
Integration of Selection Signatures and Protein Interactions Reveals NR6A1, PAPPA2, and PIK3C2B as the Promising Candidate Genes Underlying the Characteristics of Licha Black Pig
 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
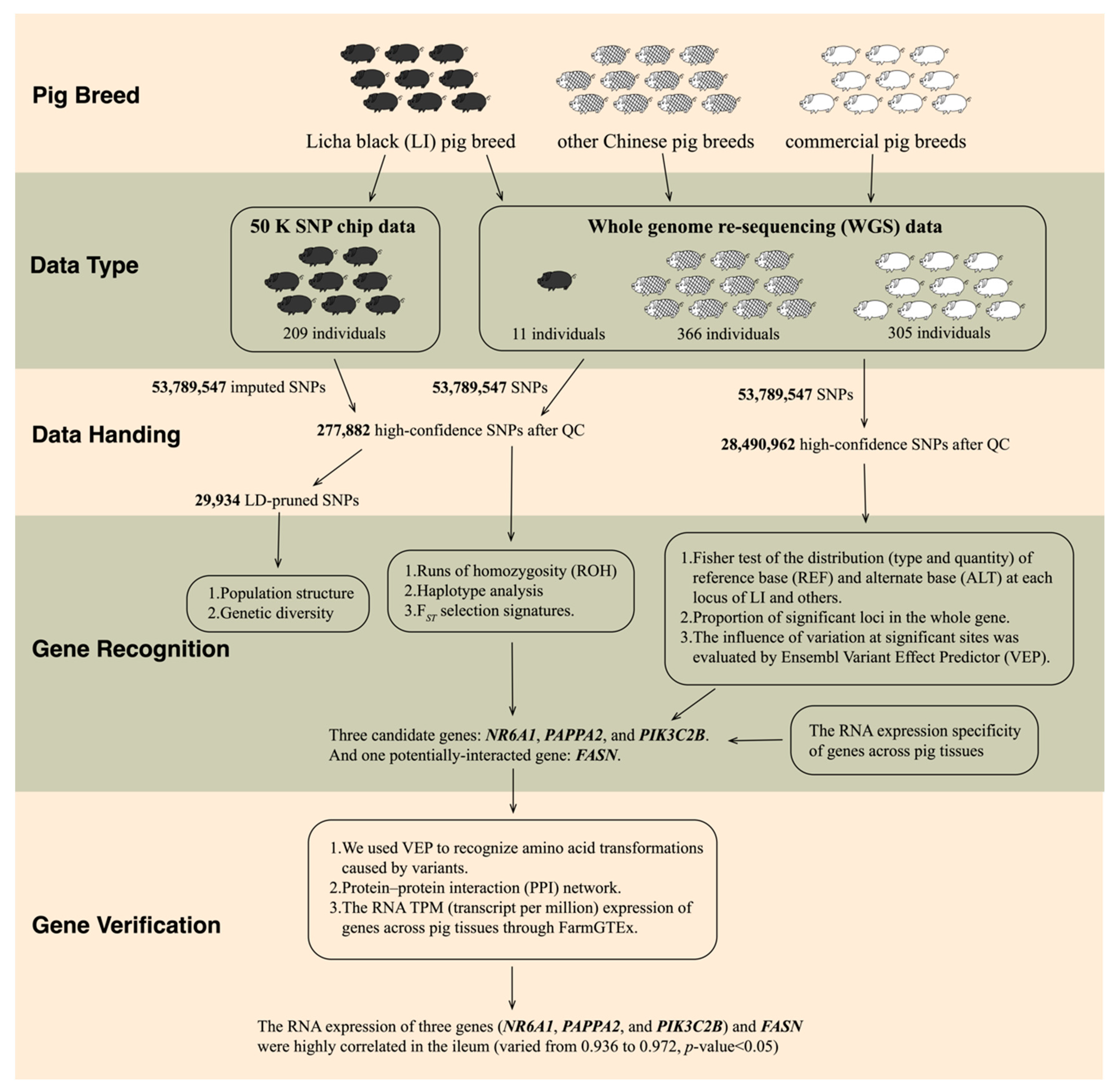
2. Materials and Methods
2.1. Populations and Data
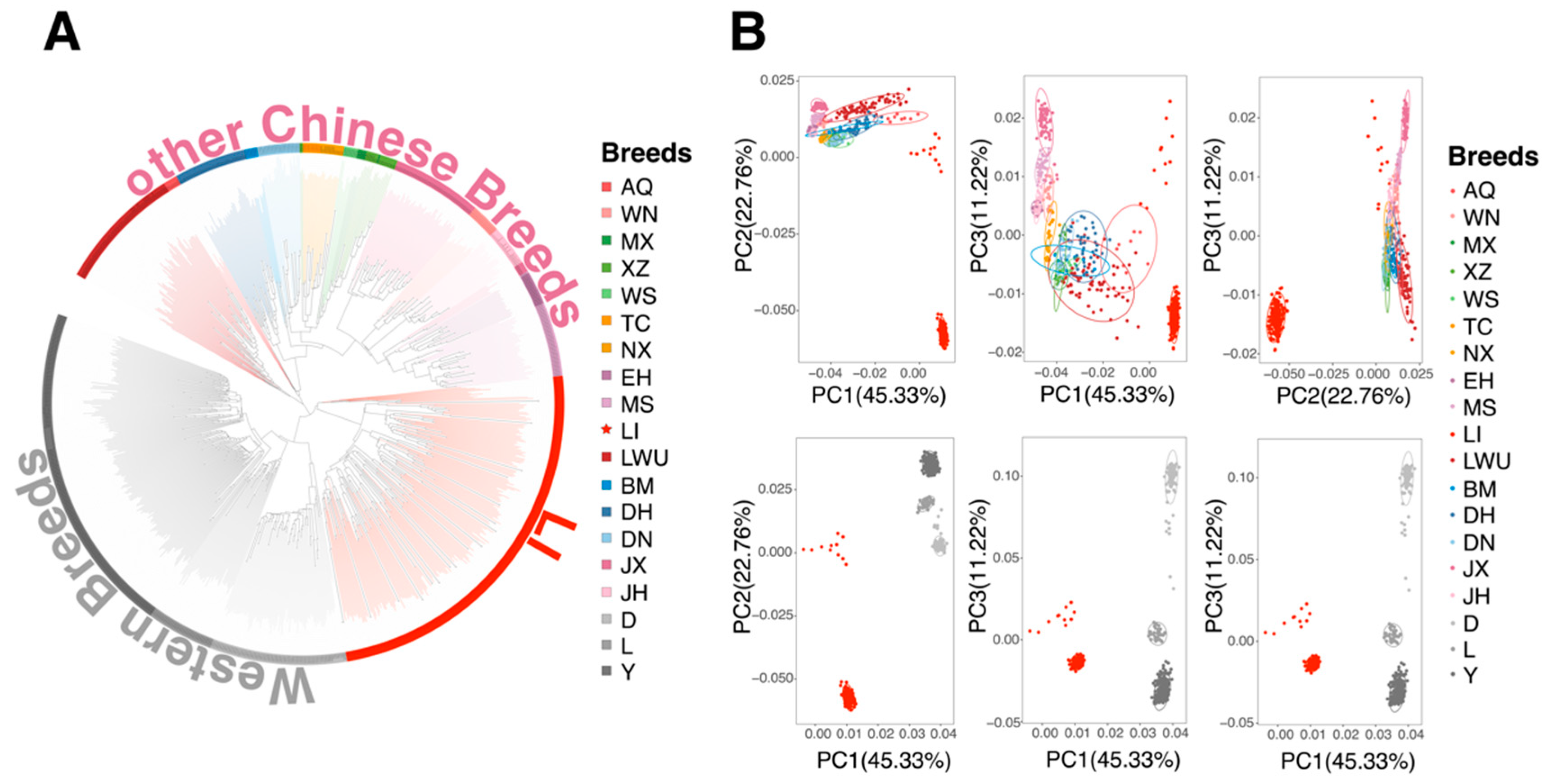
2.2. Population Structure and Genetic Diversity
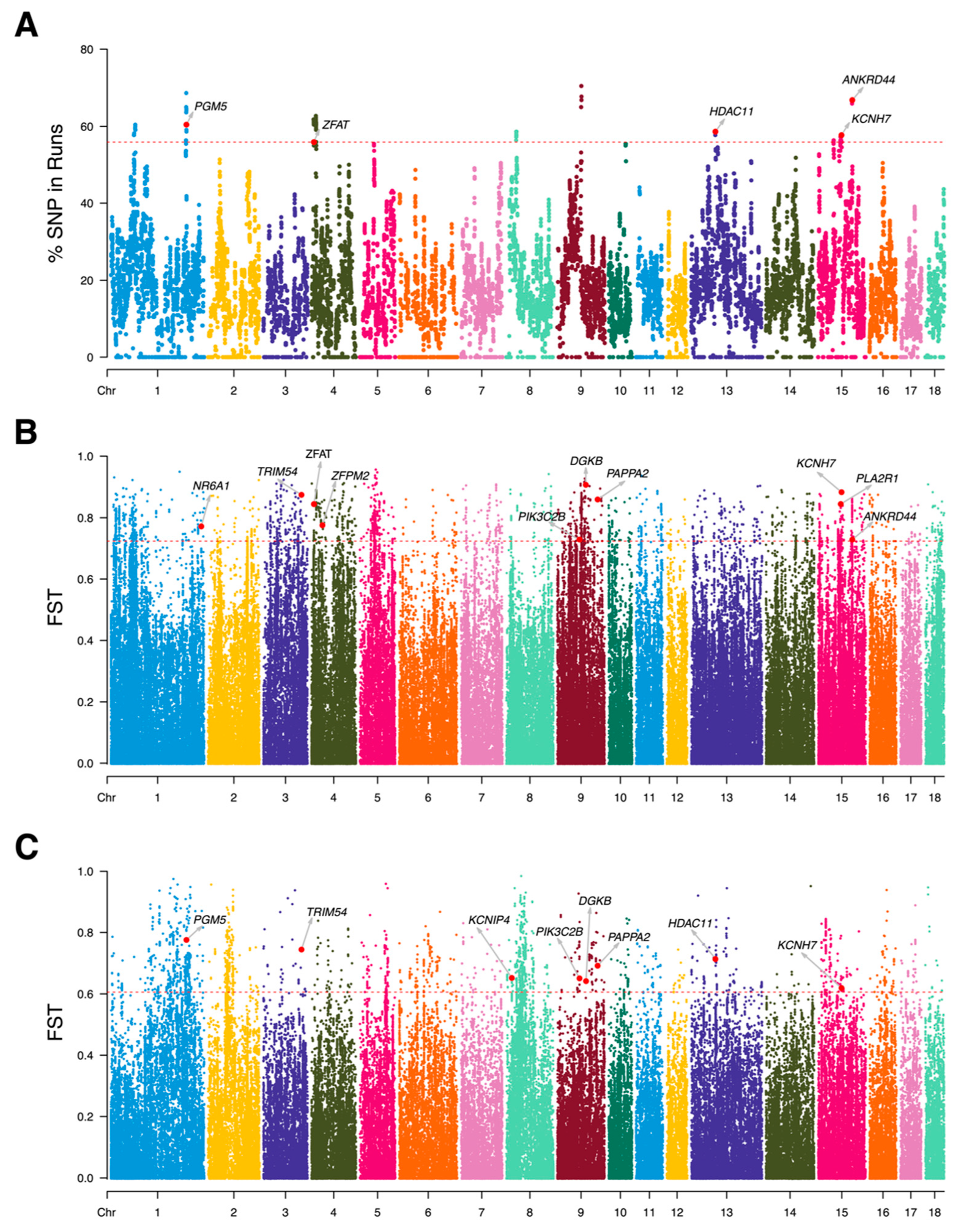
2.3. Runs of Homozygosity and Haplotype Detection among LI Pigs
2.4. Identification of Functional Annotation of Selection Signatures
2.5. RNA Expression Specificity of Candidate Genes in Human and Pig Tissues
2.6. Screening of Candidate Genes Using Whole Genome Sequencing Data
2.7. Verification of Gene Influence and Mechanism
3. Results
3.1. Population Relationship and Structure
3.2. Variety Specificity Reflected in ROH Islands, Haplotype Blocks, and FST Analyze
3.3. Candidate Genes Related to Growth, Muscle, and Fat
3.4. Allele Distribution and Variation of NR6A1, PAPPA2, and PIK3C2B
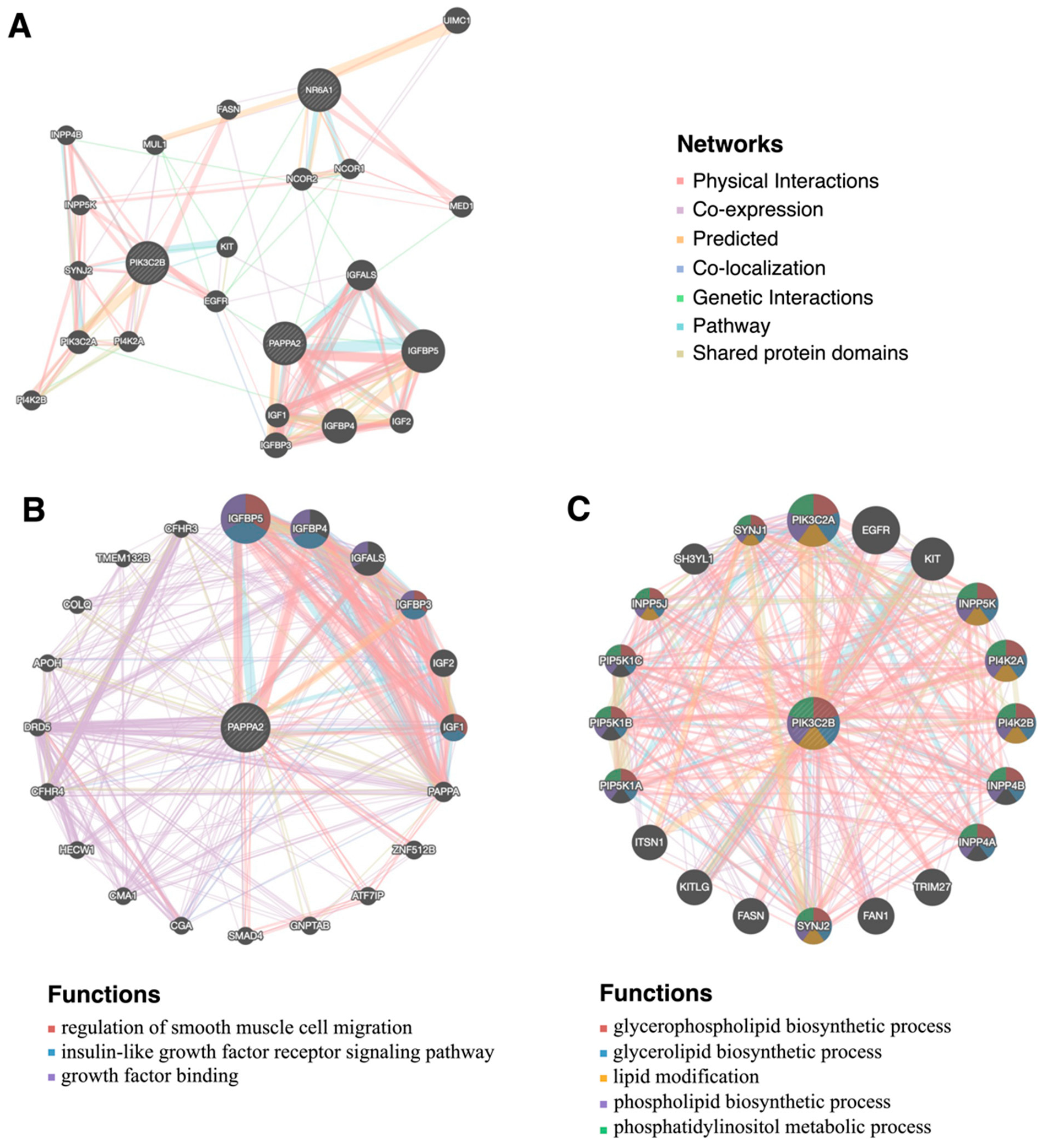
3.5. Amino Acids Transformation and Interaction of NR6A1, PAPPA2, and PIK3C2B
4. Discussion
4.1. Genetic Characteristics of Licha Black Pigs
4.2. Population-Specific Genes in Licha Black Pigs
4.3. NR6A1 Variant Is a Potential Selective Marker for Improving Pig Body Length
4.4. Interaction of NR6A1, PAPPA2, and PIK3C2B Affect Pig Fat Deposition
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Si, J.C.; Zhang, H.B.; Tang, J.J. Livestock Breeds in Shandong Province; Haitian Press: Shenzhen, China, 1999; pp. 28–31. [Google Scholar]
- Yang, G.; Ren, J.; Zhang, Z.; Huang, L. Genetic evidence for the introgression of Western NR6A1 haplotype into Chinese Licha breed associated with increased vertebral number. Anim. Genet. 2009, 40, 247–250. [Google Scholar] [CrossRef]
- Mikawa, S.; Morozumi, T.; Shimanuki, S.-I.; Hayashi, T.; Uenishi, H.; Domukai, M.; Okumura, N.; Awata, T. Fine mapping of a swine quantitative trait locus for number of vertebrae and analysis of an orphan nuclear receptor, germ cell nuclear factor (NR6A1). Genome Res. 2007, 17, 586–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Dong, R.; Li, X.; Cui, C.; Yu, G. Analysis of the Genetic Diversity and Family Structure of the Licha Black Pig Population on Jiaodong Peninsula, Shandong Province, China. Animals 2022, 12, 1045. [Google Scholar] [CrossRef] [PubMed]
- Burgos, C.; Latorre, P.; Altarriba, J.; Carrodeguas, J.A.; Varona, L.; López-Buesa, P. Allelic frequencies of NR6A1 and VRTN, two genes that affect vertebrae number in diverse pig breeds: A study of the effects of the VRTN insertion on phenotypic traits of a Duroc×Landrace–Large White cross. Meat Sci. 2015, 100, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Liu, C.; Lan, T.; Zhang, Q.; Cao, Y.; Pu, G.; Niu, P.; Zhang, Z.; Li, Q.; Zhou, J.; et al. Polymorphism of VRTN Gene g.20311_20312ins291 Was Associated with the Number of Ribs, Carcass Diagonal Length and Cannon Bone Circumference in Suhuai Pigs. Animals 2020, 10, 484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.-Y.; Xiao, S.-J.; Tu, J.-M.; Zhang, Z.-K.; Zheng, H.; Huang, L.-B.; Huang, Z.-Y.; Yan, M.; Liu, X.-D.; Guo, Y.-M. A further survey of the quantitative trait loci affecting swine body size and carcass traits in five related pig populations. Anim. Genet. 2021, 52, 621–632. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, Z.; Chen, Z.; Sun, J.; Cao, C.; Wu, F.; Xu, Z.; Zhao, W.; Sun, H.; Guo, L.; et al. PHARP: A pig haplotype reference panel for genotype imputation. Sci. Rep. 2022, 12, 12645. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Browning, B.L.; Tian, X.; Zhou, Y.; Browning, S.R. Fast two-stage phasing of large-scale sequence data. Am. J. Hum. Genet. 2021, 108, 1880–1890. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Francis, R.M. pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, J.K.; Pritchard, J.K. Inference of Population Splits and Mixtures from Genome-Wide Allele Frequency Data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, Q.; Xiao, Q.; Sun, H.; Gao, H.; Yang, Y.; Chen, J.; Li, Z.; Xue, M.; Ma, P.; et al. Distribution of runs of homozygosity in Chinese and Western pig breeds evaluated by reduced-representation sequencing data. Anim. Genet. 2018, 49, 579–591. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022. [Google Scholar]
- Fonseca, P.A.S.; Suárez-Vega, A.; Marras, G.; Cánovas, Á. GALLO: An R package for genomic annotation and integration of multiple data sources in livestock for positional candidate loci. GigaScience 2020, 9, giaa149. [Google Scholar] [CrossRef]
- Karlsson, M.; Sjöstedt, E.; Oksvold, P.; Sivertsson, Å.; Huang, J.; Álvez, M.B.; Arif, M.; Li, X.; Lin, L.; Yu, J.; et al. Genome-wide annotation of protein-coding genes in pig. BMC Biol. 2022, 20, 25. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz, M.; Rodriguez, H.; Lopes, C.; Zuberi, K.; Montojo, J.; Bader, G.D.; Morris, Q. GeneMANIA update 2018. Nucleic Acids Res. 2018, 46, W60–W64. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Chen, Z.; Teng, J.; Liu, S.; Lin, Q.; Gao, Y.; Bai, Z.; Consortium, T.F.; Li, B.; Liu, G.; et al. FarmGTEx TWAS-server: An interactive web server for customized TWAS analysis in both human and farm animals. bioRxiv 2023. [Google Scholar] [CrossRef]
- Demirkan, A.; van Duijn, C.M.; Ugocsai, P.; Isaacs, A.; Pramstaller, P.P.; Liebisch, G.; Wilson, J.F.; Johansson, Å.; Rudan, I.; Aulchenko, Y.S.; et al. Genome-wide association study identifies novel loci associated with circulating phospho- and sphingolipid concentrations. PLoS Genet. 2012, 8, e1002490. [Google Scholar] [CrossRef] [Green Version]
- Belengeanu, V.; Gamage, T.H.; Farcas, S.; Stoian, M.; Andreescu, N.; Belengeanu, A.; Frengen, E.; Misceo, D. A de novo 2.3 Mb deletion in 2q24.2q24.3 in a 20-month-old developmentally delayed girl. Gene 2014, 539, 168–172. [Google Scholar] [CrossRef]
- Ingelsson, E.; Langenberg, C.; Hivert, M.-F.; Prokopenko, I.; Lyssenko, V.; Dupuis, J.; Mägi, R.; Sharp, S.; Jackson, A.U.; Assimes, T.L.; et al. Detailed physiologic characterization reveals diverse mechanisms for novel genetic Loci regulating glucose and insulin metabolism in humans. Diabetes 2010, 59, 1266–1275. [Google Scholar] [CrossRef] [Green Version]
- Xing, K.; Wang, K.; Ao, H.; Chen, S.; Tan, Z.; Wang, Y.; Xitong, Z.; Yang, T.; Zhang, F.; Liu, Y.; et al. Comparative adipose transcriptome analysis digs out genes related to fat deposition in two pig breeds. Sci. Rep. 2019, 9, 12925. [Google Scholar] [CrossRef] [Green Version]
- Wit, J.M.; van Duyvenvoorde, H.A.; van Klinken, J.B.; Caliebe, J.; Bosch, C.A.J.; Lui, J.C.; Gijsbers, A.C.J.; Bakker, E.; Breuning, M.H.; Oostdijk, W.; et al. Copy number variants in short children born small for gestational age. Horm. Res. Paediatr. 2014, 82, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Dauber, A.; Muñoz-Calvo, M.T.; Barrios, V.; Domené, H.M.; Kloverpris, S.; Serra-Juhé, C.; Desikan, V.; Pozo, J.; Muzumdar, R.; Martos-Moreno, G.Á.; et al. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Mol. Med. 2016, 8, 363–374. [Google Scholar] [CrossRef]
- Hwa, V.; Fujimoto, M.; Zhu, G.; Gao, W.; Foley, C.; Kumbaji, M.; Rosenfeld, R.G. Genetic causes of growth hormone insensitivity beyond GHR. Rev. Endocr. Metab. Disord. 2021, 22, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Christians, J.K.; Hoeflich, A.; Keightley, P.D. PAPPA2, an enzyme that cleaves an insulin-like growth-factor-binding protein, is a candidate gene for a quantitative trait locus affecting body size in mice. Genetics 2006, 173, 1547–1553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harsanyi, S.; Zamborsky, R.; Krajciova, L.; Kokavec, M.; Danisovic, L. Genetic Study of IL6, GDF5 and PAPPA2 in Association with Developmental Dysplasia of the Hip. Genes 2021, 12, 986. [Google Scholar] [CrossRef] [PubMed]
- Sabha, N.; Volpatti, J.R.; Gonorazky, H.; Reifler, A.; Davidson, A.E.; Li, X.; Eltayeb, N.M.; Dall’Armi, C.; Di Paolo, G.; Brooks, S.V.; et al. PIK3C2B inhibition improves function and prolongs survival in myotubular myopathy animal models. J. Clin. Invest. 2016, 126, 3613–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gozzelino, L.; Kochlamazashvili, G.; Baldassari, S.; Mackintosh, A.I.; Licchetta, L.; Iovino, E.; Liu, Y.C.; Bennett, C.A.; Bennett, M.F.; Damiano, J.A.; et al. Defective lipid signalling caused by mutations in PIK3C2B underlies focal epilepsy. Brain 2022, 145, 2313–2331. [Google Scholar] [CrossRef]
- Pingel, J.; Vandenrijt, J.; Kampmann, M.-L.; Andersen, J.D. Altered gene expression levels of genes related to muscle function in adults with cerebral palsy. Tissue Cell 2022, 76, 101744. [Google Scholar] [CrossRef]
- Petit, M.M.R.; Lindskog, H.; Larsson, E.; Wasteson, P.; Athley, E.; Breuer, S.; Angstenberger, M.; Hertfelder, D.; Mattsson, E.; Nordheim, A.; et al. Smooth Muscle Expression of Lipoma Preferred Partner Is Mediated by an Alternative Intronic Promoter That Is Regulated by Serum Response Factor/Myocardin. Circ. Res. 2008, 103, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Xin, D.; Christopher, K.J.; Zeng, L.; Kong, Y.; Weatherbee, S.D. IFT56 regulates vertebrate developmental patterning by maintaining IFTB complex integrity and ciliary microtubule architecture. Development 2017, 144, 1544–1553. [Google Scholar] [CrossRef] [Green Version]
- Pettersson, M.; Vaz, R.; Hammarsjö, A.; Eisfeldt, J.; Carvalho, C.M.B.; Hofmeister, W.; Tham, E.; Horemuzova, E.; Voss, U.; Nishimura, G.; et al. Alu-Alu mediated intragenic duplications in IFT81 and MATN3 are associated with skeletal dysplasias. Hum. Mutat. 2018, 39, 1456–1467. [Google Scholar] [CrossRef]
- Xu, J.; Zeng, Y.; Si, H.; Liu, Y.; Li, M.; Zeng, J.; Shen, B. Integrating transcriptome-wide association study and mRNA expression profile identified candidate genes related to hand osteoarthritis. Arthritis Res. Ther. 2021, 23, 81. [Google Scholar] [CrossRef] [PubMed]
- N’Diaye, A.; Chen, G.K.; Palmer, C.D.; Ge, B.; Tayo, B.; Mathias, R.A.; Ding, J.; Nalls, M.A.; Adeyemo, A.; Adoue, V.; et al. Identification, Replication, and Fine-Mapping of Loci Associated with Adult Height in Individuals of African Ancestry. PLoS Genet. 2011, 7, e1002298. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.L.; Wilson, M.L.; Nilson, S.M.; Rowan, T.N.; Schnabel, R.D.; Decker, J.E.; Seabury, C.M. Genome-wide association and genotype by environment interactions for growth traits in U.S. Red Angus cattle. BMC Genom. 2022, 23, 517. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Marin de Evsikova, C.; Bian, K.; Achille, A.; Telles, E.; Pei, H.; Seto, E. Programming and Regulation of Metabolic Homeostasis by HDAC11. EBioMedicine 2018, 33, 157–168. [Google Scholar] [CrossRef]
- Yang, H.; Chen, L.; Sun, Q.; Yao, F.; Muhammad, S.; Sun, C. The role of HDAC11 in obesity-related metabolic disorders: A critical review. J. Cell Physiol. 2021, 236, 5582–5591. [Google Scholar] [CrossRef]
- Bagchi, R.A.; Ferguson, B.S.; Stratton, M.S.; Hu, T.; Cavasin, M.A.; Sun, L.; Lin, Y.-H.; Liu, D.; Londono, P.; Song, K.; et al. HDAC11 suppresses the thermogenic program of adipose tissue via BRD2. JCI Insight 2018, 3, 120159. [Google Scholar] [CrossRef] [Green Version]
- Núñez-Álvarez, Y.; Hurtado, E.; Muñoz, M.; García-Tuñon, I.; Rech, G.E.; Pluvinet, R.; Sumoy, L.; Pendás, A.M.; Peinado, M.A.; Suelves, M. Loss of HDAC11 accelerates skeletal muscle regeneration in mice. FEBS J. 2021, 288, 1201–1223. [Google Scholar] [CrossRef]
- Hannan, N.J.; Stock, O.; Spencer, R.; Whitehead, C.; David, A.L.; Groom, K.; Petersen, S.; Henry, A.; Said, J.M.; Seeho, S.; et al. Circulating mRNAs are differentially expressed in pregnancies with severe placental insufficiency and at high risk of stillbirth. BMC Med. 2020, 18, 145. [Google Scholar] [CrossRef]
- Molt, S.; Bührdel, J.B.; Yakovlev, S.; Schein, P.; Orfanos, Z.; Kirfel, G.; Winter, L.; Wiche, G.; van der Ven, P.F.M.; Rottbauer, W.; et al. Aciculin interacts with filamin C and Xin and is essential for myofibril assembly, remodeling and maintenance. J. Cell Sci. 2014, 127, 3578–3592. [Google Scholar] [CrossRef] [Green Version]
- Stenhouse, C.; Cortes-Araya, Y.; Donadeu, F.X.; Ashworth, C.J. Associations between testicular development and fetal size in the pig. J. Anim. Sci. Biotechnol. 2022, 13, 24. [Google Scholar] [CrossRef]
- Matson, C.K.; Murphy, M.W.; Sarver, A.L.; Griswold, M.D.; Bardwell, V.J.; Zarkower, D. DMRT1 prevents female reprogramming in the postnatal mammalian testis. Nature 2011, 476, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, C.; Li, X.; Liu, Z.; Ni, W.; Cao, Y.; Yao, Y.; Islamov, E.; Wei, J.; Hou, X.; et al. Association analysis of polymorphism in the NR6A1 gene with the lumbar vertebrae number traits in sheep. Genes Genom. 2019, 41, 1165–1171. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Lai, Z.; Liu, J.; Zhang, C.; Li, S.; Wu, F.; Zhou, Z.; Lei, C.; Dang, R. A Novel 13 bp Deletion within the NR6A1 Gene Is Significantly Associated with Growth Traits in Donkeys. Animals 2019, 9, 681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bovo, S.; Schiavo, G.; Utzeri, V.J.; Ribani, A.; Schiavitto, M.; Buttazzoni, L.; Negrini, R.; Fontanesi, L. A genome-wide association study for the number of teats in European rabbits (Oryctolagus cuniculus) identifies several candidate genes affecting this trait. Anim. Genet. 2021, 52, 237–243. [Google Scholar] [CrossRef]
- Fontanesi, L.; Ribani, A.; Scotti, E.; Utzeri, V.J.; Veličković, N.; Dall’Olio, S. Differentiation of meat from European wild boars and domestic pigs using polymorphisms in the MC1R and NR6A1 genes. Meat Sci. 2014, 98, 781–784. [Google Scholar] [CrossRef]
- Upners, E.N.; Ljubicic, M.L.; Busch, A.S.; Fischer, M.B.; Almstrup, K.; Petersen, J.H.; Jensen, R.B.; Hagen, C.P.; Juul, A. Dynamic Changes in Serum IGF-I and Growth During Infancy: Associations to Body Fat, Target Height, and PAPPA2 Genotype. J. Clin. Endocrinol. Metab. 2022, 107, 219–229. [Google Scholar] [CrossRef]
- Raza, S.H.A.; Gui, L.; Khan, R.; Schreurs, N.M.; Xiaoyu, W.; Wu, S.; Mei, C.; Wang, L.; Ma, X.; Wei, D.; et al. Association between FASN gene polymorphisms ultrasound carcass traits and intramuscular fat in Qinchuan cattle. Gene 2018, 645, 55–59. [Google Scholar] [CrossRef]
- Pecka-Kiełb, E.; Kowalewska-Łuczak, I.; Czerniawska-Piątkowska, E.; Króliczewska, B. FASN, SCD1 and ANXA9 gene polymorphism as genetic predictors of the fatty acid profile of sheep milk. Sci. Rep. 2021, 11, 23761. [Google Scholar] [CrossRef]
- Tang, C.; Ma, J.; Kong, F.; Li, B.; Du, Q.; Zhang, Y.; Wang, H.; Tang, Q.; Hu, S.; Liu, L.; et al. The Analysis of Transcriptomes and Microorganisms Reveals Differences between the Intestinal Segments of Guinea Pigs. Animals 2022, 12, 2925. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
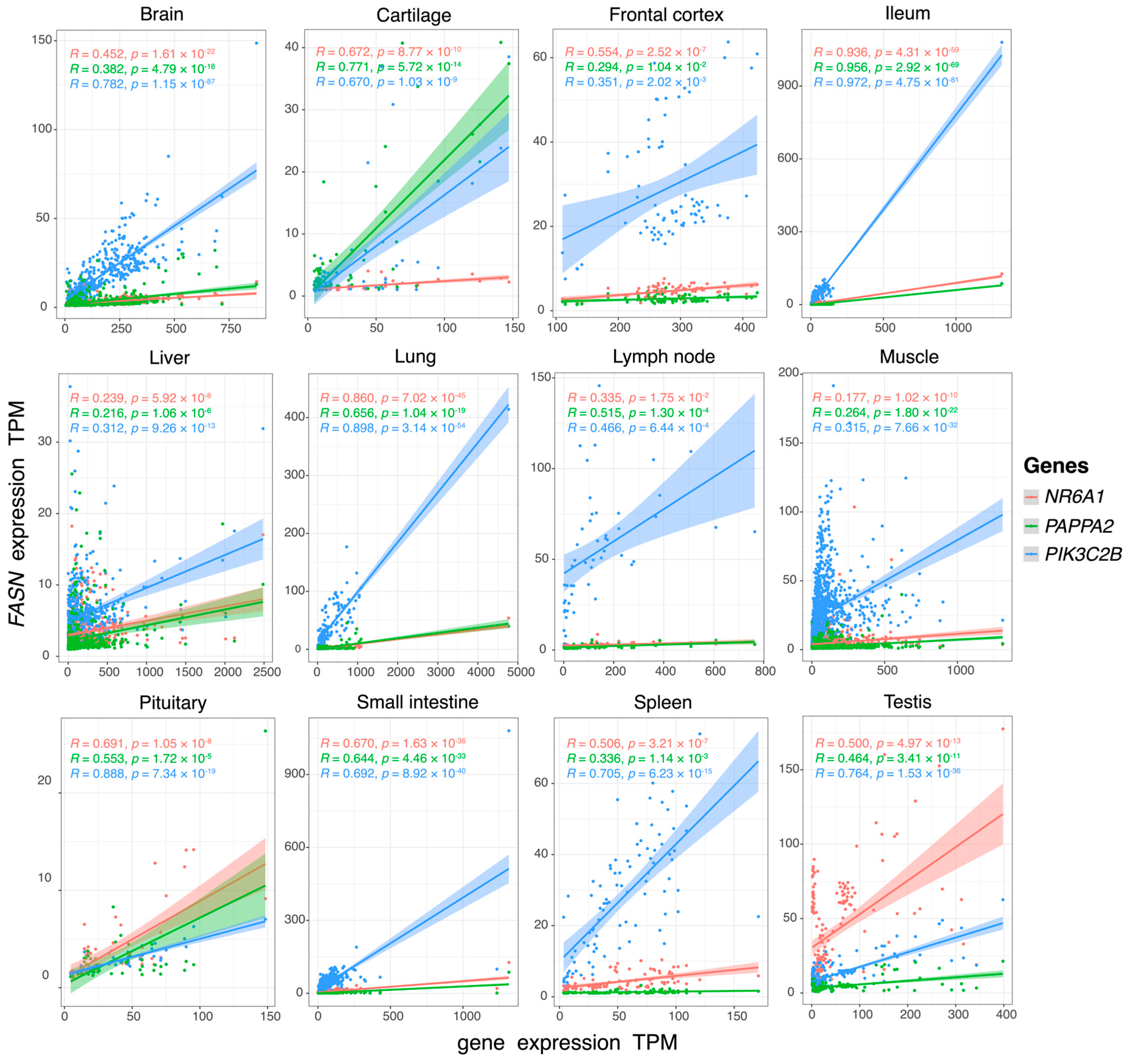{kind=link}
| Genes | Chr a | Gene Start (bp) | Gene End (bp) | Block Start (bp) | Block End (bp) | Block Length (Kb) | NSNPS b |
|---|---|---|---|---|---|---|---|
| KCNIP4 | 8 | 15,211,916 | 16,346,170 | 15,865,991 | 16,065,990 | 200,000 | 102 |
| 8 | 15,211,916 | 16,346,170 | 16,265,054 | 16,465,054 | 200,001 | 245 | |
| LPP | 13 | 125,782,471 | 126,489,650 | 126,316,668 | 126,516,664 | 199,997 | 63 |
| IFT81 | 14 | 31,368,687 | 31,657,305 | 31,422,148 | 31,622,148 | 200,001 | 119 |
| Genes | Function | RNA Expression Tissue Specificity in Pigs [23] |
|---|---|---|
| (FST-1FST-2ROH)/Haplotype | ||
| KCNH7 | Glycerophospholipid pathway [28] Developmental delay syndrome [29] | brain |
| (FST-1FST-2)/(HaplotypeROH) | ||
| DGKB | Early insulin secretion [30] Fat deposition [31] Short stature [32] | brain, salivary gland |
| PAPPA2 | Longitudinal growth [33,34] Bone formation and dysplasia of the hip [35,36] | thyroid gland |
| PIK3C2B | Muscle-specific ablation [37] Lipid signaling [38] | / |
| TRIM54 | Muscle signaling [39] | heart, mouth, skeletal muscle |
| (FST-1Haplotype)/(FST-2ROH) | ||
| LPP | Smooth muscle expression [40] | smooth muscle |
| IFT81 | Vertebrate developmental patterning [41] Skeletal dysplasias [42] | / |
| (FST-1ROH)/(FST-2Haplotype) | ||
| ANKRD44 | Skeletal muscle [43] | lymphoid tissue |
| ZFAT | Height [44] | / |
| (FST-2Haplotype)/(FST-1ROH) | ||
| KCNIP4 | Growth and development [45] | brain, retina, small intestine, smooth muscle |
| (FST-2ROH)/(FST-1Haplotype) | ||
| HDAC11 | Metabolic homeostasis and obesity [46,47] Control of adipose tissue [48] Skeletal muscle regeneration [49] | brain, testis |
| PGM5 | Fetal growth restriction [50] Myofibril assembly and repair [51] | ductus deferens, smooth muscle, urinary bladder |
| Gene | SNP | REF a/ALT b | Allele Numbers (REF/ALT) | Significance | Consequence | Impact | AA c | |||
|---|---|---|---|---|---|---|---|---|---|---|
| LI | Commercial | Chinese | LI-Commercial | LI-Chinese | ||||||
| NR6A1 | 1:265347265 | A/G | 17/5 | 600/10 | 139/593 | *** | *** | Missense variant | Moderate | L/P |
| PAPPA2 | 9:118365823 | T/C | 20/2 | 593/17 | 315/417 | *** | Missense variant | Moderate | S/P | |
| 9:118365845 | C/T | 20/2 | 593/17 | 316/416 | *** | Missense variant | Moderate | A/V | ||
| 9:118365925 | G/A | 16/6 | 610/0 | 660/72 | *** | * | Missense variant | Moderate | D/N | |
| 9:118365959 | A/G | 22/0 | 607/3 | 599/133 | * | Missense variant | Moderate | K/R | ||
| 9:118366048 | C/G | 22/0 | 609/1 | 582/150 | * | Missense variant | Moderate | P/A | ||
| 9:118549309 | G/A | 22/0 | 610/0 | 582/150 | * | Missense variant | Moderate | R/K | ||
| 9:118500711 | G/A | 22/0 | 609/1 | 609/123 | * | Missense variant | Moderate | A/T | ||
| PIK3C2B | 9:65126980 | G/C | 17/5 | 485/125 | 692/40 | ** | Missense variant | Moderate | P/A | |
| 9:65127090 | G/A | 17/5 | 482/128 | 694/38 | ** | Missense variant | Moderate | P/L | ||
| 9:65127444 | G/A | 17/5 | 485/125 | 692/40 | ** | Missense variant | Moderate | P/L | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, Q.; Zhang, Z.; Chen, Z.; Sun, J.; Li, M.; Wang, Q.; Pan, Y. Integration of Selection Signatures and Protein Interactions Reveals NR6A1, PAPPA2, and PIK3C2B as the Promising Candidate Genes Underlying the Characteristics of Licha Black Pig. Biology 2023, 12, 500. https://doi.org/10.3390/biology12040500
Xie Q, Zhang Z, Chen Z, Sun J, Li M, Wang Q, Pan Y. Integration of Selection Signatures and Protein Interactions Reveals NR6A1, PAPPA2, and PIK3C2B as the Promising Candidate Genes Underlying the Characteristics of Licha Black Pig. Biology. 2023; 12(4):500. https://doi.org/10.3390/biology12040500
Chicago/Turabian StyleXie, Qinqin, Zhenyang Zhang, Zitao Chen, Jiabao Sun, Meng Li, Qishan Wang, and Yuchun Pan. 2023. "Integration of Selection Signatures and Protein Interactions Reveals NR6A1, PAPPA2, and PIK3C2B as the Promising Candidate Genes Underlying the Characteristics of Licha Black Pig" Biology 12, no. 4: 500. https://doi.org/10.3390/biology12040500