
SPEADI: Accelerated Analysis of IDP-Ion Interactions from MD-Trajectories

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Radial Distribution Function
2.2. Time-Resolved Radial Distribution Function
2.3. Implementation
2.4. Molecular Dynamics Simulations
2.4.1. Alpha-Synuclein
2.4.2. Humanin
3. Results
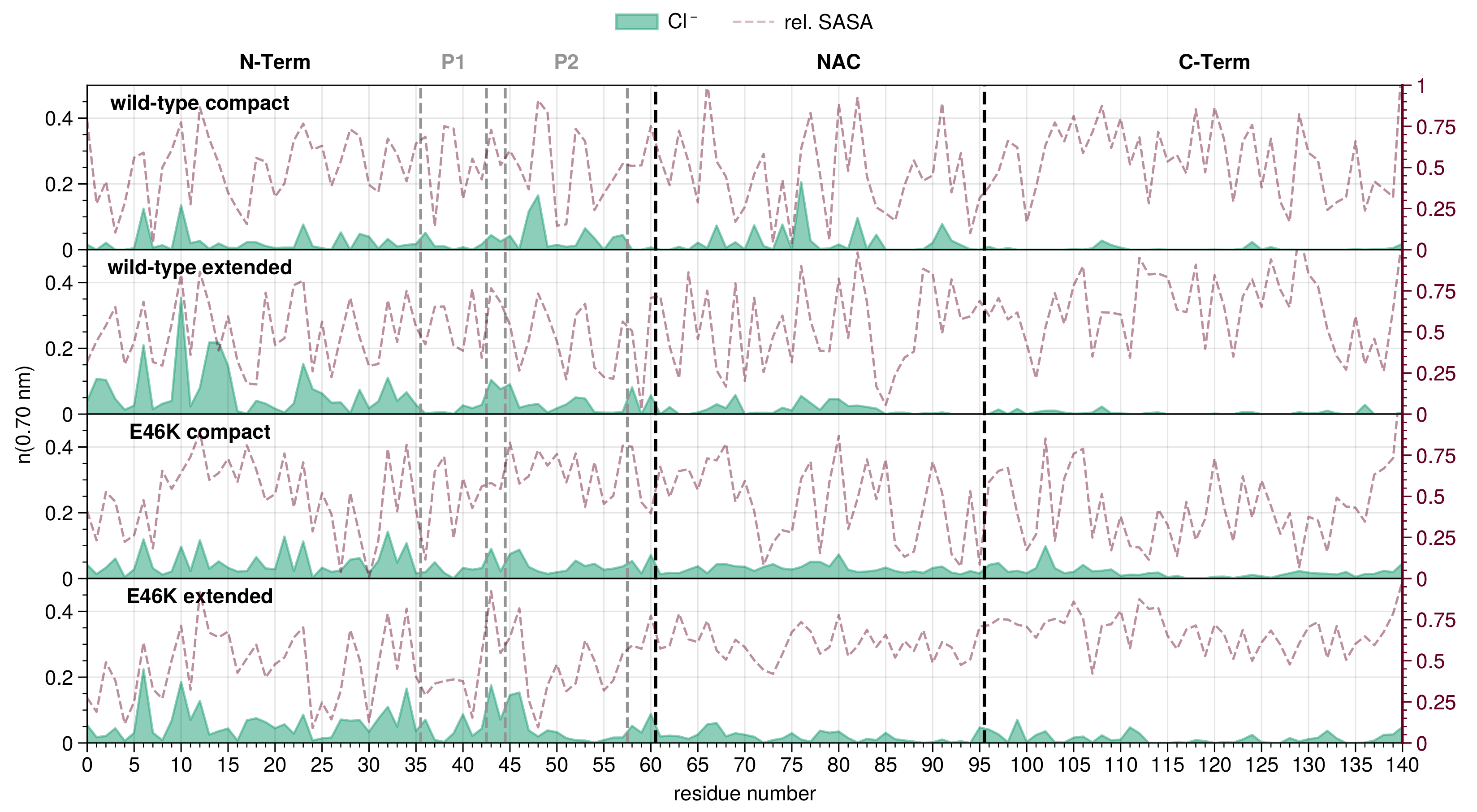
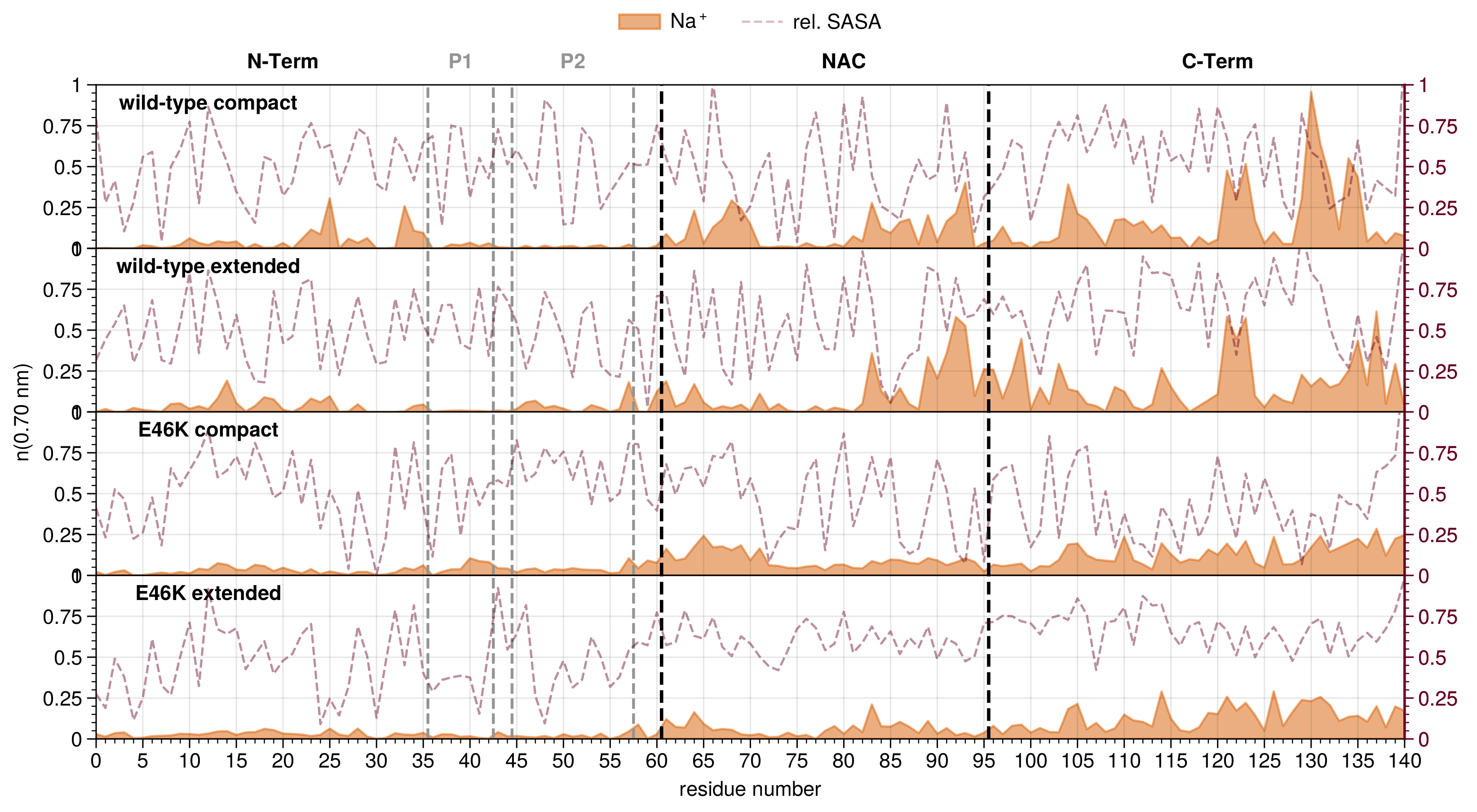
3.1. Detection of Mutant Effects in Alpha-Synuclein
3.2. Ion Equilibration Depends Strongly on Force Field Parameters
3.3. Detection of Mutant Effects in Humanin
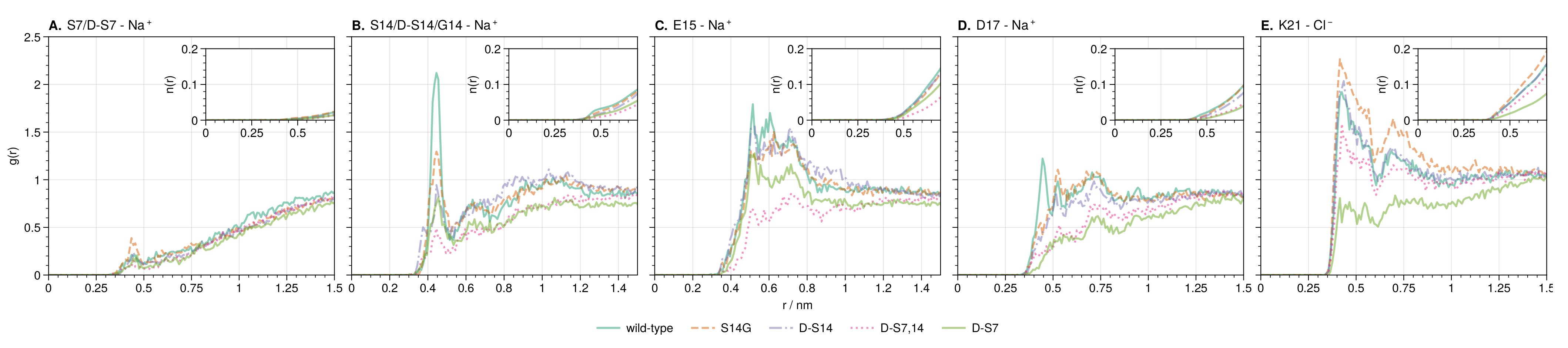
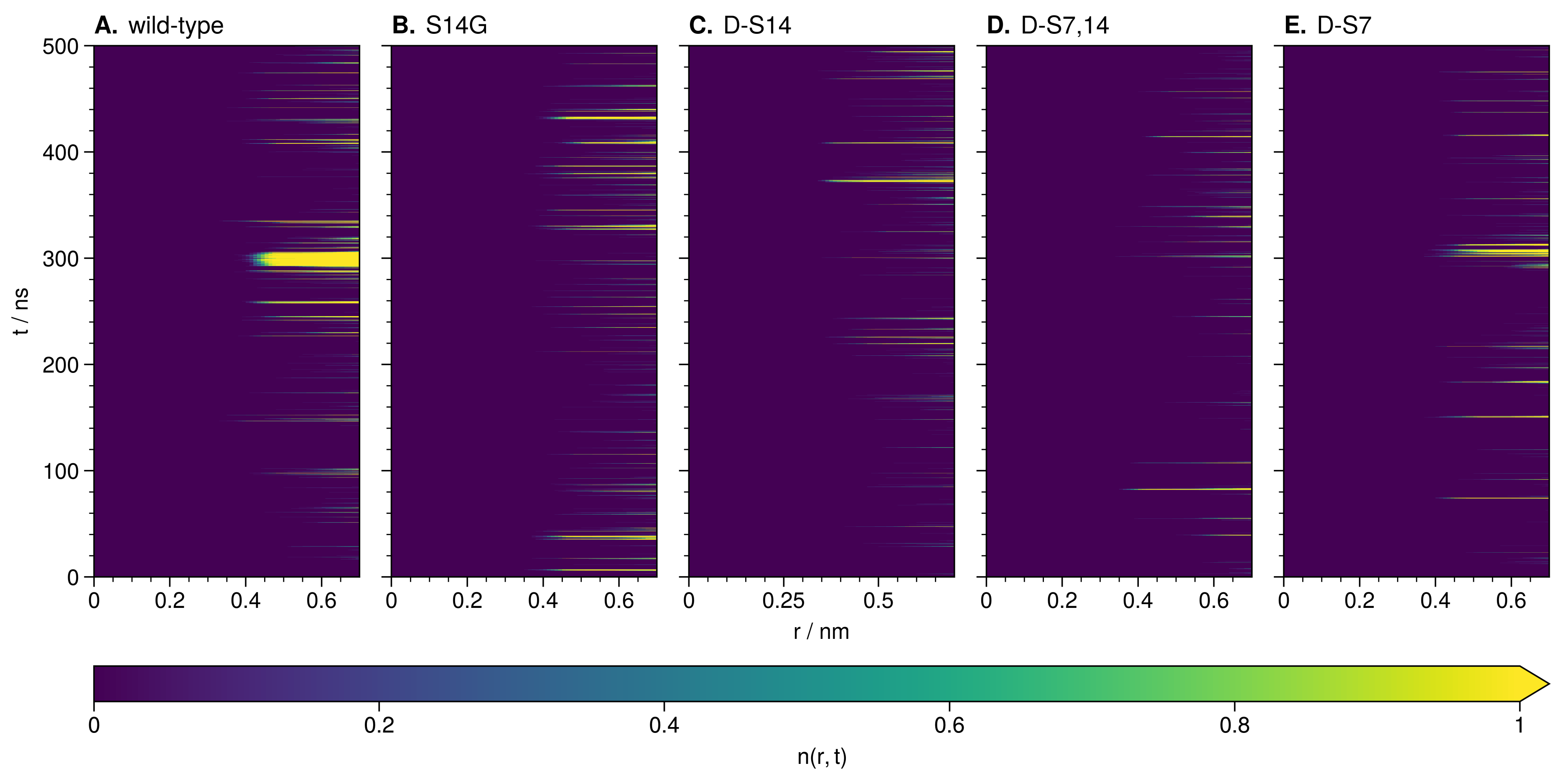
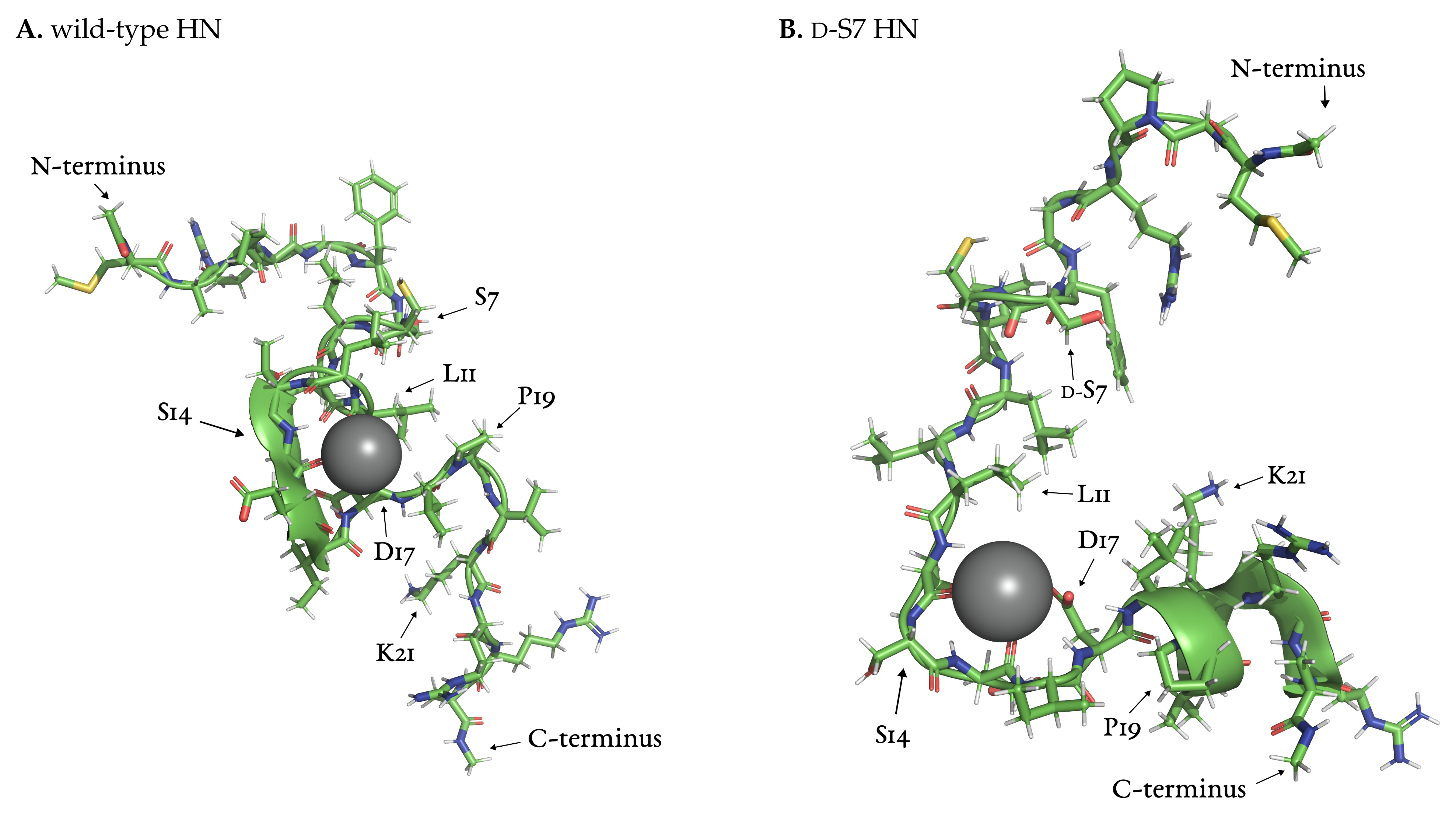
3.3.1. Local and Allosteric Effects at Positions S7 and S14
3.3.2. Ion-Stabilized Structures Centered on S14
3.3.3. Allosteric Effects of Mutations
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zangi, R.; Hagen, M.; Berne, B.J. Effect of Ions on the Hydrophobic Interaction Between Two Plates. J. Am. Chem. Soc. 2007, 129, 4678–4686. [Google Scholar] [CrossRef] [PubMed]
- Graziano, G. Hydrophobic Interaction of Two Large Plates: An Analysis of Salting-In/salting-Out Effects. Chem. Phys. Lett. 2010, 491, 54–58. [Google Scholar] [CrossRef]
- Tomé, L.I.N.; Pinho, S.P.; Jorge, M.; Gomes, J.R.B.; Coutinho, J.A.P. Salting-In With a Salting-Out Agent: Explaining the Cation Specific Effects on the Aqueous Solubility of Amino Acids. J. Phys. Chem. B 2013, 117, 6116–6128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pace, C.N.; Shirley, B.A.; McNutt, M.; Gajiwala, K. Forces Contributing To the Conformational Stability of Proteins. FASEB J. 1996, 10, 75–83. [Google Scholar] [CrossRef]
- Rose, G.D.; Fleming, P.J.; Banavar, J.R.; Maritan, A. A Backbone-Based Theory of Protein Folding. Proc. Natl. Acad. Sci. USA 2006, 103, 16623–16633. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.; Nachliel, E.; Gutman, M. Molecular Dynamics of a Protein Surface: Ion-Residues Interactions. Biophys. J. 2005, 89, 768–781. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R. Ions and the Protein Surface Revisited: Extensive Molecular Dynamics Simulations and Analysis of Protein Structures in Alkali-Chloride Solutions. J. Phys. Chem. B 2011, 115, 9213–9223. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P. Intrinsically Unstructured Proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Dunker, A.; Lawson, J.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically Disordered Protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Gough, J. Sequences and Topology: Intrinsic Disorder in the Evolving Universe of Protein Structure. Curr. Opin. Struct. Biol. 2011, 21, 379–381. [Google Scholar] [CrossRef]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing Protein Intrinsic Disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [Green Version]
- Müller-Späth, S.; Soranno, A.; Hirschfeld, V.; Hofmann, H.; Rüegger, S.; Reymond, L.; Nettels, D.; Schuler, B. Charge Interactions Can Dominate the Dimensions of Intrinsically Disordered Proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 14609–14614. [Google Scholar] [CrossRef] [Green Version]
- Soranno, A.; Koenig, I.; Borgia, M.B.; Hofmann, H.; Zosel, F.; Nettels, D.; Schuler, B. Single-Molecule Spectroscopy Reveals Polymer Effects of Disordered Proteins in Crowded Environments. Proc. Natl. Acad. Sci. USA 2014, 111, 4874–4879. [Google Scholar] [CrossRef] [Green Version]
- Choi, U.B.; Sanabria, H.; Smirnova, T.; Bowen, M.E.; Weninger, K.R. Spontaneous Switching Among Conformational Ensembles in Intrinsically Disordered Proteins. Biomolecules 2019, 9, 114. [Google Scholar] [CrossRef] [Green Version]
- Moses, D.; Yu, F.; Ginell, G.M.; Shamoon, N.M.; Koenig, P.S.; Holehouse, A.S.; Sukenik, S. Revealing the Hidden Sensitivity of Intrinsically Disordered Proteins To Their Chemical Environment. J. Phys. Chem. Lett. 2020, 11, 10131–10136. [Google Scholar] [CrossRef]
- Wohl, S.; Jakubowski, M.; Zheng, W. Salt-Dependent Conformational Changes of Intrinsically Disordered Proteins. J. Phys. Chem. Lett. 2021, 12, 6684–6691. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically Disordered Proteins and Their "Mysterious" (Meta)Physics. Front. Phys. 2019, 7, nil. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins in Human Diseases: Introducing the D2 Concept. Annu. Rev. Biophys. 2008, 37, 215–246. [Google Scholar] [CrossRef]
- Uversky, V.N. Flexible Nets of Malleable Guardians: Intrinsically Disordered Chaperones in Neurodegenerative Diseases. Chem. Rev. 2011, 111, 1134–1166. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsically Disordered Proteins and Their (disordered) Proteomes in Neurodegenerative Disorders. Front. Aging Neurosci. 2015, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. P53 Mutations in Human Cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.; Obradović, Z.; Dunker, A. Intrinsic Disorder in Cell-Signaling and Cancer-Associated Proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; LeGall, T.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. Abundance of Intrinsic Disorder in Protein Associated With Cardiovascular Disease. Biochemistry 2006, 45, 10448–10460. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, M.F.; Lolicato, F.; Mauro, G.D.; Milardi, D.; D’Urso, L.; Satriano, C.; Ramamoorthy, A.; Rosa, C.L. The Role of Cholesterol in Driving Iapp-Membrane Interactions. Biophys. J. 2016, 111, 140–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milardi, D.; Gazit, E.; Radford, S.E.; Xu, Y.; Gallardo, R.U.; Caflisch, A.; Westermark, G.T.; Westermark, P.; Rosa, C.L.; Ramamoorthy, A. Proteostasis of Islet Amyloid Polypeptide: A Molecular Perspective of Risk Factors and Protective Strategies for Type Ii Diabetes. Chem. Rev. 2021, 121, 1845–1893. [Google Scholar] [CrossRef]
- Tenchov, R.; Zhou, Q.A. Intrinsically Disordered Proteins: Perspective on Covid-19 Infection and Drug Discovery. ACS Infect. Dis. 2022, 8, 422–432. [Google Scholar] [CrossRef]
- Singh, O.; Das, B.K.; Chakraborty, D. Influence of Ion Specificity and Concentration on the Conformational Transition of Intrinsically Disordered Sheep Prion Peptide. ChemPhysChem 2022, 23, nil. [Google Scholar] [CrossRef]
- Ponomarev, S.Y.; Thayer, K.M.; Beveridge, D.L. Ion Motions in Molecular Dynamics Simulations on Dna. Proc. Natl. Acad. Sci. USA 2004, 101, 14771–14775. [Google Scholar] [CrossRef] [Green Version]
- Makarov, V.A.; Feig, M.; Andrews, B.K.; Pettitt, B.M. Diffusion of Solvent Around Biomolecular Solutes: A Molecular Dynamics Simulation Study. Biophys. J. 1998, 75, 150–158. [Google Scholar] [CrossRef] [Green Version]
- Dahanayake, J.N.; Mitchell-Koch, K.R. Entropy Connects Water Structure and Dynamics in Protein Hydration Layer. Phys. Chem. Chem. Phys. 2018, 20, 14765–14777. [Google Scholar] [CrossRef]
- Patra, M.; Karttunen, M.; Hyvönen, M.; Falck, E.; Lindqvist, P.; Vattulainen, I. Molecular Dynamics Simulations of Lipid Bilayers: Major Artifacts Due To Truncating Electrostatic Interactions. Biophys. J. 2003, 84, 3636–3645. [Google Scholar] [CrossRef] [Green Version]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. Mdanalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Gowers, R.; Linke, M.; Barnoud, J.; Reddy, T.; Melo, M.; Seyler, S.; Domański, J.; Dotson, D.; Buchoux, S.; Kenney, I.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016. [Google Scholar] [CrossRef] [Green Version]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernández, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. Mdtraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. Gromacs: High Performance Molecular Simulations Through Multi-Level Parallelism From Laptops To Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Tuckerman, M. Statistical Mechanics: Theory and Molecular Simulation; Oxford University Press: Oxford, UK, 2010. [Google Scholar]
- Brooks, C.; Karplus, M.; Pettitt, B.; Prigogine, I.; Rice, S. Proteins: A Theoretical Perspective of Dynamics, Structure, and Thermodynamics; Advances in Chemical Physics; Wiley: New York, NY, USA, 1988; Volume 71. [Google Scholar]
- Lyubartsev, A.P.; Laaksonen, A. Calculation of Effective Interaction Potentials From Radial Distribution Functions: A Reverse Monte Carlo Approach. Phys. Rev. E 1995, 52, 3730–3737. [Google Scholar] [CrossRef]
- Onufriev, A.V.; Izadi, S. Water Models for Biomolecular Simulations. WIREs Comput. Mol. Sci. 2017, 8, nil. [Google Scholar] [CrossRef]
- Dahanayake, J.N.; Shahryari, E.; Roberts, K.M.; Heikes, M.E.; Kasireddy, C.; Mitchell-Koch, K.R. Protein Solvent Shell Structure Provides Rapid Analysis of Hydration Dynamics. J. Chem. Inf. Model. 2019, 59, 2407–2422. [Google Scholar] [CrossRef]
- Wicky, B.I.M.; Shammas, S.L.; Clarke, J. Affinity of Idps To Their Targets Is Modulated By Ion-Specific Changes in Kinetics and Residual Structure. Proc. Natl. Acad. Sci. USA 2017, 114, 9882–9887. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.G.; Stone, J.E.; Kohlmeyer, A. Fast Analysis of Molecular Dynamics Trajectories With Graphics Processing Units-Radial Distribution Function Histogramming. J. Comput. Phys. 2011, 230, 3556–3569. [Google Scholar] [CrossRef] [Green Version]
- Van Hove, L. Correlations in Space and Time and Born Approximation Scattering in Systems of Interacting Particles. Phys. Rev. 1954, 95, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Zernike, F.; Prins, J.A. Die Beugung Von Röntgenstrahlen in Flüssigkeiten Als Effekt Der Molekülanordnung. Z. Für Phys. A Hadron. Nucl. 1927, 41, 184–194. [Google Scholar] [CrossRef]
- Kirkwood, J.G.; Maun, E.K.; Alder, B.J. Radial Distribution Functions and the Equation of State of a Fluid Composed of Rigid Spherical Molecules. J. Chem. Phys. 1950, 18, 1040–1047. [Google Scholar] [CrossRef]
- Bjerrum, N. Der Einfluss der Ionenassoziation auf die Aktivität der Ionen bei mittleren Assoziationsgraden; (Bjerrum: Untersuchungen über Ionenassoziation); Høst in Komm.: Copenhagen, Denmark, 1926. [Google Scholar]
- Shinohara, Y.; Matsumoto, R.; Thompson, M.W.; Ryu, C.W.; Dmowski, W.; Iwashita, T.; Ishikawa, D.; Baron, A.Q.R.; Cummings, P.T.; Egami, T. Identifying Water-Anion Correlated Motion in Aqueous Solutions Through Van Hove Functions. J. Phys. Chem. Lett. 2019, 10, 7119–7125. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, Y.; Dmowski, W.; Iwashita, T.; Ishikawa, D.; Baron, A.Q.R.; Egami, T. Local Correlated Motions in Aqueous Solution of Sodium Chloride. Phys. Rev. Mater. 2019, 3, 065604. [Google Scholar] [CrossRef]
- Egami, T.; Shinohara, Y. Correlated Atomic Dynamics in Liquid Seen in Real Space and Time. J. Chem. Phys. 2020, 153, 180902. [Google Scholar] [CrossRef]
- Hopkins, P.; Fortini, A.; Archer, A.J.; Schmidt, M. The Van Hove Distribution Function for Brownian Hard Spheres: Dynamical Test Particle Theory and Computer Simulations for Bulk Dynamics. J. Chem. Phys. 2010, 133, 224505. [Google Scholar] [CrossRef] [Green Version]
- Ghannad, Z. Fickian Yet Non-Gaussian Diffusion in Two-Dimensional Yukawa Liquids. Phys. Rev. E 2019, 100, 033211. [Google Scholar] [CrossRef]
- Lettinga, M.P.; Alvarez, L.; Korculanin, O.; Grelet, E. When Bigger Is Faster: A Self-Van Hove Analysis of the Enhanced Self-Diffusion of Non-Commensurate Guest Particles in Smectics. J. Chem. Phys. 2021, 154, 204901. [Google Scholar] [CrossRef]
- Van der Walt, S.; Colbert, S.C.; Varoquaux, G. The Numpy Array: A Structure for Efficient Numerical Computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array Programming With Numpy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Hoyer, S.; Hamman, J. Xarray: N-D Labeled Arrays and Datasets in Python. J. Open Res. Softw. 2017, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. Gromacs 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A Molecular Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics: Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Andersen, H.C. Molecular Dynamics Simulations At Constant Pressure And/or Temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Friesner, R.A.; Berne, B.J. Replica Exchange With Solute Scaling: A More Efficient Version of Replica Exchange With Solute Tempering (REST2). J. Phys. Chem. B 2011, 115, 9431–9438. [Google Scholar] [CrossRef] [Green Version]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. Plumed: A Portable Plugin for Free-Energy Calculations With Molecular Dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef] [Green Version]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. Plumed 2: New Feathers for an Old Bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Patriksson, A.; van der Spoel, D. A Temperature Predictor for Parallel Tempering Simulations. Phys. Chem. Chem. Phys. 2008, 10, 2073. [Google Scholar] [CrossRef]
- Rossetti, G.; Musiani, F.; Abad, E.; Dibenedetto, D.; Mouhib, H.; Fernandez, C.O.; Carloni, P. Conformational Ensemble of Human α-synuclein Physiological Form Predicted By Molecular Simulations. Phys. Chem. Chem. Phys. 2016, 18, 5702–5706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fauvet, B.; Fares, M.B.; Samuel, F.; Dikiy, I.; Tandon, A.; Eliezer, D.; Lashuel, H.A. Characterization of Semisynthetic and Naturally Nα-Acetylated α-Synuclein in Vitro and in Intact Cells. J. Biol. Chem. 2012, 287, 28243–28262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burré, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Südhof, T.C. Properties of Native Brain α-synuclein. Nature 2013, 498, E4–E6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 2.5.0. Available online: http://www.pymol.org/pymol (accessed on 17 March 2023).
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a Molecular Dynamics Force Field for Both Folded and Disordered Protein States. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, M.R.; Piana, S.; Tan, D.; LeVine, M.V.; Shaw, D.E. Development of Force Field Parameters for the Simulation of Single- and Double-Stranded Dna Molecules and Dna-Protein Complexes. J. Phys. Chem. B 2022, 126, 4442–4457. [Google Scholar] [CrossRef] [PubMed]
- Benaki, D.; Zikos, C.; Evangelou, A.; Livaniou, E.; Vlassi, M.; Mikros, E.; Pelecanou, M. Solution Structure of Humanin, a Peptide Against Alzheimer’s Disease-Related Neurotoxicity. Biochem. Biophys. Res. Commun. 2005, 329, 152–160. [Google Scholar] [CrossRef]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.B.; Meyer, E.F.; Rodgers, M.D.B.J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank. a Computer-Based Archival File for Macromolecular Structures. Eur. J. Biochem. 1977, 80, 319–324. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H.; Markley, J.L. The Worldwide Protein Data Bank (wwPDB): Ensuring a Single, Uniform Archive of Pdb Data. Nucleic Acids Res. 2007, 35, D301–D303. [Google Scholar] [CrossRef] [Green Version]
- wwPDB Consortium; Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Costanzo, L.D.; Christie, C.; Duarte, J.M.; Dutta, S.; et al. Protein Data Bank: The Single Global Archive for 3d Macromolecular Structure Data. Nucleic Acids Res. 2018, 47, D520–D528. [Google Scholar] [CrossRef] [Green Version]
- Dauer, W.; Przedborski, S. Parkinson’s Disease. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Emamzadeh, F. Alpha-Synuclein Structure, Functions, and Interactions. J. Res. Med. Sci. 2016, 21, 29. [Google Scholar] [CrossRef]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different Species of -synuclein Oligomers Induce Calcium Influx and Seeding. J. Neurosci. 2007, 27, 9220–9232. [Google Scholar] [CrossRef] [Green Version]
- Bengoa-Vergniory, N.; Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Alpha-Synuclein Oligomers: A New Hope. Acta Neuropathol. 2017, 134, 819–838. [Google Scholar] [CrossRef] [Green Version]
- Ahn, B.H.; Rhim, H.; Kim, S.Y.; Sung, Y.M.; Lee, M.Y.; Choi, J.Y.; Wolozin, B.; Chang, J.S.; Lee, Y.H.; Kwon, T.K.; et al. α-Synuclein Interacts With Phospholipase D Isozymes and Inhibits Pervanadate-Induced Phospholipase D Activation in Human Embryonic Kidney-293 Cells. J. Biol. Chem. 2002, 277, 12334–12342. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, S.; Andersen, J.K. Alpha Synuclein Aggregation: Is It the Toxic Gain of Function Responsible for Neurodegeneration in Parkinson’s Disease? Mech. Ageing Dev. 2001, 122, 1499–1510. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Ivanova, M.I.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Sangwan, S.; Guenther, E.L.; Johnson, L.M.; Zhang, M.; et al. Structure of the Toxic Core of α-synuclein From Invisible Crystals. Nature 2015, 525, 486–490. [Google Scholar] [CrossRef] [Green Version]
- Ly, T.; Julian, R.R. Protein-Metal Interactions of Calmodulin and α-synuclein Monitored By Selective Noncovalent Adduct Protein Probing Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2008, 19, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Ahn, M.; Kim, S.; Kang, M.; Ryu, Y.; Kim, T.D. Chaperone-Like Activities of α-synuclein: α-Synuclein Assists Enzyme Activities of Esterases. Biochem. Biophys. Res. Commun. 2006, 346, 1142–1149. [Google Scholar] [CrossRef]
- Hoyer, W.; Antony, T.; Cherny, D.; Heim, G.; Jovin, T.M.; Subramaniam, V. Dependence of α-Synuclein Aggregate Morphology on Solution Conditions. J. Mol. Biol. 2002, 322, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Doherty, C.P.A.; Ulamec, S.M.; Maya-Martinez, R.; Good, S.C.; Makepeace, J.; Khan, G.N.; van Oosten-Hawle, P.; Radford, S.E.; Brockwell, D.J. A Short Motif in the N-Terminal Region of α-synuclein Is Critical for Both Aggregation and Function. Nat. Struct. Mol. Biol. 2020, 27, 249–259. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Van der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Appadurai, R.; Koneru, J.K.; Bonomi, M.; Robustelli, P.; Srivastava, A. Demultiplexing the heterogeneous conformational ensembles of intrinsically disordered proteins into structurally similar clusters. bioRxiv 2022. [Google Scholar] [CrossRef]
- Tien, M.Z.; Meyer, A.G.; Sydykova, D.K.; Spielman, S.J.; Wilke, C.O. Maximum Allowed Solvent Accessibilites of Residues in Proteins. PLoS ONE 2013, 8, e80635. [Google Scholar] [CrossRef] [Green Version]
- Zarbiv, Y.; Simhi-Haham, D.; Israeli, E.; Elhadi, S.A.; Grigoletto, J.; Sharon, R. Lysine Residues At the First and Second Ktkegv Repeats Mediate α-Synuclein Binding To Membrane Phospholipids. Neurobiol. Dis. 2014, 70, 90–98. [Google Scholar] [CrossRef]
- Lee, J.H.; Ying, J.; Bax, A. Nuclear Magnetic Resonance Observation of α-Synuclein Membrane Interaction By Monitoring the Acetylation Reactivity of Its Lysine Side Chains. Biochemistry 2016, 55, 4949–4959. [Google Scholar] [CrossRef]
- Cho, M.K.; Kim, H.Y.; Fernandez, C.O.; Becker, S.; Zweckstetter, M. Conserved Core of Amyloid Fibrils of Wild Type and A30p Mutant α-synuclein. Protein Sci. 2011, 20, 387–395. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Han, W.; Ma, W.; Schulten, K. Transient β-hairpin Formation in α-synuclein Monomer Revealed By Coarse-Grained Molecular Dynamics Simulation. J. Chem. Phys. 2015, 143, 243142. [Google Scholar] [CrossRef] [Green Version]
- Waxman, E.A.; Mazzulli, J.R.; Giasson, B.I. Characterization of Hydrophobic Residue Requirements for α-Synuclein Fibrillization. Biochemistry 2009, 48, 9427–9436. [Google Scholar] [CrossRef] [Green Version]
- Guzzo, A.; Delarue, P.; Rojas, A.; Nicolaï, A.; Maisuradze, G.G.; Senet, P. Wild-Type α-Synuclein and Variants Occur in Different Disordered Dimers and Pre-Fibrillar Conformations in Early Stage of Aggregation. Front. Mol. Biosci. 2022, 9, nil. [Google Scholar] [CrossRef]
- Binolfi, A.; Rasia, R.M.; Bertoncini, C.W.; Ceolin, M.; Zweckstetter, M.; Griesinger, C.; Jovin, T.M.; Fernández, C.O. Interaction of α-Synuclein With Divalent Metal Ions Reveals Key Differences: A Link Between Structure, Binding Specificity and Fibrillation Enhancement. J. Am. Chem. Soc. 2006, 128, 9893–9901. [Google Scholar] [CrossRef]
- Golts, N.; Snyder, H.; Frasier, M.; Theisler, C.; Choi, P.; Wolozin, B. Magnesium Inhibits Spontaneous and Iron-Induced Aggregation of α-Synuclein. J. Biol. Chem. 2002, 277, 16116–16123. [Google Scholar] [CrossRef] [Green Version]
- Semenyuk, P.I. Remd Simulations of Full-Length Alpha-Synuclein Together With Ligands Reveal Binding Region and Effect on Amyloid Conversion. Int. J. Mol. Sci. 2022, 23, 11545. [Google Scholar] [CrossRef]
- Grabenauer, M.; Bernstein, S.L.; Lee, J.C.; Wyttenbach, T.; Dupuis, N.F.; Gray, H.B.; Winkler, J.R.; Bowers, M.T. Spermine Binding To Parkinson’s Protein α-Synuclein and Its Disease-Related A30p and A53t Mutants. J. Phys. Chem. B 2008, 112, 11147–11154. [Google Scholar] [CrossRef] [Green Version]
- Roeters, S.J.; Iyer, A.; Pletikapić, G.; Kogan, V.; Subramaniam, V.; Woutersen, S. Evidence for Intramolecular Antiparallel Beta-Sheet Structure in Alpha-Synuclein Fibrils From a Combination of Two-Dimensional Infrared Spectroscopy and Atomic Force Microscopy. Sci. Rep. 2017, 7, 41051. [Google Scholar] [CrossRef]
- Bai, J.; Cheng, K.; Liu, M.; Li, C. Impact of the α-Synuclein Initial Ensemble Structure on Fibrillation Pathways and Kinetics. J. Phys. Chem. B 2016, 120, 3140–3147. [Google Scholar] [CrossRef]
- Fujiwara, S.; Kono, F.; Matsuo, T.; Sugimoto, Y.; Matsumoto, T.; Narita, A.; Shibata, K. Dynamic Properties of Human α-Synuclein Related To Propensity To Amyloid Fibril Formation. J. Mol. Biol. 2019, 431, 3229–3245. [Google Scholar] [CrossRef]
- Ramis, R.; Ortega-Castro, J.; Vilanova, B.; Adrover, M.; Frau, J. Unraveling the Nacl Concentration Effect on the First Stages of α-Synuclein Aggregation. Biomacromolecules 2020, 21, 5200–5212. [Google Scholar] [CrossRef]
- Gupta, A.; Dey, S.; Hicks, A.; Zhou, H.X. Artificial Intelligence Guided Conformational Mining of Intrinsically Disordered Proteins. Commun. Biol. 2022, 5, 610. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Niikura, T.; Tajima, H.; Yasukawa, T.; Sudo, H.; Ito, Y.; Kita, Y.; Kawasumi, M.; Kouyama, K.; Doyu, M.; et al. A Rescue Factor Abolishing Neuronal Cell Death By a Wide Spectrum of Familial Alzheimer’s Disease Genes and Aβ. Proc. Natl. Acad. Sci. USA 2001, 98, 6336–6341. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yen, K.; Cohen, P. Humanin: A Harbinger of Mitochondrial-Derived Peptides? Trends Endocrinol. Metab. 2013, 24, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yen, K.; Lee, C.; Mehta, H.; Cohen, P. The Emerging Role of the Mitochondrial-Derived Peptide Humanin in Stress Resistance. J. Mol. Endocrinol. 2012, 50, R11–R19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.T.; Zhao, L.; Li, J.H. Neuroprotective Peptide Humanin Inhibits Inflammatory Response in Astrocytes Induced By Lipopolysaccharide. Neurochem. Res. 2013, 38, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Klein, L.E.; Cui, L.; Gong, Z.; Su, K.; Muzumdar, R. A Humanin Analog Decreases Oxidative Stress and Preserves Mitochondrial Integrity in Cardiac Myoblasts. Biochem. Biophys. Res. Commun. 2013, 440, 197–203. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.; Wang, S.; Liu, Z.; Cao, Y.; Wang, F.; Wang, J.; Liu, C.; Xie, Y.; Zhang, Y. Gly[14]-humanin Inhibits ox-LDL Uptake and Stimulates Cholesterol Efflux in Macrophage-Derived Foam Cells. Biochem. Biophys. Res. Commun. 2017, 482, 93–99. [Google Scholar] [CrossRef]
- Terashita, K.; Hashimoto, Y.; Niikura, T.; Tajima, H.; Yamagishi, Y.; Ishizaka, M.; Kawasumi, M.; Chiba, T.; Kanekura, K.; Yamada, M.; et al. Two Serine Residues Distinctly Regulate the Rescue Function of Humanin, an Inhibiting Factor of Alzheimer’s Disease-Related Neurotoxicity: Functional Potentiation By Isomerization and Dimerization. J. Neurochem. 2003, 85, 1521–1538. [Google Scholar] [CrossRef] [Green Version]
- Kariya, S.; Hirano, M.; Furiya, Y.; Sugie, K.; Ueno, S. Humanin Detected in Skeletal Muscles of Melas Patients: A Possible New Therapeutic Agent. Acta Neuropathol. 2005, 109, 367–372. [Google Scholar] [CrossRef]
- Hayashi, K.; Sasabe, J.; Chiba, T.; Aiso, S.; Utsunomiya-Tate, N. D-Ser-Containing Humanin Shows Promotion of Fibril Formation. Amino Acids 2011, 42, 2293–2297. [Google Scholar] [CrossRef]
- Maftei, M.; Tian, X.; Manea, M.; Exner, T.E.; Schwanzar, D.; Arnim, C.A.F.; Przybylski, M. Interaction Structure of the Complex Between Neuroprotective Factor Humanin and Alzheimer’s β-amyloid Peptide Revealed By Affinity Mass Spectrometry and Molecular Modeling. J. Pept. Sci. 2012, 18, 373–382. [Google Scholar] [CrossRef]
- Ikonen, M.; Liu, B.; Hashimoto, Y.; Ma, L.; Lee, K.W.; Niikura, T.; Nishimoto, I.; Cohen, P. Interaction Between the Alzheimer’s Survival Peptide Humanin and Insulin-Like Growth Factor-Binding Protein 3 Regulates Cell Survival and Apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 13042–13047. [Google Scholar] [CrossRef] [Green Version]
- Weber, O.C.; Uversky, V.N. How Accurate Are Your Simulations? Effects of Confined Aqueous Volume and Amber Ff99sb and Charmm22/cmap Force Field Parameters on Structural Ensembles of Intrinsically Disordered Proteins: Amyloid-β42 in Water. Intrinsically Disord. Proteins 2017, 5, e1377813. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.K.; Pitrou, A.; Seibert, S. Numba. In Proceedings of the Second Workshop on the LLVM Compiler Infrastructure in HPC-LLVM ’15, Austin, TX, USA, 15 November 2015. [Google Scholar] [CrossRef]
- Bradbury, J.; Frostig, R.; Hawkins, P.; Johnson, M.J.; Leary, C.; Maclaurin, D.; Necula, G.; Paszke, A.; VanderPlas, J.; Wanderman-Milne, S.; et al. JAX: Composable Transformations of Python+NumPy Programs (v0.2.5). Available online: http://github.com/google/jax (accessed on 17 March 2023).
- De Bruyn, E. SPEADI: Scalable Protein Environment Analysis for Dynamics and Ions (v1.0.0). Available online: https://github.com/FZJ-JSC/speadi (accessed on 17 March 2023). [CrossRef]
- Krause, D. Juwels: Modular Tier-0/1 Supercomputer At Jülich Supercomputing Centre. J. Large-Scale Res. Facil. JLSRF 2019, 5, A135. [Google Scholar] [CrossRef] [Green Version]
- Kesselheim, S.; Herten, A.; Krajsek, K.; Ebert, J.; Jitsev, J.; Cherti, M.; Langguth, M.; Gong, B.; Stadtler, S.; Mozaffari, A.; et al. JUWELS Booster-A Supercomputer for Large-Scale AI Research. In Lecture Notes in Computer Science; Springer International Publishing: Zurich, Switzerland, 2021; pp. 453–468. [Google Scholar] [CrossRef]
- Alvarez, D. Juwels Cluster and Booster: Exascale Pathfinder With Modular Supercomputing Architecture At Juelich Supercomputing Centre. J. Large-Scale Res. Facil. JLSRF 2021, 7, A183. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trajectory | Conformation | Cluster | % of Converged Trajectory | /nm |
|---|---|---|---|---|
| wild-type | compact | 14 | 8.76 | 2.52 (0.05) |
| extended | 11 | 7.76 | 3.25 (0.11) | |
| E46K | compact | 4 | 9.88 | 2.95 (0.09) |
| extended | 16 | 3.52 | 3.71 (0.17) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Bruyn, E.; Dorn, A.E.; Zimmermann, O.; Rossetti, G. SPEADI: Accelerated Analysis of IDP-Ion Interactions from MD-Trajectories. Biology 2023, 12, 581. https://doi.org/10.3390/biology12040581
de Bruyn E, Dorn AE, Zimmermann O, Rossetti G. SPEADI: Accelerated Analysis of IDP-Ion Interactions from MD-Trajectories. Biology. 2023; 12(4):581. https://doi.org/10.3390/biology12040581
Chicago/Turabian Stylede Bruyn, Emile, Anton Emil Dorn, Olav Zimmermann, and Giulia Rossetti. 2023. "SPEADI: Accelerated Analysis of IDP-Ion Interactions from MD-Trajectories" Biology 12, no. 4: 581. https://doi.org/10.3390/biology12040581