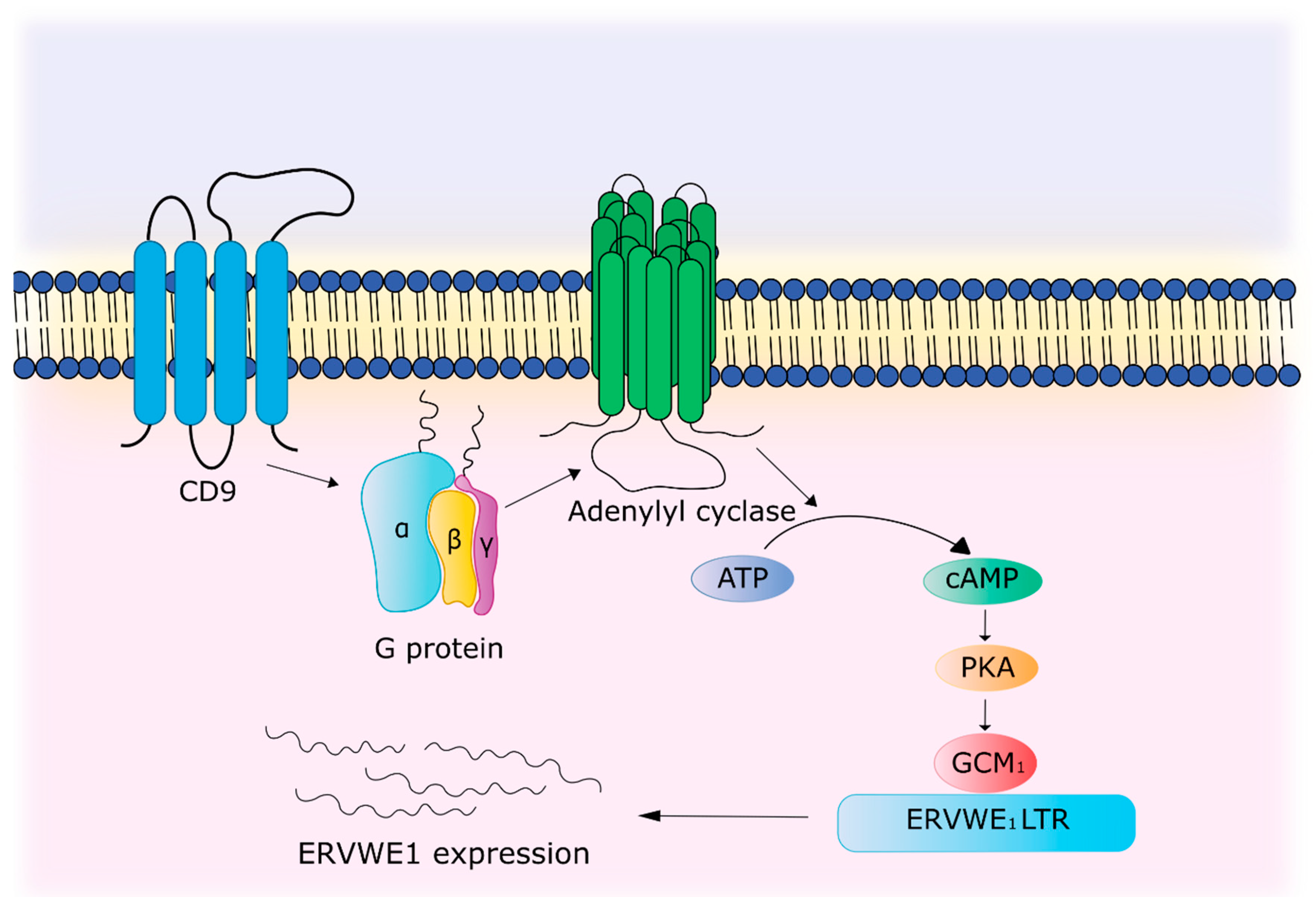
- Muroi, Y.; Sakurai, T.; Hanashi, A.; Kubota, K.; Nagaoka, K.; Imakawa, K. CD9 regulates transcription factor GCM1 and ERVWE1 expression through the cAMP/protein kinase A signaling pathway. Reproduction 2009, 138, 945–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, F.; Li, C.; Wang, X.; Du, J.; Liu, K.; Li, B.; Yao, P.; Li, Y.; Zhang, S. Syncytin 1, CD9, and CD47 regulating cell fusion to form PGCCs associated with cAMP/PKA and JNK signaling pathway. Cancer Med. 2019, 8, 3047–3058. [Google Scholar] [CrossRef] [PubMed]
- Uleri, E.; Mei, A.; Mameli, G.; Poddighe, L.; Serra, C.; Dolei, A. HIV Tat acts on endogenous retroviruses of the W family and this occurs via Toll-like receptor 4: Inference for neuroAIDS. Aids 2014, 28, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Saylor, D. Multiple sclerosis and HIV: A case of multiple sclerosis-immune reconstitution inflammatory syndrome associated with antiretroviral therapy initiation. Int. J. STD AIDS 2018, 29, 929–932. [Google Scholar] [CrossRef] [PubMed]
- Assinger, A.; Yaiw, K.-C.; Göttesdorfer, I.; Leib-Mösch, C.; Söderberg-Nauclér, C. Human Cytomegalovirus (HCMV) induces Human Endogenous Retrovirus (HERV) transcription. Retrovirology 2013, 10, 132. [Google Scholar] [CrossRef] [Green Version]
- Ruprecht, K.; Obojes, K.; Wengel, V.; Gronen, F.; Kim, K.S.; Perron, H.; Schneider-Schaulies, J.; Rieckmann, P. Regulation of human endogenous retrovirus W protein expression by herpes simplex virus type 1: Implications for multiple sclerosis. J. Neurovirol. 2006, 12, 65–71. [Google Scholar] [CrossRef]
- Charvet, B.; Reynaud, J.M.; Gourru-Lesimple, G.; Perron, H.; Marche, P.N.; Horvat, B. Induction of Proinflammatory Multiple Sclerosis-Associated Retrovirus Envelope Protein by Human Herpesvirus-6A and CD46 Receptor Engagement. Front. Immunol. 2018, 9, 2803. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors | Downregulation Mechanism | References |
|---|---|---|
| RNA interference | miRNA with homology to HERV-W: hsa-miR-376b, hsa-miR-22, hsa-miR-574, hsa-miR-570, hsa-miR-198, MIMAT0000228, hsa-miR-493-3p, MIMAT0003161, hsa-miR-214, MIMAT0000271, hsa-miR-101, hsa-miR-296, hsa-miR-31, hsa-miR-659, hsa-miR-185, hsa-miR-202, hsa-miR-122a, hsa-miR-24, hsa-miR-506, hsa-miR-632, hsa-miR-376a, hsa-miR-326. siRNA to the p65 gene of the NF-κB subunit reduces URE-LTR activation of the ERVWE1 promoter by TNFα. | [68,69] |
| DNA methylation | Hypermethylation in the LTR region of HERV-W leads to repression of HERV-W expression. | [83] |
| Upregulation mechanism | ||
| Citrullination | Increased level of PADI4 leads to formation of H3cit8K9me3 histone in promotor region of ERVWE1. Local increased citrullination of H3R8 histone localized above H3K9 was associated with increased expression of HERV-W/ERVWE1. | [70] |
| Changes in regulatory region | The 142T > C mutation promotes the formation of an additional binding site with c-Myb in 3′-LTR of ERVWE1 and due to this activation of its promoter activity. | [97] |
| Activation by immune system | TNFα and IFN-γ activate the ERVWE1 promoter. TNFα promotes increased binding of p65 of the NF-κB subunit to a sensitive element located in the enhancer region of the ERVWE1 promoter. CD9 through cAMP/PKA signaling pathway leads to the activation of GCM1, which binds to the regulatory region of ERVWE1 and enhances gene expression. | [69,102,103] |
| Exogenous viruses | Tat protein of HIV activate transcription of MSRV and ERVWE1 in in astrocytes through TLR-4 and TNFα. HCMV activate reverse transcriptase necessary for transcription of HERV. ICP0 protein of HSV-1 activate transcription of the HERV-Wenv and MSRVenv genes. HHV-6A activates MSRVenv expression by interacting with the isoform of CD46-Cyt1 receptor. | [104,106,107,108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lezhnyova, V.R.; Martynova, E.V.; Khaiboullin, T.I.; Urbanowicz, R.A.; Khaiboullina, S.F.; Rizvanov, A.A. The Relationship of the Mechanisms of the Pathogenesis of Multiple Sclerosis and the Expression of Endogenous Retroviruses. Biology 2020, 9, 464. https://doi.org/10.3390/biology9120464
Lezhnyova VR, Martynova EV, Khaiboullin TI, Urbanowicz RA, Khaiboullina SF, Rizvanov AA. The Relationship of the Mechanisms of the Pathogenesis of Multiple Sclerosis and the Expression of Endogenous Retroviruses. Biology. 2020; 9(12):464. https://doi.org/10.3390/biology9120464
Chicago/Turabian StyleLezhnyova, Vera R., Ekaterina V. Martynova, Timur I. Khaiboullin, Richard A. Urbanowicz, Svetlana F. Khaiboullina, and Albert A. Rizvanov. 2020. "The Relationship of the Mechanisms of the Pathogenesis of Multiple Sclerosis and the Expression of Endogenous Retroviruses" Biology 9, no. 12: 464. https://doi.org/10.3390/biology9120464