

Role of Cytokines and Growth Factors in the Manufacturing of iPSC-Derived Allogeneic Cell Therapy Products


Abstract
:Simple Summary
Abstract
1. Background
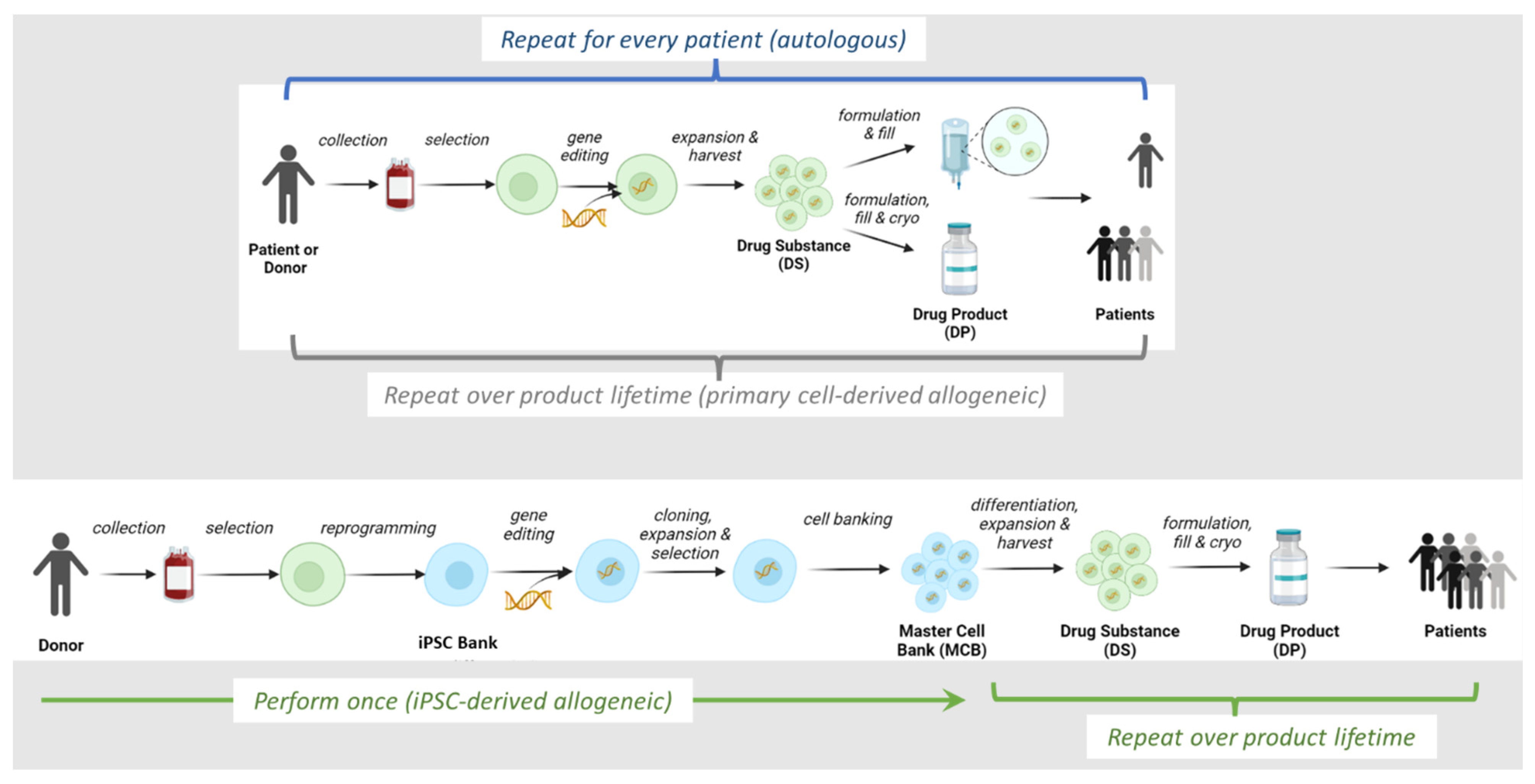
2. Overview of Allogeneic IPSC-Derived Effector Cell Manufacturing Process
3. Use of Transcription Factors to Generate Pluripotent Starting Material
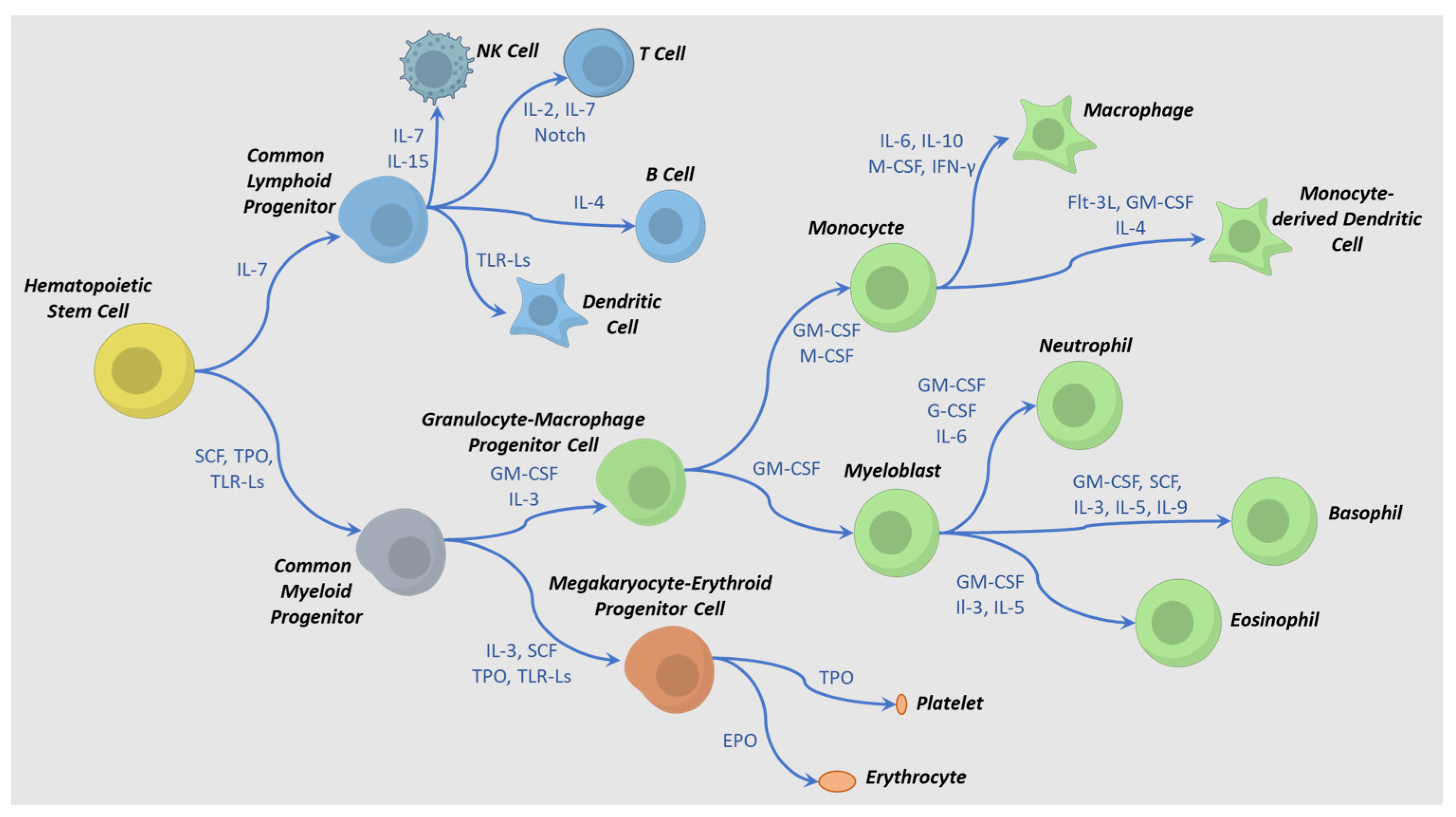
4. IPSC to HPC Differentiation Process
5. Differentiation and Activation Requirements for Production of Immune-Effector Cells
6. Cryopreservation
7. Engineering Cytokine Support into Allogeneic Cell Therapies
8. Use of Exogenous Cytokines in Cell Therapy Trials
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burke, C.J.; Zylberberg, C. Sources of Variability in Manufacturing of Cell Therapeutics. Regen. Eng. Transl. Med. 2019, 5, 332–340. [Google Scholar] [CrossRef]
- Borges, L.; Levitsky, H. The Challenge of Immune Rejection for Allogeneic Cell Therapies. Bioprocess Online 2022. Available online: https://www.cellandgene.com/doc/the-challenge-of-immune-rejection-for-allogeneic-cell-therapies-0001 (accessed on 23 December 2022).
- Koga, K.; Wang, B.; Kaneko, S. Current Status and Future Perspectives of HLA-Edited Induced Pluripotent Stem Cells. Inflamm Regen 2020, 40, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hurton, L.V.; Singh, H.; Najjar, A.M.; Switzer, K.C.; Mi, T.; Maiti, S.; Olivares, S.; Rabinovich, B.; Huls, H.; Forget, M.A.; et al. Tethered IL-15 Augments Antitumor Activity and Promotes a Stem-Cell Memory Subset in Tumor-Specific T Cells. Proc. Natl. Acad. Sci. USA 2016, 113, E7788–E7797. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, I.; Ho, W.J.; Marple, A.; Ravich, J.W.; Tam, A.; Rahnama, R.; Fearnow, A.; Rietberg, C.; Yanik, S.; Solomou, E.E.; et al. Engineering CAR-NK Cells to Secrete IL-15 Sustains Their Anti-AML Functionality but Is Associated with Systemic Toxicities. J. Immunother. Cancer 2021, 9, e003894. [Google Scholar] [CrossRef]
- Mesquitta, W.T.; Wandsnider, M.; Kang, H.J.; Thomson, J.; Moskvin, O.; Suknuntha, K.; Slukvin, I.I. UM171 Expands Distinct Types of Myeloid and NK Progenitors from Human Pluripotent Stem Cells. Sci. Rep. 2019, 9, 6622. [Google Scholar] [CrossRef]
- Ni, Z.; Knorr, D.A.; Kaufman, D.S. Hematopoietic and Nature Killer Cell Development from Human Pluripotent Stem Cells. Methods Mol. Biol. 2013, 1029, 33–41. [Google Scholar] [CrossRef]
- Wang, Z.; McWilliams-Koeppen, H.P.; Reza, H.; Ostberg, J.R.; Chen, W.; Wang, X.; Huynh, C.; Vyas, V.; Chang, W.C.; Starr, R.; et al. 3D-Organoid Culture Supports Differentiation of Human CAR+ IPSCs into Highly Functional CAR T Cells. Cell Stem Cell 2022, 29, 515–527. [Google Scholar] [CrossRef]
- Jing, R.; Scarfo, I.; Najia, M.A.; Lummertz da Rocha, E.; Han, A.; Sanborn, M.; Bingham, T.; Kubaczka, C.; Jha, D.K.; Falchetti, M.; et al. EZH1 Repression Generates Mature IPSC-Derived CAR T Cells with Enhanced Antitumor Activity. Cell Stem Cell 2022, 29, 1181–1196.e6. [Google Scholar] [CrossRef]
- Li, Y.R.; Zhou, Y.; Kim, Y.J.; Zhu, Y.; Ma, F.; Yu, J.; Wang, Y.C.; Chen, X.; Li, Z.; Zeng, S.; et al. Development of Allogeneic HSC-Engineered INKT Cells for off-the-Shelf Cancer Immunotherapy. Cell Rep. Med. 2021, 2, 100449. [Google Scholar] [CrossRef]
- Liu, G.; David, B.T.; Trawczynski, M.; Fessler, R.G. Advances in Pluripotent Stem Cells: History, Mechanisms, Technologies, and Applications. Stem. Cell Rev. Rep. 2020, 16, 3–32. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Scesa, G.; Adami, R.; Bottai, D. IPSC Preparation and Epigenetic Memory: Does the Tissue Origin Matter? Cells 2021, 10, 1470. [Google Scholar] [CrossRef] [PubMed]
- Malik, N.; Rao, M.S. A Review of the Methods for Human IPSC Derivation. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2013; Volume 997. [Google Scholar]
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of Induced Pluripotent Stem Cells from Human Terminally Differentiated Circulating t Cells. Cell Stem Cell 2010, 7, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Seki, T.; Yuasa, S.; Fukuda, K. Derivation of Induced Pluripotent Stem Cells from Human Peripheral Circulating T Cells. Curr. Protoc. Stem Cell Biol. 2011, 18, 4A.3.1–4A.3.9. [Google Scholar] [CrossRef]
- Haridhasapavalan, K.K.; Borgohain, M.P.; Dey, C.; Saha, B.; Narayan, G.; Kumar, S.; Thummer, R.P. An Insight into Non-Integrative Gene Delivery Approaches to Generate Transgene-Free Induced Pluripotent Stem Cells. Gene 2019, 686, 146–159. [Google Scholar] [CrossRef]
- Hirsch, M.L.; Wolf, S.J.; Samulski, R.J. Delivering Transgenic DNA Exceeding the Carrying Capacity of AAV Vectors. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2016; Volume 1382. [Google Scholar]
- Li, J.; Song, W.; Pan, G.; Zhou, J. Advances in Understanding the Cell Types and Approaches Used for Generating Induced Pluripotent Stem Cells. J. Hematol. Oncol. 2014, 7, 50. [Google Scholar] [CrossRef]
- Rao, M.S.; Malik, N. Assessing IPSC Reprogramming Methods for Their Suitability in Translational Medicine. J. Cell Biochem. 2012, 113, 3061–3068. [Google Scholar] [CrossRef]
- al Abbar, A.; Ngai, S.C.; Nograles, N.; Alhaji, S.Y.; Abdullah, S. Induced Pluripotent Stem Cells: Reprogramming Platforms and Applications in Cell Replacement Therapy. Biores. Open Access 2020, 9, 121–136. [Google Scholar] [CrossRef]
- Ray, A.; Joshi, J.M.; Sundaravadivelu, P.K.; Raina, K.; Lenka, N.; Kaveeshwar, V.; Thummer, R.P. An Overview on Promising Somatic Cell Sources Utilized for the Efficient Generation of Induced Pluripotent Stem Cells. Stem Cell Rev. Rep. 2021, 17, 1954–1974. [Google Scholar] [CrossRef]
- Raab, S.; Klingenstein, M.; Liebau, S.; Linta, L. A Comparative View on Human Somatic Cell Sources for IPSC Generation. Stem Cells Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Shamsian, A.; Sahebnasagh, R.; Norouzy, A.; Hussein, S.H.; Ghahremani, M.H.; Azizi, Z. Cancer Cells as a New Source of Induced Pluripotent Stem Cells. Stem Cell Res. Ther. 2022, 13, 459. [Google Scholar] [CrossRef]
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.E.; Diol, N.R.; Propson, N.E.; et al. Chemically Defined Conditions for Human IPSC Derivation and Culture. Nat. Methods 2011, 8, 424–429. [Google Scholar] [CrossRef]
- Yasuda, S.Y.; Ikeda, T.; Shahsavarani, H.; Yoshida, N.; Nayer, B.; Hino, M.; Vartak-Sharma, N.; Suemori, H.; Hasegawa, K. Chemically Defined and Growth-Factor-Free Culture System for the Expansion and Derivation of Human Pluripotent Stem Cells. Nat. Biomed. Eng. 2018, 2, 173–182. [Google Scholar] [CrossRef]
- Kuo, H.H.; Gao, X.; DeKeyser, J.M.; Fetterman, K.A.; Pinheiro, E.A.; Weddle, C.J.; Fonoudi, H.; Orman, M.V.; Romero-Tejeda, M.; Jouni, M.; et al. Negligible-Cost and Weekend-Free Chemically Defined Human IPSC Culture. Stem Cell Rep. 2020, 14, 256–270. [Google Scholar] [CrossRef]
- Borys, B.S.; So, T.; Colter, J.; Dang, T.; Roberts, E.L.; Revay, T.; Larijani, L.; Krawetz, R.; Lewis, I.; Argiropoulos, B.; et al. Optimized Serial Expansion of Human Induced Pluripotent Stem Cells Using Low-Density Inoculation to Generate Clinically Relevant Quantities in Vertical-Wheel Bioreactors. Stem Cells Transl. Med. 2020, 9, 1036–1052. [Google Scholar] [CrossRef]
- Elanzew, A.; Sommer, A.; Pusch-Klein, A.; Brüstle, O.; Haupt, S. A Reproducible and Versatile System for the Dynamic Expansion of Human Pluripotent Stem Cells in Suspension. Biotechnol. J. 2015, 10, 1589–1599. [Google Scholar] [CrossRef]
- Nogueira, D.E.S.; Rodrigues, C.A.V.; Carvalho, M.S.; Miranda, C.C.; Hashimura, Y.; Jung, S.; Lee, B.; Cabral, J.M.S. Strategies for the Expansion of Human Induced Pluripotent Stem Cells as Aggregates in Single-Use Vertical-WheelTM Bioreactors. J. Biol. Eng. 2019, 13, 1–14. [Google Scholar] [CrossRef]
- Gordeeva, O. TGFβ Family Signaling Pathways in Pluripotent and Teratocarcinoma Stem Cells’ Fate Decisions: Balancing between Self-Renewal, Differentiation, and Cancer. Cells 2019, 8, 1500. [Google Scholar] [CrossRef]
- Tursky, M.L.; Loi, T.H.; Artuz, C.M.; Alateeq, S.; Wolvetang, E.J.; Tao, H.; Ma, D.D.; Molloy, T.J. Direct Comparison of Four Hematopoietic Differentiation Methods from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2020, 15, 735–748. [Google Scholar] [CrossRef]
- Fang, F.; Xie, S.; Chen, M.; Li, Y.; Yue, J.; Ma, J.; Shu, X.; He, Y.; Xiao, W.; Tian, Z. Advances in NK Cell Production. Cell Mol. Immunol. 2022, 19, 460–481. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; D’Souza, S.; Uenishi, G.; Park, M.; Lee, J.; Slukvin, I. Generation of T Cells from Human and Nonhuman Primate Pluripotent Stem Cells. Bio. Protoc. 2020, 10, e3675. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Yu, Q.; Long, Y.; Luo, Q.; Li, H.; Han, Y.; Xu, Y.; Fu, S.; Zeng, X.; Wei, C.; et al. Enhanced HSC-like Cell Generation from Mouse Pluripotent Stem Cells in a 3D Induction System Cocultured with Stromal Cells. Stem Cell Res. Ther. 2021, 12, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.F.; Kuo, H.C.; Chien, C.L.; Shun, C.T.; Yao, Y.L.; Ip, P.L.; Chuang, C.Y.; Wang, C.C.; Yang, Y.S.; Ho, H.N. Derivation, Characterization and Differentiation of Human Embryonic Stem Cells: Comparing Serum-Containing versus Serum-Free Media and Evidence of Germ Cell Differentiation. Hum. Reprod. 2007, 22, 567–577. [Google Scholar] [CrossRef]
- Bruveris, F.F.; Ng, E.S.; Stanley, E.G.; Elefanty, A.G. VEGF, FGF2, and BMP4 Regulate Transitions of Mesoderm to Endothelium and Blood Cells in a Human Model of Yolk Sac Hematopoiesis. Exp. Hematol. 2021, 103, 30–39.e2. [Google Scholar] [CrossRef]
- Mills, J.A.; Paluru, P.; Weiss, M.J.; Gadue, P.; French, D.L. Hematopoietic Differentiation of Pluripotent Stem Cells in Culture. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2014; Volume 1185. [Google Scholar] [CrossRef]
- Ackermann, M.; Rafiei Hashtchin, A.; Manstein, F.; Carvalho Oliveira, M.; Kempf, H.; Zweigerdt, R.; Lachmann, N. Continuous Human IPSC-Macrophage Mass Production by Suspension Culture in Stirred Tank Bioreactors. Nat. Protoc. 2022, 17, 513–519. [Google Scholar] [CrossRef]
- Pandey, P.R.; Tomney, A.; Woon, M.T.; Uth, N.; Shafighi, F.; Ngabo, I.; Vallabhaneni, H.; Levinson, Y.; Abraham, E.; Ben-Nun, I.F. End-to-End Platform for Human Pluripotent Stem Cell Manufacturing. Int. J. Mol. Sci. 2020, 21, 89. [Google Scholar] [CrossRef]
- Rieger, M.A.; Hoppe, P.S.; Smejkal, B.M.; Eitelhuber, A.C.; Schroeder, T. Hematopoietic Cytokines Can Instruct Lineage Choice. Science 2009, 325, 217–218. [Google Scholar] [CrossRef]
- Carlyle, J.R.; Zúñiga-Pflücker, J.C. Lineage Commitment and Differentiation of T and Natural Killer Lymphocytes in the Fetal Mouse. Immunol. Rev. 1998, 165, 63–74. [Google Scholar] [CrossRef]
- Smith, M.J.; Webber, B.R.; Mohtashami, M.; Stefanski, H.E.; Zúñiga-Pflücker, J.C.; Blazar, B.R. In Vitro T-Cell Generation from Adult, Embryonic, and Induced Pluripotent Stem Cells: Many Roads to One Destination. Stem Cells 2015, 33, 3174–3180. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A Guide to Cancer Immunotherapy: From T Cell Basic Science to Clinical Practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Iriguchi, S.; Yasui, Y.; Kawai, Y.; Arima, S.; Kunitomo, M.; Sato, T.; Ueda, T.; Minagawa, A.; Mishima, Y.; Yanagawa, N.; et al. A Clinically Applicable and Scalable Method to Regenerate T-Cells from IPSCs for off-the-Shelf T-Cell Immunotherapy. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Vizcardo, R.; Masuda, K.; Yamada, D.; Ikawa, T.; Shimizu, K.; Fujii, S.I.; Koseki, H.; Kawamoto, H. Regeneration of Human Tumor Antigen-Specific T Cells from IPSCs Derived from Mature CD8+ T Cells. Cell Stem Cell 2013, 12, 31–36. [Google Scholar] [CrossRef]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of Rejuvenated Antigen-Specific T Cells by Reprogramming to Pluripotency and Redifferentiation. Cell Stem Cell 2013, 12, 114–126. [Google Scholar] [CrossRef]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef]
- Euchner, J.; Sprissler, J.; Cathomen, T.; Fürst, D.; Schrezenmeier, H.; Debatin, K.M.; Schwarz, K.; Felgentreff, K. Natural Killer Cells Generated From Human Induced Pluripotent Stem Cells Mature to CD56brightCD16+NKp80+/- In-Vitro and Express KIR2DL2/DL3 and KIR3DL1. Front. Immunol. 2021, 12, 640672. [Google Scholar] [CrossRef]
- Grzywacz, B.; Kataria, N.; Kataria, N.; Blazar, B.R.; Miller, J.S.; Verneris, M.R. Natural Killer-Cell Differentiation by Myeloid Progenitors. Blood 2011, 117, 3548–3558. [Google Scholar] [CrossRef]
- Luevano, M.; Madrigal, A.; Saudemont, A. Generation of Natural Killer Cells from Hematopoietic Stem Cells in Vitro for Immunotherapy. Cell Mol. Immunol. 2012, 9, 310–320. [Google Scholar] [CrossRef]
- Pixley, F.J.; Stanley, E.R. Cytokines and Cytokine Receptors Regulating Cell Survival, Proliferation, and Differentiation in Hematopoiesis. In Handbook of Cell Signaling, 2nd ed.; Academic Press: Cambridge, MA, USA, 2010; Volume 3. [Google Scholar]
- Fiedler, K.; Brunner, C. Mechanisms Controlling Hematopoiesis. In Hematology—Science and Practice; IntechOpen: London, UK, 2012. [Google Scholar]
- Zhang, M.; Wen, B.; Anton, O.M.; Yao, Z.; Dubois, S.; Ju, W.; Sato, N.; DiLillo, D.J.; Bamford, R.N.; Ravetch, J.V.; et al. IL-15 Enhanced Antibody-Dependent Cellular Cytotoxicity Mediated by NK Cells and Macrophages. Proc. Natl. Acad. Sci. USA 2018, 115, E10924. [Google Scholar] [CrossRef]
- Parihar, R.; Dierksheide, J.; Hu, Y.; Carson, W.E. IL-12 Enhances the Natural Killer Cell Cytokine Response to Ab-Coated Tumor Cells. J. Clin. Investig. 2002, 110, 983–992. [Google Scholar] [CrossRef]
- Lusty, E.; Poznanski, S.M.; Kwofie, K.; Mandur, T.S.; Lee, D.A.; Ashkar, A.A. IL-18/IL-15/IL-12 Synergy Induces Elevated and Prolonged IFN-γ Production by Ex Vivo Expanded NK Cells Which Is Not Due to Enhanced STAT4 Activation. Mol. Immunol. 2017, 88, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Zwirner, N.W.; Ziblat, A. Regulation of NK Cell Activation and Effector Functions by the IL-12 Family of Cytokines: The Case of IL-27. Front. Immunol. 2017, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Kiniwa, T.; Enomoto, Y.; Terazawa, N.; Omia, A.; Miyataa, N.; Ishiwata, K.; Miyajima, A. NK Cells Activated by Interleukin-4 in Cooperation with Interleukin-15 Exhibit Distinctive Characteristics. Proc. Natl. Acad. Sci. USA 2016, 113, 10139–10144. [Google Scholar] [CrossRef] [PubMed]
- Gang, M.; Marin, N.D.; Wong, P.; Neal, C.C.; Marsala, L.; Foster, M.; Schappe, T.; Meng, W.; Tran, J.; Schaettler, M.; et al. CAR-Modified Memory-like NK Cells Exhibit Potent Responses to NK-Resistant Lymphomas. Blood 2020, 136, 2308–2318. [Google Scholar] [CrossRef]
- Mahapatra, S.; Mace, E.M.; Minard, C.G.; Forbes, L.R.; Vargas-Hernandez, A.; Duryea, T.K.; Makedonas, G.; Banerjee, P.P.; Shearer, W.T.; Orange, J.S. High-Resolution Phenotyping Identifies NK Cell Subsets That Distinguish Healthy Children from Adults. PLoS ONE 2017, 12, e0181134. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef]
- Freeman, B.E.; Raué, H.-P.; Hill, A.B.; Slifka, M.K. Cytokine-Mediated Activation of NK Cells during Viral Infection. J. Virol. 2015, 89, 7922–7931. [Google Scholar] [CrossRef]
- Schluns, K.S.; Lefrançois, L. Cytokine Control of Memory T-Cell Development and Survival. Nat. Rev. Immunol. 2003, 3, 269–279. [Google Scholar] [CrossRef]
- Zhang, Y.; Guan, X.Y.; Jiang, P. Cytokine and Chemokine Signals of T-Cell Exclusion in Tumors. Front. Immunol. 2020, 11, 594609. [Google Scholar] [CrossRef]
- Bevington, S.L.; Cauchy, P.; Withers, D.R.; Lane, P.J.L.; Cockerill, P.N. T Cell Receptor and Cytokine Signaling Can Function at Different Stages to Establish and Maintain Transcriptional Memory and Enable T Helper Cell Differentiation. Front. Immunol. 2017, 8, 204. [Google Scholar] [CrossRef]
- Geginat, J.; Sallusto, F.; Lanzavecchia, A. Cytokine-Driven Proliferation and Differentiation of Human Naive, Central Memory, and Effector Memory CD4+ T Cells. J. Exp. Med. 2001, 194, 1711–1720. [Google Scholar] [CrossRef]
- Raué, H.-P.; Brien, J.D.; Hammarlund, E.; Slifka, M.K. Activation of Virus-Specific CD8+ T Cells by Lipopolysaccharide-Induced IL-12 and IL-18. J. Immunol. 2004, 173, 6873–6881. [Google Scholar] [CrossRef]
- Smeltz, R.B. Profound Enhancement of the IL-12/IL-18 Pathway of IFN-γ Secretion in Human CD8+ Memory T Cell Subsets via IL-15. J. Immunol. 2007, 178, 4786–4792. [Google Scholar] [CrossRef]
- Freeman, B.E.; Hammarlund, E.; Raué, H.P.; Slifka, M.K. Regulation of Innate CD8+ T-Cell Activation Mediated by Cytokines. Proc. Natl. Acad. Sci. USA 2012, 109, 9971–9976. [Google Scholar] [CrossRef]
- Huang, W.; August, A. The Signaling Symphony: T Cell Receptor Tunes Cytokine-Mediated T Cell Differentiation. J. Leukoc. Biol. 2015, 97, 477–485. [Google Scholar] [CrossRef]
- Rochman, Y.; Spolski, R.; Leonard, W.J. New Insights into the Regulation of T Cells by Γc Family Cytokines. Nat. Rev. Immunol. 2009, 9, 480–490. [Google Scholar] [CrossRef]
- Dwyer, C.J.; Knochelmann, H.M.; Smith, A.S.; Wyatt, M.M.; Rivera, G.O.R.; Arhontoulis, D.C.; Bartee, E.; Li, Z.; Rubinstein, M.P.; Paulos, C.M. Fueling Cancer Immunothery with Common Gamma Chain Cytokines. Front. Immunol. 2019, 10, 263. [Google Scholar] [CrossRef]
- Maeda, T.; Nagano, S.; Ichise, H.; Kataoka, K.; Yamada, D.; Ogawa, S.; Koseki, H.; Kitawaki, T.; Kadowaki, N.; Takaori-Kondo, A.; et al. Regeneration of CD8αβ T Cells from T-Cell-Derived IPSC Imparts Potent Tumor Antigen-Specific Cytotoxicity. Cancer Res. 2016, 76, 6839–6850. [Google Scholar] [CrossRef]
- Cichocki, F.; Bjordahl, R.; Gaidarova, S.; Mahmood, S.; Abujarour, R.; Wang, H.; Tuininga, K.; Felices, M.; Davis, Z.B.; Bendzick, L.; et al. IPSC-Derived NK Cells Maintain High Cytotoxicity and Enhance in Vivo Tumor Control in Concert with T Cells and Anti–PD-1 Therapy. Sci. Transl. Med. 2020, 12, 5618. [Google Scholar] [CrossRef] [PubMed]
- Knorr, D.A.; Ni, Z.; Hermanson, D.; Hexum, M.K.; Bendzick, L.; Cooper, L.J.N.; Lee, D.A.; Kaufman, D.S. Clinical-Scale Derivation of Natural Killer Cells From Human Pluripotent Stem Cells for Cancer Therapy. Stem Cells Transl. Med. 2013, 2, 274–283. [Google Scholar] [CrossRef]
- LI, R.; JOHNSON, R.; YU, G.; MCKENNA, D.H.; HUBEL, A. Preservation of Cell-Based Immunotherapies for Clinical Trials. Cytotherapy 2019, 21, 943–957. [Google Scholar] [CrossRef]
- Cottle, C.; Porter, A.P.; Lipat, A.; Turner-Lyles, C.; Nguyen, J.; Moll, G.; Chinnadurai, R. Impact of Cryopreservation and Freeze-Thawing on Therapeutic Properties of Mesenchymal Stromal/Stem Cells and Other Common Cellular Therapeutics. Curr. Stem Cell Rep. 2022, 8, 72–92. [Google Scholar] [CrossRef] [PubMed]
- Uhrig, M.; Ezquer, F.; Ezquer, M. Improving Cell Recovery: Freezing and Thawing Optimization of Induced Pluripotent Stem Cells. Cells 2022, 11, 799. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Yang, D.; Li, Q.; Tian, W.; Guo, W. The Establishment of a Chemically Defined Serum-Free Culture System for Human Dental Pulp Stem Cells. Stem Cell Res. Ther. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Liu, W.; Chen, G. Cryopreservation of Human Pluripotent Stem Cells in Defined Medium. Curr. Protoc. Stem Cell Biol. 2014, 2014, 1C.17.1–1C.17.13. [Google Scholar] [CrossRef] [PubMed]
- Panch, S.R.; Srivastava, S.K.; Elavia, N.; McManus, A.; Liu, S.; Jin, P.; Highfill, S.L.; Li, X.; Dagur, P.; Kochenderfer, J.N.; et al. Effect of Cryopreservation on Autologous Chimeric Antigen Receptor T Cell Characteristics. Mol. Ther. 2019, 27, 1275–1285. [Google Scholar] [CrossRef]
- Jennes, W.; Kestens, L.; Nixon, D.F.; Shacklett, B.L. Enhanced ELISPOT Detection of Antigen-Specific T Cell Responses from Cryopreserved Specimens with Addition of Both IL-7 and IL-15—The Amplispot Assay. J. Immunol. Methods 2002, 270, 99–108. [Google Scholar] [CrossRef]
- Mark, C.; Czerwinski, T.; Roessner, S.; Mainka, A.; Hörsch, F.; Heublein, L.; Winterl, A.; Sanokowski, S.; Richter, S.; Bauer, N.; et al. Cryopreservation Impairs 3-D Migration and Cytotoxicity of Natural Killer Cells. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Berg, M.; Lundqvist, A.; McCoy, P.; Samsel, L.; Fan, Y.; Tawab, A.; Childs, R. Clinical-Grade Ex Vivo-Expanded Human Natural Killer Cells up-Regulate Activating Receptors and Death Receptor Ligands and Have Enhanced Cytolytic Activity against Tumor Cells. Cytotherapy 2009, 11, 341–355. [Google Scholar] [CrossRef]
- Hassan, H.T.; Zander, A. Stem Cell Factor as a Survival and Growth Factor in Human Normal and Malignant Hematopoiesis. Acta Haematol. 1996, 95, 257–262. [Google Scholar] [CrossRef]
- Nelson, B.H. IL-2, Regulatory T Cells, and Tolerance. J. Immunol. 2004, 172, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.K.; Bhoopathi, P.; Maji, S.; Kumar, A.; Guo, C.; Mannangatti, P.; Li, J.; Wang, X.Y.; Sarkar, D.; Emdad, L.; et al. Enhanced Cancer Therapy Using an Engineered Designer Cytokine Alone and in Combination With an Immune Checkpoint Inhibitor. Front. Oncol. 2022, 12, 812560. [Google Scholar] [CrossRef]
- Kim, M.Y.; Jayasinghe, R.; Devenport, J.M.; Ritchey, J.K.; Rettig, M.P.; O’Neal, J.; Staser, K.W.; Kennerly, K.M.; Carter, A.J.; Gao, F.; et al. A Long-Acting Interleukin-7, RhIL-7-HyFc, Enhances CAR T Cell Expansion, Persistence, and Anti-Tumor Activity. Nat. Commun. 2022, 13, 3296. [Google Scholar] [CrossRef]
- Bell, M.; Gottschalk, S. Engineered Cytokine Signaling to Improve CAR T Cell Effector Function. Front. Immunol. 2021, 12, 684642. [Google Scholar] [CrossRef] [PubMed]
- Koneru, M.; Purdon, T.J.; Spriggs, D.; Koneru, S.; Brentjens, R.J. IL-12 Secreting Tumor-Targeted Chimeric Antigen Receptor T Cells Eradicate Ovarian Tumors in Vivo. Oncoimmunology 2015, 4, e994446. [Google Scholar] [CrossRef]
- Hombach, A.; Barden, M.; Hannappel, L.; Chmielewski, M.; Rappl, G.; Sachinidis, A.; Abken, H. IL12 Integrated into the CAR Exodomain Converts CD8+ T Cells to Poly-Functional NK-like Cells with Superior Killing of Antigen-Loss Tumors. Mol. Ther. 2022, 30, 593–605. [Google Scholar] [CrossRef]
- Li, L.; Li, Q.; Yan, Z.-X.; Sheng, L.-S.; Fu, D.; Xu, P.; Wang, L.; Zhao, W.-L. Transgenic Expression of IL-7 Regulates CAR-T Cell Metabolism and Enhances in Vivo Persistence against Tumor Cells. Sci. Rep. 2022, 12, 12506. [Google Scholar] [CrossRef] [PubMed]
- Shum, T.; Omer, B.; Tashiro, H.; Kruse, R.L.; Wagner, D.L.; Parikh, K.; Yi, Z.; Sauer, T.; Liu, D.; Parihar, R.; et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov. 2017, 7, 1238–1247. [Google Scholar] [CrossRef]
- Sockolosky, J.T.; Trotta, E.; Parisi, G.; Picton, L.; Su, L.L.; Le, A.C.; Chhabra, A.; Silveria, S.L.; George, B.M.; King, I.C.; et al. Selective Targeting of Engineered T Cells Using Orthogonal IL-2 Cytokine-Receptor Complexes. Science 2018, 359, 1037–1042. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhuang, Q.; Wang, F.; Zhang, C.; Xu, C.; Gu, A.; Zhong, W.H.; Hu, Y.; Zhong, X. Co-Expression IL-15 Receptor Alpha with IL-15 Reduces Toxicity via Limiting IL-15 Systemic Exposure during CAR-T Immunotherapy. J. Transl. Med. 2022, 20, 432. [Google Scholar] [CrossRef]
- Rowley, J.; Monie, A.; Hung, C.F.; Wu, T.C. Expression of IL-15RA or an IL-15/IL-15RA Fusion on CD8+ T Cells Modifies Adoptively Transferred T-Cell Function in Cis. Eur. J. Immunol. 2009, 39, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, Q.; Han, Z.; Zhu, Y.; Shen, H.; Liu, Z.; Zhou, Z.; Ding, W.; Han, S.; He, J.; et al. IL-7 and CCR2b Co-Expression-Mediated Enhanced CAR-T Survival and Infiltration in Solid Tumors. Front. Oncol. 2021, 11, 734593. [Google Scholar] [CrossRef]
- Goto, S.; Sakoda, Y.; Adachi, K.; Sekido, Y.; Yano, S.; Eto, M.; Tamada, K. Enhanced Anti-Tumor Efficacy of IL-7/CCL19-Producing Human CAR-T Cells in Orthotopic and Patient-Derived Xenograft Tumor Models. Cancer Immunol. Immunother. 2021, 70, 2503–2515. [Google Scholar] [CrossRef]
- Tan, A.H.J.; Vinanica, N.; Campana, D. Chimeric Antigen Receptor-T Cells with Cytokine Neutralizing Capacity. Blood Adv. 2020, 4, 1419–1431. [Google Scholar] [CrossRef]
- Qiu, Y.; Su, M.; Liu, L.; Tang, Y.; Pan, Y.; Sun, J. Clinical Application of Cytokines in Cancer Immunotherapy. Drug Des. Devel. Ther. 2021, 15, 2269–2287. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in Cancer Immunotherapy. Oncoimmunology 2016, 5, e1163462. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Type of Cytokine/Cell Engineering | Cell Type | Engineering Method | In Vitro/In Vivo | Effect | Ref. |
|---|---|---|---|---|---|
| Engineering individual cytokines for exogenous use | |||||
| Superkine/IL-24S, engineered IL-24 | N/A | Adenoviruses | In vivo | Enhanced secretion and increased stability | [88] |
| rhIL-7-hyFc (homodimeric genetically modified IL-7) | N/A | CRISPR | In vivo and Clinical | Prolong IL-7 half-life in vivo. Promoted proliferation, persistence, and cytotoxicity of human CAR-T | [89] |
| Transgenic expression of cytokine or cytokine-receptor pairs/synthetic cytokine | |||||
| Co-expression of CAR and IL-12 | CAR-T | Retroviral Transduction | In vitro/In vivo | Improved T cell proliferation, cytokine secretion, and in vivo anti-tumor efficacy | [90] |
| Co-expression of CAR and IL-12 | CAR-T | Retroviral Transduction | In vitro/In vivo | Enhanced tumor recognition and elimination. NK-like phenotype | [91] |
| Transgenic expression of IL-7 | CAR-T | Lentivirus | In vitro/In vivo | Improved in vivo persistence and anti-tumor efficacy | [92] |
| Constitutive expression of IL-7 Receptor | CAR-T | Retroviral Transduction | In vitro/In vivo | Improved T cell proliferation, survival, and antitumor activity | [93] |
| Ortho2/Ortho2R (synthetic IL-2/IL-2R) | T cell | Retroviral Transduction | In vitro/In vivo | The orthogonal pairs of Ortho2/2R increase specificity in engineered CAR-T cell activation and reduce side effect of toxicity by native IL-2 | [94] |
| Co-expression of CAR, IL-7, CCR2b | CAR-T | Retroviral Transduction | In vitro/In vivo | Enhanced CAR-T survival, migration, and anti-tumor activity | [95] |
| Co-expression of CAR, IL-7, CCL19 | CAR-T | Retroviral Transduction | In vivo | significant inhibition of tumor growth and prolonged survival of pancreatic cancer mice model, following treatment with IL-7/CCL19-producing CAR-T cells | [96] |
| nonsignaling membrane-bound IL-6R | T cell | Retroviral Transduction | In vitro/In vivo | engineered T cells constitutively expressing a nonsignaling membrane-bound IL-6R to effectively deplete IL-6 produced by macrophage and thus reduce IL-6–mediated toxicity in mice | [97] |
| Type of Therapy | Indications * | Cytokines/Factors ** | IL-2 Dose Timing/Frequency | Status *** | Study Number |
|---|---|---|---|---|---|
| Autologous NK cells | AML, ALL, CML, NHL, CLL, others | IL-2 | With cell infusion, then 3× weekly or 2× weekly for up to 4 w | 2022–not yet recruiting | NCT05400122 |
| Hematopoietic Stem Cells | hematological disease | IL-2 | 5d/w from Day 15–40 post-graft | 2022–recruiting | NCT03862833 |
| Allogeneic, iPSC-derived CAR-NK | B-cell malignancies, NHL | IL-2 | During 3 w cell injection period | 2022–not yet recruiting | NCT05336409 |
| Allogeneic, iPSC-derived CAR-T cells (FT819) | BCL, CLL, ALL | IL-2 | Single dose in combination with cells | 2022–recruiting | NCT04629729 |
| Allogeneic, Tumor-infiltrating lymphocytes | NSCLC, melanoma | IL-2 | Dose following cell infusion | 2022–recruiting | NCT05361174 |
| Autologous, Peripheral Blood Lymphocytes | CLL, SLL | IL-2 | 6 doses following cell infusion | 2022–recruiting | NCT04155710 |
| Autologous, T cells | B-cell malignancies | IL-2 | Every other day for 2 weeks and then rest for 2 weeks for up to 6 months | 2021–suspended | NCT03098355 |
| Autologous, EBV-CTL cells | DLBCL, T cell lymphoma, gastric/nasopharyngeal carcinoma, Hodgkin’s lymphoma | IL-2 | Daily for 5 days after cell infusion | 2017–recruiting | NCT03044743 |
| Peripheral stem cells | Breast/kidney/ovarian cancers, lymphoma, sarcoma, others | IL-2 GM-CSF IFN-α | Daily Days 17–21 post cell administration | 2013–completed | NCT00003408 |
| Peripheral stem cells | Breast cancer, leukemia, lymphoma, MM, others | GM-CSF flt3 ligand TPO IL-3 | IL-2 treated SCs administered, then continuous IV for 5 d; repeat every 7 days for 4 courses | 2012–completed | NCT00006225 |
| Peripheral stem cells | Lymphoma, solid tumors | IL-2 G-CSF GM-CSF | IL-2 treated SCs administered, then continuous IV for 5 d; repeat every 7 days for 4 courses | 2010–completed | NCT00027937 |
| Peripheral stem cells | Breast/kidney/ovarian cancers, lymphoma, sarcoma, others | IL-11 G-CSF | n/a | 2010–completed | NCT00004157 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kao, C.-Y.; Mills, J.A.; Burke, C.J.; Morse, B.; Marques, B.F. Role of Cytokines and Growth Factors in the Manufacturing of iPSC-Derived Allogeneic Cell Therapy Products. Biology 2023, 12, 677. https://doi.org/10.3390/biology12050677
Kao C-Y, Mills JA, Burke CJ, Morse B, Marques BF. Role of Cytokines and Growth Factors in the Manufacturing of iPSC-Derived Allogeneic Cell Therapy Products. Biology. 2023; 12(5):677. https://doi.org/10.3390/biology12050677
Chicago/Turabian StyleKao, Chen-Yuan, Jason A. Mills, Carl J. Burke, Barry Morse, and Bruno F. Marques. 2023. "Role of Cytokines and Growth Factors in the Manufacturing of iPSC-Derived Allogeneic Cell Therapy Products" Biology 12, no. 5: 677. https://doi.org/10.3390/biology12050677