
How AlphaFold2 Predicts Conditionally Folding Regions Annotated in an Intrinsically Disordered Protein Database, IDEAL

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Dataset
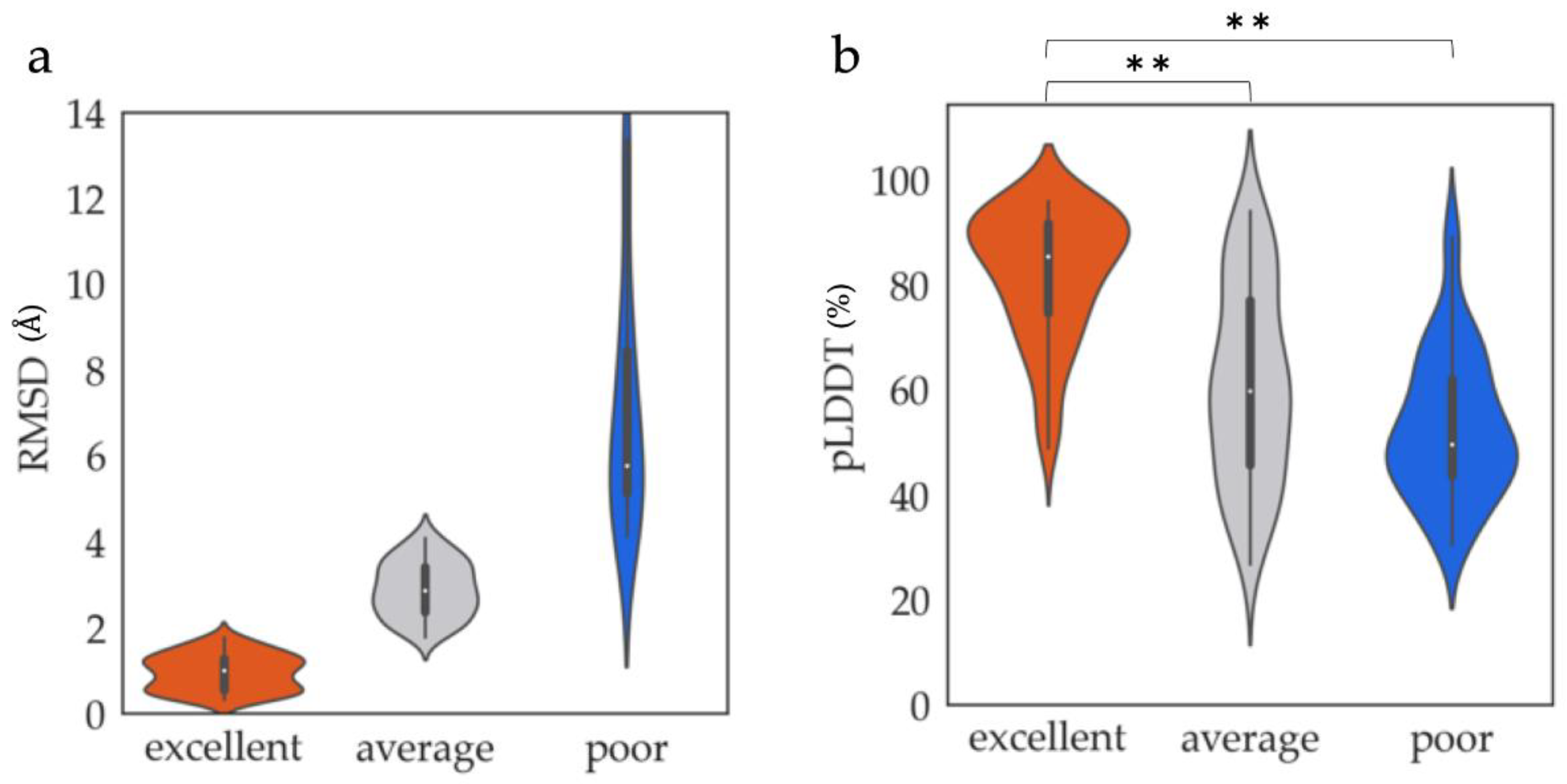
2.2. Classification of ProS Structures
2.3. Structural and Sequential Features
2.4. Characterization of ProSs in the Excellent Class
2.5. Software
3. Results and Discussions
3.1. ProSs Agreeing with AF2 Models
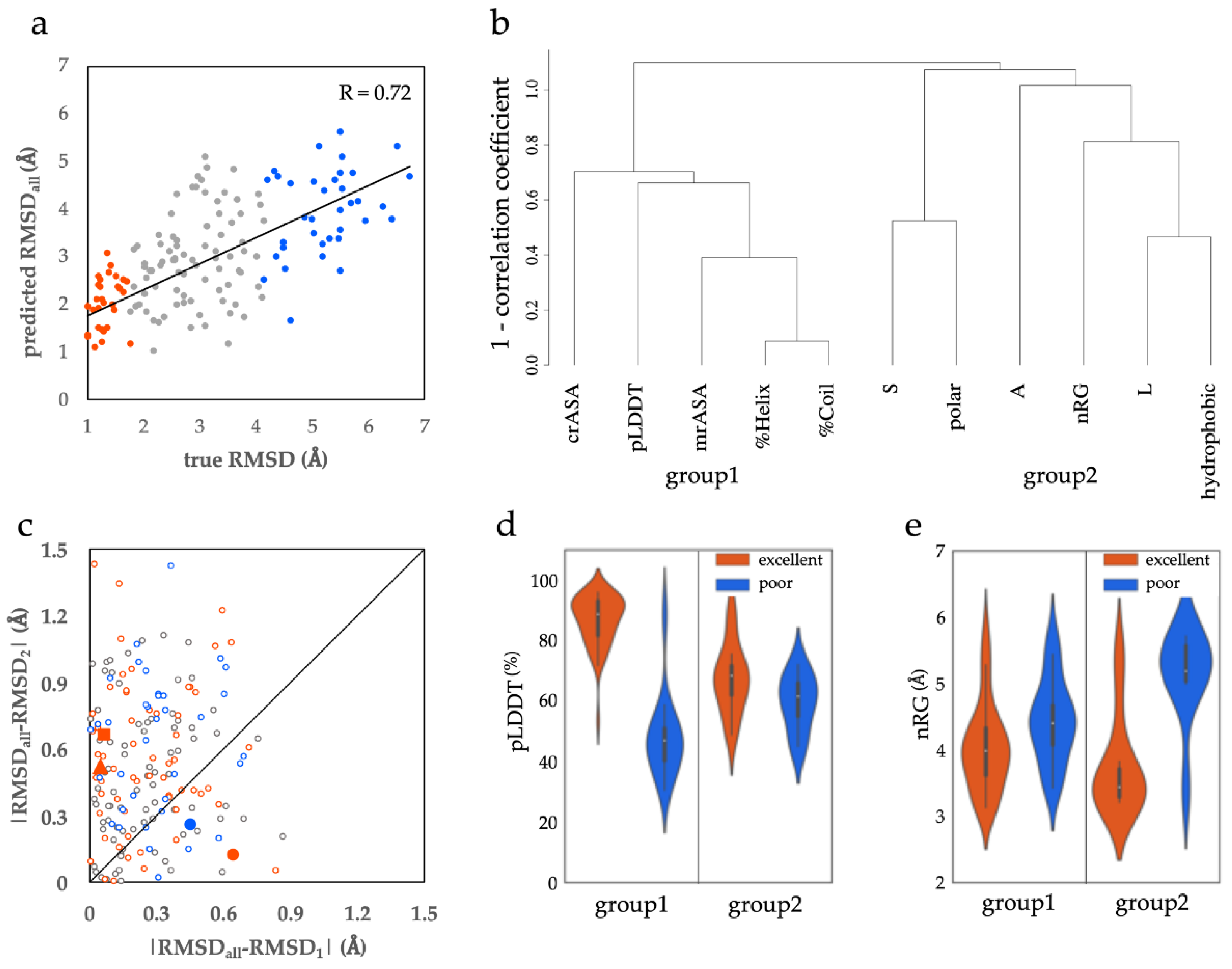
3.1.1. Structural and Sequential Features to Differentiate between Excellent and Poor Classes
3.1.2. Two Types of ProSs in the Excellent Class
3.1.3. Features of the Poor Class ProSs
3.1.4. Examples of ProSs in the Excellent Class
3.1.5. Examples of ProSs in the Poor Class
3.2. Comparison with Other Assessments of Conditionally Folding Regions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix B
References
- Branden, C.I.; Tooze, J. Introduction to Protein Structure; Garland Science: New York, NY, USA, 2012. [Google Scholar]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The Single Global Macromolecular Structure Archive. Methods Mol. Biol. 2017, 1607, 627–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kryshtafovych, A.; Schwede, T.; Topf, M.; Fidelis, K.; Moult, J. Critical assessment of methods of protein structure prediction (CASP)-Round XIV. Proteins 2021, 89, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- David, A.; Islam, S.; Tankhilevich, E.; Sternberg, M.J.E. The AlphaFold Database of Protein Structures: A Biologist’s Guide. J. Mol. Biol. 2022, 434, 167336. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Dunker, A.K.; Brown, C.J.; Lawson, J.D.; Iakoucheva, L.M.; Obradović, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar] [CrossRef] [Green Version]
- Dyson, H.J.; Wright, P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005, 6, 197–208. [Google Scholar] [CrossRef]
- Tompa, P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005, 579, 3346–3354. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iakoucheva, L.M.; Brown, C.J.; Lawson, J.D.; Obradović, Z.; Dunker, A.K. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 2002, 323, 573–584. [Google Scholar] [CrossRef]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef]
- Minezaki, Y.; Homma, K.; Kinjo, A.R.; Nishikawa, K. Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. J. Mol. Biol. 2006, 359, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Uversky, V.N.; Chen, Z.; Dunker, A.K.; Obradovic, Z. Short Linear Motifs recognized by SH2, SH3 and Ser/Thr Kinase domains are conserved in disordered protein regions. BMC Genom. 2008, 9 (Suppl. 2), S26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Schad, E.; Ficho, E.; Pancsa, R.; Simon, I.; Dosztanyi, Z.; Meszaros, B. DIBS: A repository of disordered binding sites mediating interactions with ordered proteins. Bioinformatics 2018, 34, 535–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuchi, S.; Sakamoto, S.; Nobe, Y.; Murakami, S.D.; Amemiya, T.; Hosoda, K.; Koike, R.; Hiroaki, H.; Ota, M. IDEAL: Intrinsically Disordered proteins with Extensive Annotations and Literature. Nucleic Acids Res. 2012, 40, D507–D511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuchi, S.; Amemiya, T.; Sakamoto, S.; Nobe, Y.; Hosoda, K.; Kado, Y.; Murakami, S.D.; Koike, R.; Hiroaki, H.; Ota, M. IDEAL in 2014 illustrates interaction networks composed of intrinsically disordered proteins and their binding partners. Nucleic Acids Res. 2014, 42, D320–D325. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Dondoshansky, I.; Wolf, Y. Blastclust (NCBI Software Development Toolkit); NCBI: Bethesda, MD, USA, 2002.
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Van Rossum, G.a.D.; Fred, L. CreateSpace; CreateSpace: Scotts Valley, CA, USA, 2009. [Google Scholar]
- Waskom, M. Seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Schrödinger, L. The PyMOL Molecular Graphics System, Version 2.5.0; PyMOL: Portland, OR, USA, 2015. [Google Scholar]
- Willis, S.N.; Fletcher, J.I.; Kaufmann, T.; van Delft, M.F.; Chen, L.; Czabotar, P.E.; Ierino, H.; Lee, E.F.; Fairlie, W.D.; Bouillet, P.; et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science 2007, 315, 856–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinds, M.G.; Smits, C.; Fredericks-Short, R.; Risk, J.M.; Bailey, M.; Huang, D.C.; Day, C.L. Bim, Bad and Bmf: Intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ. 2007, 14, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaustov, L.; Ouyang, H.; Amaya, M.; Lemak, A.; Nady, N.; Duan, S.; Wasney, G.A.; Li, Z.; Vedadi, M.; Schapira, M.; et al. Recognition and specificity determinants of the human cbx chromodomains. J. Biol. Chem. 2011, 286, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Ballas, N.; Battaglioli, E.; Atouf, F.; Andres, M.E.; Chenoweth, J.; Anderson, M.E.; Burger, C.; Moniwa, M.; Davie, J.R.; Bowers, W.J.; et al. Regulation of neuronal traits by a novel transcriptional complex. Neuron 2001, 31, 353–365. [Google Scholar] [CrossRef] [Green Version]
- Naruse, Y.; Aoki, T.; Kojima, T.; Mori, N. Neural restrictive silencer factor recruits mSin3 and histone deacetylase complex to repress neuron-specific target genes. Proc. Natl. Acad. Sci. USA 1999, 96, 13691–13696. [Google Scholar] [CrossRef] [Green Version]
- Nomura, M.; Uda-Tochio, H.; Murai, K.; Mori, N.; Nishimura, Y. The neural repressor NRSF/REST binds the PAH1 domain of the Sin3 corepressor by using its distinct short hydrophobic helix. J. Mol. Biol. 2005, 354, 903–915. [Google Scholar] [CrossRef]
- Gulbis, J.M.; Kelman, Z.; Hurwitz, J.; O’Donnell, M.; Kuriyan, J. Structure of the C-terminal region of p21(WAF1/CIP1) complexed with human PCNA. Cell 1996, 87, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Warbrick, E.; Lane, D.P.; Glover, D.M.; Cox, L.S. A small peptide inhibitor of DNA replication defines the site of interaction between the cyclin-dependent kinase inhibitor p21WAF1 and proliferating cell nuclear antigen. Curr. Biol. 1995, 5, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Alderson, T.R.; Pritišanac, I.; Moses, A.M.; Forman-Kay, J.D. Systematic identification of conditionally folded intrinsically disordered regions by AlphaFold 2. bioRxiv 2022. [Google Scholar] [CrossRef]
- Meszaros, B.; Simon, I.; Dosztanyi, Z. Prediction of protein binding regions in disordered proteins. PLoS Comput. Biol. 2009, 5, e1000376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Disfani, F.M.; Hsu, W.L.; Mizianty, M.J.; Oldfield, C.J.; Xue, B.; Dunker, A.K.; Uversky, V.N.; Kurgan, L. MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics 2012, 28, i75–i83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akdel, M.; Pires, D.E.V.; Porta Pardo, E.; Jänes, J.; Zalevsky, A.O.; Mészáros, B.; Bryant, P.; Good, L.L.; Laskowski, R.A.; Pozzati, G.; et al. A structural biology community assessment of AlphaFold 2 applications. bioRxiv 2021. [Google Scholar] [CrossRef]
- Di Cola, E.; Yakubov, G.E.; Waigh, T.A. Double-globular structure of porcine stomach mucin: A small-angle X-ray scattering study. Biomacromolecules 2008, 9, 3216–3222. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| coef | t | |
|---|---|---|
| pLDDT | 0.050 | 8.979 |
| nRG | 0.514 | 4.143 |
| constant term | 5.007 | 3.614 |
| mrASA | −3.931 | −2.997 |
| crASA | 2.261 | 2.081 |
| %Coil | 1.273 | 1.682 |
| L | 2.302 | 1.572 |
| S | 1.819 | 1.387 |
| polar | −0.374 | −0.440 |
| hydrophobic | 0.516 | 0.428 |
| %Helix | −0.242 | −0.337 |
| A | 0.380 | 0.218 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anbo, H.; Sakuma, K.; Fukuchi, S.; Ota, M. How AlphaFold2 Predicts Conditionally Folding Regions Annotated in an Intrinsically Disordered Protein Database, IDEAL. Biology 2023, 12, 182. https://doi.org/10.3390/biology12020182
Anbo H, Sakuma K, Fukuchi S, Ota M. How AlphaFold2 Predicts Conditionally Folding Regions Annotated in an Intrinsically Disordered Protein Database, IDEAL. Biology. 2023; 12(2):182. https://doi.org/10.3390/biology12020182
Chicago/Turabian StyleAnbo, Hiroto, Koya Sakuma, Satoshi Fukuchi, and Motonori Ota. 2023. "How AlphaFold2 Predicts Conditionally Folding Regions Annotated in an Intrinsically Disordered Protein Database, IDEAL" Biology 12, no. 2: 182. https://doi.org/10.3390/biology12020182