
Genomic Signature in Evolutionary Biology: A Review




Abstract
:Simple Summary
Abstract
1. Introduction
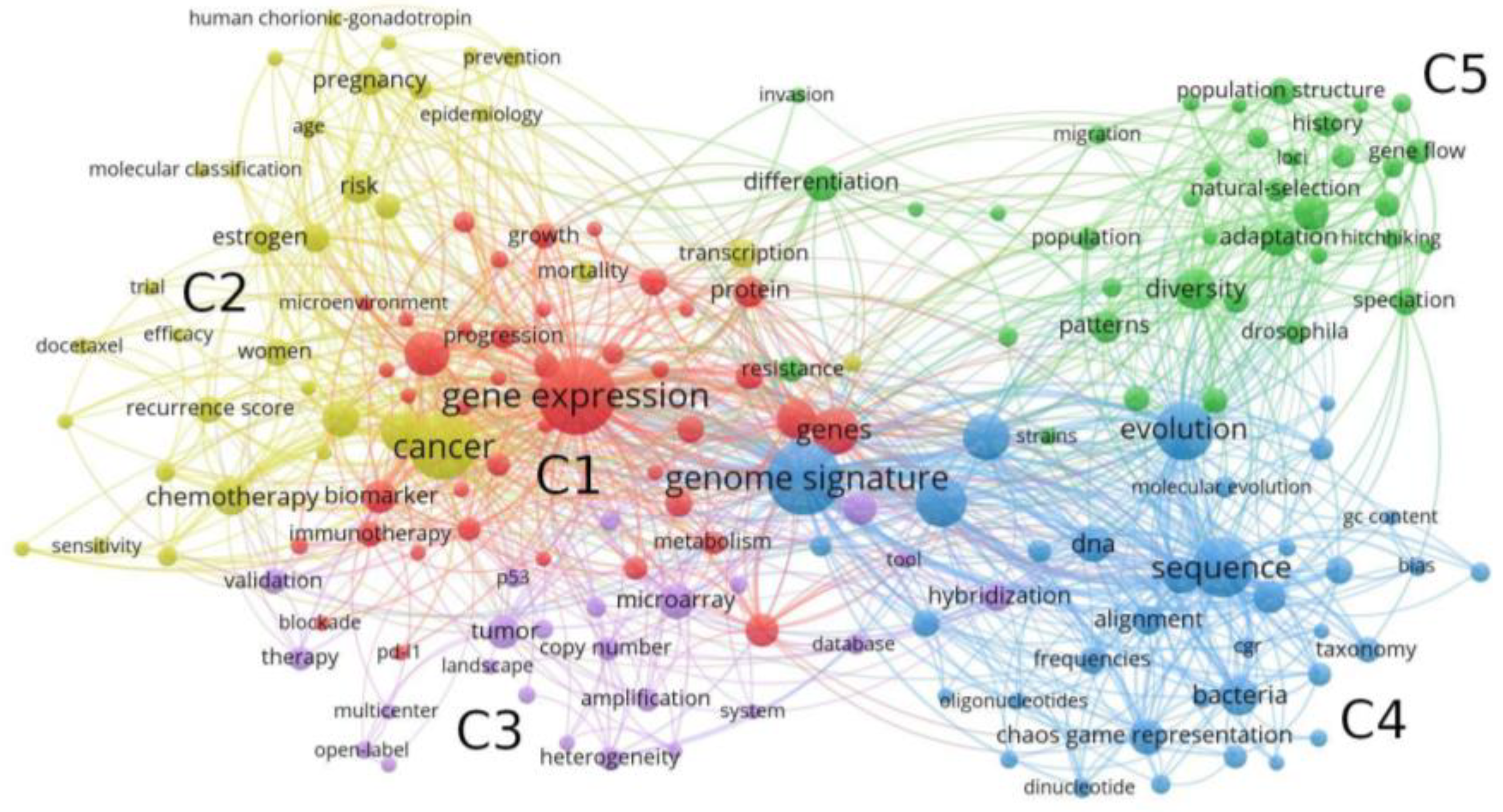
2. Bibliometric Analysis of the Genomic Signature
2.1. Methods
2.2. Co-Word Bibliometric Analysis
2.3. Bibliographic Coupling
2.3.1. Applications in Medicine
2.3.2. Applications in Comparative Genomics and Evolutionary Biology
3. Revealing Patterns in Genome Sequences
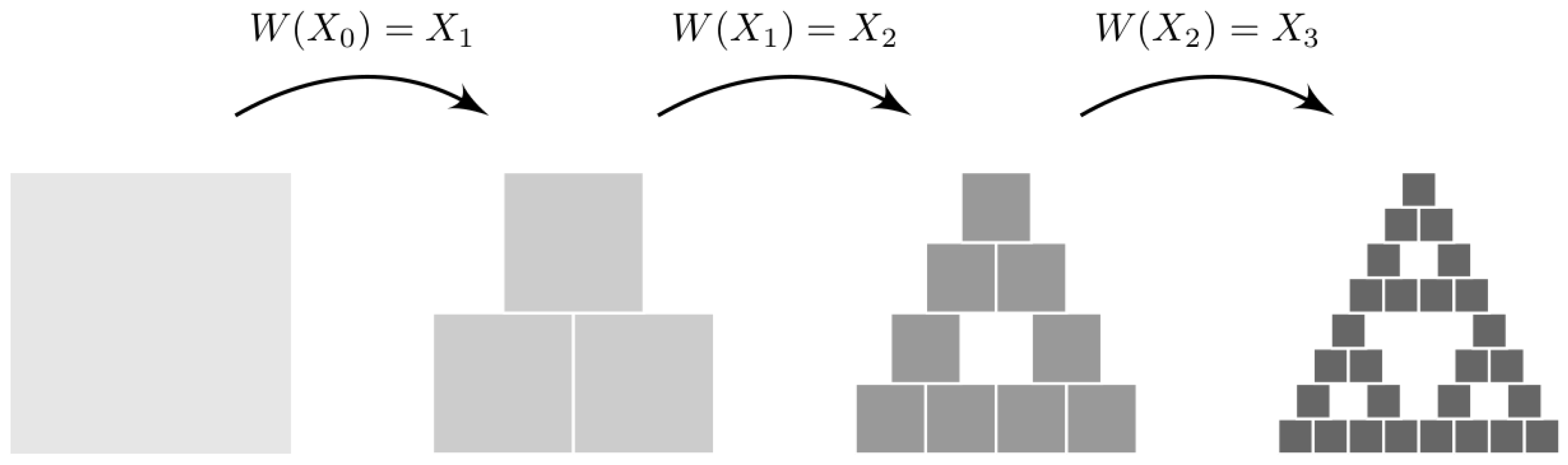
3.1. Iterated Function Systems
- 1. Take the four vertices (0, 0), (0, 1), (1, 0), and (1, 1) defining the unit square box.
- 2. Start from an initial random point in the unit square.
- 3. Select one vertex randomly, and compute the halfway point between the previous point and the vertex.
- 4. Repeat step 3 as many times as you want.
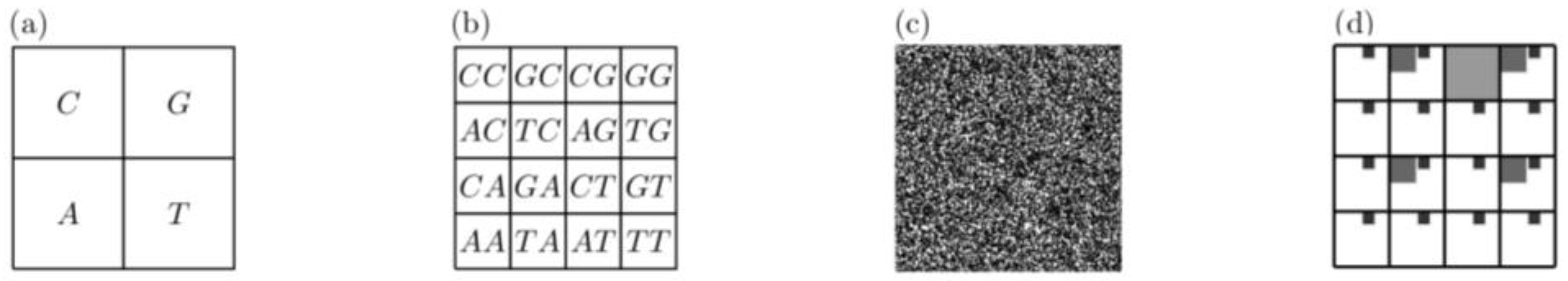
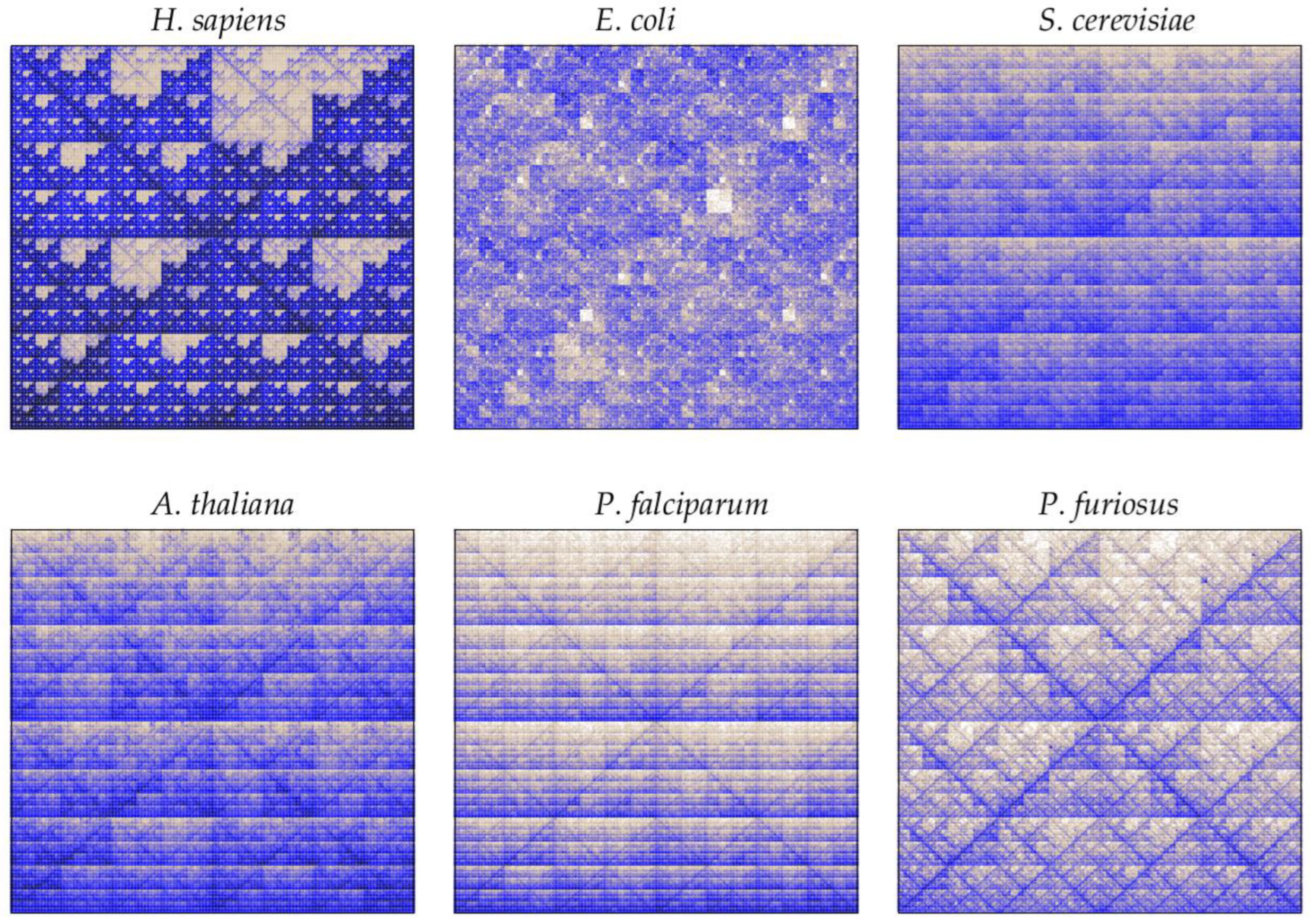
3.2. Underlying Patterns in DNA Sequences
4. Genomic Signature in Evolutionary Biology
4.1. What Is Causing the Organismal Signature?
4.2. Dinucleotide Biases
4.3. Taxonomic Inference from Word-Based Metrics
4.4. Examples of Case Studies
4.4.1. Horizontal Gene Transfer
4.4.2. Phage–Host Relations
4.4.3. Phylogenetics and SARS-CoV-2
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Phillips, A.J. Homology assessment and molecular sequence alignment. J. Biomed. Inform. 2006, 39, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.; Janies, D.; Wheeler, W. Multiple Sequence Alignment in Phylogenetic Analysis. Mol. Phylogenet. Evol. 2000, 16, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.A. Multiple sequence alignment for phylogenetic purposes. Aust. Syst. Bot. 2006, 19, 479–539. [Google Scholar] [CrossRef] [Green Version]
- Ochoterena, H.; Vrijdaghs, A.; Smets, E.; Claßen-Bockhoff, R. The Search for Common Origin: Homology Revisited. Syst. Biol. 2019, 68, 767–780. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F. Amino-acid substitution matrices from an information theoretic perspective. J. Mol. Biol. 1991, 219, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Karlin, S.; Altschul, S.F. Methods for assessing the statistical significance of molecular sequence features by using general scoring chemes. Proc. Natl. Acad. Sci. USA 1990, 87, 2264–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipman, D.J.; Altschul, S.F.; Kececioglu, J.D. A tool for multiple sequence alignment. Proc. Natl. Acad. Sci. USA 1989, 86, 4412–4415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotoh, O. An improved algorithm for matching biological sequences. J. Mol. Biol. 1982, 162, 705–708. [Google Scholar] [CrossRef]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- O’Meara, B.C. Evolutionary Inferences from Phylogenies: A Review of Methods. Annu. Rev. Ecol. Evol. Syst. 2012, 43, 267–285. [Google Scholar] [CrossRef]
- Huelsenbeck, J.; Crandall, K. Phylogeny estimation and hypothesis testing using maximum likelihood. Annu. Rev. Ecol. Syst. 1997, 28, 437–466. [Google Scholar] [CrossRef] [Green Version]
- Zielezinski, A.; Vinga, S.; Almeida, J.; Karlowski, W.M. Alignment-free sequence comparison: Benefits, applications, and tools. Genome Biol. 2017, 18, 186. [Google Scholar] [CrossRef] [Green Version]
- Jukes, T.H.; Cantor, C.R. Evolution of protein molecules. Mamm. Protein Metab. 1969, 3, 21–132. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Felsenstein, J. Taking variation of evolutionary rates between sites into account in inferring phylogenies. J. Mol. Evol. 2001, 53, 447–455. [Google Scholar] [CrossRef]
- Goldman, N.; Yang, Z. A codon-based model of nucleotide substitution for protein-coding DNA sequences. Mol. Biol. Evol. 1994, 11, 725–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Nielsen, R.; Hasegawa, M. Models of amino acid substitution and applications to mitochondrial protein evolution. Mol. Biol. Evol. 1998, 15, 1600–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Kishino, H.; Yano, T.A. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- McGuire, G.; Denham, M.; Balding, D. Models of sequence evolution for DNA sequences containing gaps. Mol. Biol. Evol. 2001, 18, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lartillot, N.; Philippe, H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol. Biol. Evol. 2004, 21, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Mayrose, I.; Friedman, N.; Pupko, T. A Gamma mixture model better accounts for among site rate heterogeneity. Bioinformatics 2005, 21, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Susko, E.; Roger, A.J. PROCOV: Maximum likelihood estimation of protein phylogeny under covarion models and site-specific covarion pattern analysis. BMC Evol. Biol. 2009, 9, 225. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Li, W. Estimation of evolutionary distances under stationary and nonstationary models of nucleotide substitution. Proc. Natl. Acad. Sci. USA 1998, 95, 5899–5905. [Google Scholar] [CrossRef] [Green Version]
- Whelan, S.; Blackburne, B.P.; Spencer, M. Phylogenetic Substitution Models for Detecting Heterotachy during Plastid Evolution. Mol. Biol. Evol. 2011, 28, 449–458. [Google Scholar] [CrossRef] [Green Version]
- Lopez, P.; Casane, D.; Philippe, H. Heterotachy, an important process of protein evolution. Mol. Biol. Evol. 2002, 19, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Pagel, M.; Meade, A. Modelling heterotachy in phylogenetic inference by reversible-jump Markov chain Monte Carlo. Philosofical Trans. R. Soc. B Biol. Sci. 2008, 363, 3955–3964. [Google Scholar] [CrossRef] [Green Version]
- Kuritzin, A.; Kischka, T.; Schmitz, J.; Churakov, G. Incomplete Lineage Sorting and Hybridization Statistics for Large-Scale Retroposon Insertion Data. PLoS Comput. Biol. 2016, 12, e1004812. [Google Scholar] [CrossRef] [Green Version]
- Doronina, L.; Churakov, G.; Kuritzin, A.; Shi, J.; Baertsch, R.; Clawson, H.; Schmitz, J. Speciation network in Laurasiatheria: Retrophylogenomic signals. Genome Res. 2017, 27, 997–1003. [Google Scholar] [CrossRef]
- Vitales, D.; Garcia, S.; Dodsworth, S. Reconstructing phylogenetic relationships based on repeat sequence similarities. Mol. Phylogenet. Evol. 2020, 147, 106766. [Google Scholar] [CrossRef] [PubMed]
- Dodsworth, S.; Chase, M.W.; Kelly, L.J.; Leitch, I.J.; Macas, J.; Novák, P.; Piednoël, M.; Weiss-Schneeweiss, H.; Leitch, A.R. Genomic Repeat Abundances Contain Phylogenetic Signal. Syst. Biol. 2014, 64, 112–126. [Google Scholar] [CrossRef] [Green Version]
- Martín-Peciña, M.; Ruiz-Ruano, F.J.; Camacho, J.P.M.; Dodsworth, S. Phylogenetic signal of genomic repeat abundances can be distorted by random homoplasy: A case study from hominid primates. Zool. J. Linn. Soc. 2018, 185, 543–554. [Google Scholar] [CrossRef]
- Piednoël, M.; Sousa, A.; Renner, S.S. Transposable elements in a clade of three tetraploids and a diploid relative, focusing on Gypsy amplification. Mob. DNA 2015, 6, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kariin, S.; Burge, C. Dinucleotide relative abundance extremes: A genomic signature. Trends Genet. 1995, 11, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Karlin, S.; Ladunga, I. Comparisons of Eukaryotic genomic sequences. Proc. Natl. Acad. Sci. USA 1994, 91, 12832–12836. [Google Scholar] [CrossRef] [Green Version]
- Deschavanne, P.; Giron, A.; Vilain, J.; Fagot, G.; Fertil, B. Genomic signature: Characterization and classification of species assessed by chaos game representation of sequences. Mol. Biol. Evol. 1999, 16, 1391–1399. [Google Scholar] [CrossRef]
- Kari, L.; Hill, K.A.; Sayem, A.S.; Karamichalis, R.; Bryans, N.; Davis, K.; Dattani, N.S. Mapping the Space of Genomic Signatures. PLoS ONE 2015, 10, e0119815. [Google Scholar] [CrossRef] [Green Version]
- Karamichalis, R.; Kari, L.; Konstantinidis, S.; Kopecki, S. An investigation into inter- and intragenomic variations of graphic genomic signatures. BMC Bioinform. 2015, 16, 246. [Google Scholar] [CrossRef] [Green Version]
- Moya, A.; Oliver, J.L.; Verdu, M.; Delaye, L.; Arnau, V.; Bernaola-Galvan, P.; de la Fuente, R.; Diaz, W.; Gomez-Martin, C.; Gonzalez, F.M.; et al. Driven progressive evolution of genome sequence complexity in Cyanobacteria. Sci. Rep. 2020, 10, 19073. [Google Scholar] [CrossRef]
- Donthu, N.; Kumar, S.; Mukherjee, D.; Pandey, N.; Lim, W.M. How to conduct a bibliometric analysis: An overview and guidelines. J. Bus. Res. 2021, 133, 285–296. [Google Scholar] [CrossRef]
- Muñoz-Leiva, F.; Viedma-del Jesús, M.I.; Sánchez-Fernández, J.; López-Herrera, A.G. An application of co-word analysis and bibliometric maps for detecting the most highlighting themes in the consumer behaviour research from a longitudinal perspective. Qual. Quant. 2012, 46, 1077–1095. [Google Scholar] [CrossRef]
- Kessler, M.M. Bibliographic coupling between scientific papers. Am. Doc. 1963, 14, 10–25. [Google Scholar] [CrossRef]
- Van Eck, N.J.; Waltman, L. Software survey: VOSviewer, a computer program for bibliometric mapping. Scientometrics 2010, 84, 523–538. [Google Scholar] [CrossRef] [Green Version]
- Hurt, E.M.; Kawasaki, B.T.; Klarmann, G.J.; Thomas, S.B.; Farrar, W.L. CD44(+)CD24(-) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br. J. Cancer 2008, 98, 756–765. [Google Scholar] [CrossRef] [Green Version]
- Rebucci, M.; Michiels, C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem. Pharmacol. 2013, 85, 1219–1226. [Google Scholar] [CrossRef]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]
- Kim, J.; Mouw, K.W.; Polak, P.; Braunstein, L.Z.; Kamburov, A.; Tiao, G.; Kwiatkowski, D.J.; Rosenberg, J.E.; Van Allen, E.M.; D’Andrea, A.D.; et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet. 2016, 48, 600–606. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, A.M.; Soldi, R.; Anderlind, C.; Scholand, M.B.; Qian, J.; Zhang, X.; Cooper, K.; Walker, D.; McWilliams, A.; Liu, G.; et al. Airway PI3K Pathway Activation Is an Early and Reversible Event in Lung Cancer Development. Sci. Transl. Med. 2010, 2, 26ra25. [Google Scholar] [CrossRef] [Green Version]
- Popova, T.; Manie, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and Large-Scale Genomic Instability Consistently Identify Basal-like Breast Carcinomas with BRCA1/2 Inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [Green Version]
- Clifford, S.C.; Lusher, M.E.; Lindsey, J.C.; Langdon, J.A.; Gilbertson, R.J.; Straughton, D.; Ellison, D.W. Wnt/Wingless Pathway Activation and Chromosome 6 Loss Characterise a Distinct Molecular Sub-Group of Medulloblastomas Associated with a Favourable Prognosis. Cell Cycle 2006, 5, 2666–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haslinger, C.; Schweifer, N.; Stilgenbauer, S.; Döhner, H.; Lichter, P.; Kraut, N.; Stratowa, C.; Abseher, R. Microarray Gene Expression Profiling of B-Cell Chronic Lymphocytic Leukemia Subgroups Defined by Genomic Aberrations and VH Mutation Status. J. Clin. Oncol. 2004, 22, 3937–3949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maak, M.; Simon, I.; Nitsche, U.; Roepman, P.; Snel, M.; Glas, A.M.; Schuster, T.; Keller, G.; Zeestraten, E.; Goossens, I.; et al. Independent Validation of a Prognostic Genomic Signature (ColoPrint) for Patients with Stage II Colon Cancer. Ann. Surg. 2013, 257, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Asztalos, S.; Gann, P.H.; Hayes, M.K.; Nonn, L.; Beam, C.A.; Dai, Y.; Wiley, E.L.; Tonetti, D.A. Gene Expression Patterns in the Human Breast after Pregnancy. Cancer Prev. Res. 2010, 3, 301–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moral, R.; Wang, R.; Russo, I.H.; Lamartiniere, C.A.; Pereira, J.; Russo, J. Effect of prenatal exposure to the endocrine disruptor bisphenol A on mammary gland morphology and gene expression signature. J. Endocrinol. 2008, 196, 101–112. [Google Scholar] [CrossRef] [Green Version]
- Rutgers, E.; Piccart-Gebhart, M.J.; Bogaerts, J.; Delaloge, S.; ‘t Veer, L.V.; Rubio, I.T.; Viale, G.; Thompson, A.M.; Passalacqua, R.; Nitz, U.; et al. The EORTC 10041/BIG 03-04 MINDACT trial is feasible: Results of the pilot phase. Eur. J. Cancer 2011, 47, 2742–2749. [Google Scholar] [CrossRef]
- Perez, E.A.; Thompson, E.A.; Ballman, K.V.; Anderson, S.K.; Asmann, Y.W.; Kalari, K.R.; Eckel-Passow, J.E.; Dueck, A.C.; Tenner, K.S.; Jen, J.; et al. Genomic Analysis Reveals That Immune Function Genes Are Strongly Linked to Clinical Outcome in the North Central Cancer Treatment Group N9831 Adjuvant Trastuzumab Trial. J. Clin. Oncol. 2015, 33, 701–708. [Google Scholar] [CrossRef]
- Nitz, U.; Gluz, O.; Christgen, M.; Kates, R.E.; Clemens, M.; Malter, W.; Nuding, B.; Aktas, B.; Kuemmel, S.; Reimer, T.; et al. Reducing chemotherapy use in clinically high-risk, genomically low-risk pN0 and pN1 early breast cancer patients: Five-year data from the prospective, randomised phase 3 West German Study Group (WSG) PlanB trial. Breast Cancer Res. Treat. 2019, 175, 265–266. [Google Scholar] [CrossRef] [Green Version]
- Impey, S.; McCorkle, S.; Cha-Molstad, H.; Dwyer, J.; Yochum, G.; Boss, J.; McWeeney, S.; Dunn, J.; Mandel, G.; Goodman, R. Defining the CREB regulon: A genome-wide analysis of transcription factor regulatory regions. Cell 2004, 119, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Gomez, E.; Ruiz-Alonso, M.; Miravet, J.; Simon, C. Human Endometrial Transcriptomics: Implications for Embryonic Implantation. Cold Spring Harb. Perspect. Med. 2015, 5, a022996. [Google Scholar] [CrossRef]
- Brunner, P.M.; Khattri, S.; Garcet, S.; Finney, R.; Oliva, M.; Dutt, R.; Fuentes-Duculan, J.; Zheng, X.; Li, X.; Bonifacio, K.M.; et al. A mild topical steroid leads to progressive anti-inflammatory effects in the skin of patients with moderate-to-severe atopic dermatitis. J. Allergy Clin. Immunol. 2016, 138, 169–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haider, A.S.; Lowes, M.A.; Suarez-Farinas, M.; Zaba, L.C.; Cardinale, I.; Blumenberg, M.; Krueger, J.G. Cellular genomic maps help dissect pathology in human skin disease. J. Investig. Dermatol. 2008, 128, 606–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afreixo, V.; Bastos, C.A.C.; Pinho, A.J.; Garcia, S.P.; Ferreira, P.J.S.G. Genome analysis with inter-nucleotide distances. Bioinformatics 2009, 25, 3064–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapus, C.; Dufraigne, C.; Edwards, S.; Giron, A.; Fertil, B.; Deschavanne, P. Exploration of phylogenetic data using a global sequence analysis method. BMC Evol. Biol. 2005, 5, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHardy, A.C.; Garcia Martin, H.; Tsirigos, A.; Hugenholtz, P.; Rigoutsos, I. Accurate phylogenetic classification of variable-length DNA fragments. Nat. Methods 2007, 4, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Mrázek, J. Phylogenetic Signals in DNA Composition: Limitations and Prospects. Mol. Biol. Evol. 2009, 26, 1163–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Passel, M.W.J.; Kuramae, E.E.; Luyf, A.C.M.; Bart, A.; Boekhout, T. The reach of the genome signature in prokaryotes. BMC Evol. Biol. 2006, 6, 84. [Google Scholar] [CrossRef] [Green Version]
- Karlin, S.; Mrázek, J. Compositional differences within and between eukaryotic genomes. Proc. Natl. Acad. Sci. USA 1997, 94, 10227–10232. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Wada, K.; Iwasaki, Y.; Ikemura, T. Novel bioinformatics for inter- and intraspecies comparison of genome signatures in plant genomes. Plant Biotechnol. 2009, 26, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Bohlin, J.; Skjerve, E. Examination of Genome Homogeneity in Prokaryotes Using Genomic Signatures. PLoS ONE 2009, 4, e8113. [Google Scholar] [CrossRef] [Green Version]
- Bohlin, J.; Skjerve, E.; Ussery, D.W. Analysis of genomic signatures in prokaryotes using multinomial regression and hierarchical clustering. BMC Genom. 2009, 10, 487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, A.; Mrázek, J.; Karlin, S. Genome signature comparisons among prokaryote, plasmid, and mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1999, 96, 9184–9189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jernigan, R.; Baran, R. Pervasive properties of the genomic signature. BMC Genom. 2002, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Karlin, S.; Mrázek, J.; Campbell, A.M. Compositional biases of bacterial genomes and evolutionary implications. J. Bacteriol. 1997, 179, 3899–3913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satapathy, S.S.; Powdel, B.R.; Dutta, M.; Buragohain, A.K.; Ray, S.K. Constraint on di-nucleotides by codon usage bias in bacterial genomes. Gene 2014, 536, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Bohlin, J. Genomic Signatures in Microbes—Properties and Applications. Sci. World J. 2011, 11, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Dick, G.J.; Andersson, A.F.; Baker, B.J.; Simmons, S.L.; Yelton, A.P.; Banfield, J.F. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 2009, 10, R85. [Google Scholar] [CrossRef] [Green Version]
- Hooper, S.; Berg, O. Detection of genes with atypical nucleotide sequence in microbial genomes. J. Mol. Evol. 2002, 54, 365–375. [Google Scholar] [CrossRef]
- Willner, D.; Thurber, R.V.; Rohwer, F. Metagenomic signatures of 86 microbial and viral metagenomes. Environ. Microbiol. 2009, 11, 1752–1766. [Google Scholar] [CrossRef]
- Deschavanne, P.; DuBow, M.S.; Regeard, C. The use of genomic signature distance between bacteriophages and their hosts displays evolutionary relationships and phage growth cycle determination. Virol. J. 2010, 7, 163. [Google Scholar] [CrossRef] [Green Version]
- Simmons, M.P. Potential use of host-derived genome signatures to root virus phylogenies. Mol. Phylogenet. Evol. 2008, 49, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Van Passel, M.; Bart, A.; Luyf, A.; van Kampen, A.; van der Ende, A. Compositional discordance between prokaryotic plasmids and host chromosomes. BMC Genom. 2006, 7, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Yano, H.; Brown, C.J.; Top, E.M. Predicting Plasmid Promiscuity Based on Genomic Signature. J. Bacteriol. 2010, 192, 6045–6055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baran, R.H.; Ko, H. Detecting Horizontally Transferred and Essential Genes Based on Dinucleotide Relative Abundance. DNA Res. 2008, 15, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufraigne, C.; Fertil, B.; Lespinats, S.; Giron, A.; Deschavanne, P. Detection and characterization of horizontal transfers in prokaryotes using genomic signature. Nucleic Acids Res. 2005, 33, e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quirke, A.M.; Reen, F.J.; Claesson, M.J.; Boyd, E.F. Genomic island identification in Vibrio vulnificus reveals significant genome plasticity in this human pathogen. Bioinformatics 2006, 22, 905–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Passel, M.; Bart, A.; Thygesen, H.; Luyf, A.; van Kampen, A.; van der Ende, A. An acquisition account of genomic islands based on genome signature comparisons. BMC Genom. 2005, 6, 163. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hill, K.; Singh, S.; Kari, L. The spectrum of genomic signatures: From dinucleotides to chaos game representation. Gene 2005, 346, 173–185. [Google Scholar] [CrossRef]
- Apostolou-Karampelis, K.; Polychronopoulos, D.; Almirantis, Y. Introduction of ‘Generalized Genomic Signatures’ for the quantification of neighbour preferences leads to taxonomy- and functionality-based distinction among sequences. Sci. Rep. 2019, 9, 1700. [Google Scholar] [CrossRef] [Green Version]
- Karamichalis, R.; Kari, L.; Konstantinidis, S.; Kopecki, S.; Solis-Reyes, S. Additive methods for genomic signatures. BMC Bioinform. 2016, 17, 313. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Cao, C.C.; Sun, X. Intrinsic correlation of oligonucleotides: A novel genomic signature for metagenome analysis. J. Theor. Biol. 2014, 353, 9–18. [Google Scholar] [CrossRef]
- Bauer, M.; Schuster, S.M.; Sayood, K. The average mutual information profile as a genomic signature. BMC Bioinform. 2008, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Vinga, S. Information theory applications for biological sequence analysis. Brief. Bioinform. 2013, 15, 376–389. [Google Scholar] [CrossRef] [Green Version]
- Dehnert, M.; Helm, W.E.; Hütt, M.T. Information theory reveals large-scale synchronisation of statistical correlations in eukaryote genomes. Gene 2005, 345, 81–90. [Google Scholar] [CrossRef]
- Lin, T.; Zhu, G.; Zhang, J.; Xu, X.; Yu, Q.; Zheng, Z.; Zhang, Z.; Lun, Y.; Li, S.; Wang, X.; et al. Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 2014, 46, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, E.; Ratnakumar, A.; Arendt, M.L.; Maqbool, K.; Webster, M.T.; Perloski, M.; Liberg, O.; Arnemo, J.M.; Hedhammar, A.; Lindblad-Toh, K. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature 2013, 495, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Hufford, M.B.; Lubinksy, P.; Pyhaejaervi, T.; Devengenzo, M.T.; Ellstrand, N.C.; Ross-Ibarra, J. The Genomic Signature of Crop-Wild Introgression in Maize. PLoS Genet. 2013, 9, e1003477. [Google Scholar] [CrossRef]
- Narayanasamy, S.; Jarosz, Y.; Muller, E.E.L.; Heintz-Buschart, A.; Herold, M.; Kaysen, A.; Laczny, C.C.; Pinel, N.; May, P.; Wilmes, P. IMP: A pipeline for reproducible reference-independent integrated metagenomic and metatranscriptomic analyses. Genome Biol. 2016, 17, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holert, J.; Cardenas, E.; Bergstrand, L.H.; Zaikova, E.; Hahn, A.S.; Hallam, S.J.; Mohn, W.W. Metagenomes Reveal Global Distribution of Bacterial Steroid Catabolism in Natural, Engineered, and Host Environments. MBio 2018, 9, e02345-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebeskind, B.J.; Hillis, D.M.; Zakon, H.H. Convergence of ion channel genome content in early animal evolution. Proc. Natl. Acad. Sci. USA 2015, 112, E846–E851. [Google Scholar] [CrossRef] [Green Version]
- De Almeida, O.G.G.; Furlan, J.P.R.; Stehling, E.G.; De Martinis, E.C.P. Comparative phylo-pangenomics reveals generalist lifestyles in representative Acinetobacter species and proposes candidate gene markers for species identification. Gene 2021, 791, 145707. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Magallanes, V.; Deschavanne, P.; Quintana-Murci, L.; Brosch, R.; Gicquel, B.; Neyrolles, O. Horizontal Transfer of a Virulence Operon to the Ancestor of Mycobacterium tuberculosis. Mol. Biol. Evol. 2006, 23, 1129–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendriksen, R.S.; Leekitcharoenphon, P.; Lukjancenko, O.; Lukwesa-Musyani, C.; Tambatamba, B.; Mwaba, J.; Kalonda, A.; Nakazwe, R.; Kwenda, G.; Jensen, J.D.; et al. Genomic Signature of Multidrug-Resistant Salmonella enterica Serovar Typhi Isolates Related to a Massive Outbreak in Zambia between 2010 and 2012. J. Clin. Microbiol. 2015, 53, 262–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Wang, A. Genomic islands mediate environmental adaptation and the spread of antibiotic resistance in multiresistant enterococci-evidence from genomic sequences. BMC Microbiol. 2021, 21, 55. [Google Scholar] [CrossRef]
- Thompson, C.C.; Chimetto, L.; Edwards, R.A.; Swings, J.; Stackebrandt, E.; Thompson, F.L. Microbial genomic taxonomy. BMC Genom. 2013, 14, 913. [Google Scholar] [CrossRef] [Green Version]
- Tortoli, E.; Fedrizzi, T.; Meehan, C.J.; Trovato, A.; Grottola, A.; Giacobazzi, E.; Serpini, G.F.; Tagliazucchi, S.; Fabio, A.; Bettua, C.; et al. The new phylogeny of the genus Mycobacterium: The old and the news. Infect. Genet. Evol. 2017, 56, 19–25. [Google Scholar] [CrossRef]
- Thompson, C.C.; Vicente, A.C.P.; Souza, R.C.; Vasconcelos, A.T.R.; Vesth, T.; Alves, N., Jr.; Ussery, D.W.; Iida, T.; Thompson, F.L. Genomic taxonomy of vibrios. BMC Evol. Biol. 2009, 9, 258. [Google Scholar] [CrossRef] [Green Version]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef]
- Randhawa, G.S.; Soltysiak, M.P.M.; El Roz, H.; de Souza, C.P.E.; Hill, K.A.; Kari, L. Machine learning using intrinsic genomic signatures for rapid classification of novel pathogens: COVID-19 case study. PLoS ONE 2021, 16, e0232391. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, H.L.; Conover, M.S.; Chou, W.C.; Hibbing, M.E.; Manson, A.L.; Dodson, K.W.; Hannan, T.J.; Roberts, P.L.; Stapleton, A.E.; Hooton, T.M.; et al. Bacterial virulence phenotypes of Escherichia coli and host susceptibility determine risk for urinary tract infections. Sci. Transl. Med. 2017, 9, eaaf1283. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Jelsbak, L.; Marvig, R.L.; Damkiær, S.; Workman, C.T.; Rau, M.H.; Hansen, S.K.; Folkesson, A.; Johansen, H.K.; Ciofu, O.; et al. Evolutionary dynamics of bacteria in a human host environment. Proc. Natl. Acad. Sci. USA 2011, 108, 7481–7486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkins, C.; Kotewicz, M.; Jackson, S.; Lacher, D.; Abu-Ali, G.; Patel, I. Genomic paradigms for food-borne enteric pathogen analysis at the USFDA: Case studies highlighting method utility, integration and resolution. Food Addit. Contam. Part A 2013, 30, 1422–1436. [Google Scholar] [CrossRef] [PubMed]
- Bridier-Nahmias, A.; Launay, A.; Bleibtreu, A.; Magnan, M.; Walewski, V.; Chatel, J.; Dion, S.; Robbe-Saule, V.; Clermont, O.; Norel, F.; et al. Escherichia coli Genomic Diversity within Extraintestinal Acute Infections Argues for Adaptive Evolution at Play. mSphere 2021, 6, e01176-20. [Google Scholar] [CrossRef] [PubMed]
- McClain, M.T.; Nicholson, B.P.; Park, L.P.; Liu, T.Y.; Hero, A.O., III; Tsalik, E.L.; Zaas, A.K.; Veldman, T.; Hudson, L.L.; Lambkin-Williams, R.; et al. A Genomic Signature of Influenza Infection Shows Potential for Presymptomatic Detection, Guiding Early Therapy, and Monitoring Clinical Responses. Open Forum Infect. Dis. 2016, 3, ofw007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makkoch, J.; Suwannakarn, K.; Payungporn, S.; Prachayangprecha, S.; Cheiocharnsin, T.; Linsuwanon, P.; Theamboonlers, A.; Poovorawan, Y. Whole Genome Characterization, Phylogenetic and Genome Signature Analysis of Human Pandemic H1N1 Virus in Thailand, 2009–2012. PLoS ONE 2012, 7, e51275. [Google Scholar] [CrossRef]
- Xu, W.; Dai, Y.; Hua, C.; Wang, Q.; Zou, P.; Deng, Q.; Jiang, S.; Lu, L. Genomic signature analysis of the recently emerged highly pathogenic A(H5N8) avian influenza virus: Implying an evolutionary trend for bird-to-human transmission. Microbes Infect. 2017, 19, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Cheung, B.; Tan, S.; Li, C.; Li, L.; Liu, S.; Jiang, S. Genomic Signature and Mutation Trend Analysis of Pandemic (H1N1) 2009 Influenza A Virus. PLoS ONE 2010, 5, A31–A37. [Google Scholar] [CrossRef] [PubMed]
- Jaron, K.S.; Moravec, J.C.; Martínková, N. SigHunt: Horizontal gene transfer finder optimized for eukaryotic genomes. Bioinformatics 2013, 30, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Kong, R.; Xu, X.; Liu, X.; He, P.; Zhang, M.Q.; Dai, Q. 2SigFinder: The combined use of small-scale and large-scale statistical testing for genomic island detection from a single genome. BMC Bioinform. 2020, 21, 159. [Google Scholar] [CrossRef]
- Da Silva Filho, A.C.; Raittz, R.T.; Guizelini, D.; De Pierri, C.R.; Augusto, D.W.; dos Santos-Weiss, I.C.R.; Marchaukoski, J.N. Comparative Analysis of Genomic Island Prediction Tools. Front. Genet. 2018, 9, 619. [Google Scholar] [CrossRef] [Green Version]
- Dai, Q.; Bao, C.; Hai, Y.; Ma, S.; Zhou, T.; Wang, C.; Wang, Y.; Huo, W.; Liu, X.; Yao, Y.; et al. MTGIpick allows robust identification of genomic islands from a single genome. Brief. Bioinform. 2016, 19, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Liu, X.; Yang, S.; Bao, C.; He, P.; Dai, Q. An efficient genomic signature ranking method for genomic island prediction from a single genome. J. Theor. Biol. 2019, 467, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, H. Chaos game representation of gene structure. Nucleic Acids Res. 1990, 18, 2163–2170. [Google Scholar] [CrossRef] [Green Version]
- Barnsley, M.F. Fractals Everywhere, 2nd ed.; Academic Press: Boston, MA, USA, 1993. [Google Scholar] [CrossRef]
- Hoang, T.; Yin, C.; Yau, S.S.T. Numerical encoding of DNA sequences by chaos game representation with application in similarity comparison. Genomics 2016, 108, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Poor, N.R.; Yaghoobi, M. A new approach in DNA sequence compression: Fast DNA sequence compression using parallel chaos game representation. Expert Syst. Appl. 2019, 116, 487–493. [Google Scholar] [CrossRef]
- Yin, C. Encoding and Decoding DNA Sequences by Integer Chaos Game Representation. J. Comput. Biol. 2019, 26, 143–151. [Google Scholar] [CrossRef]
- Josse, J.; Kornberg, A.; Kaiser, A. Enzymatic synthesis of desoxiribonucleic acid. Frequencies of nearest neighbor base sequences in desoxyribonucleic acid. J. Biol. Chem. 1961, 236, 864–875. [Google Scholar] [CrossRef]
- Iversen, G.R.; Gergen, M. Statistics: The Conceptual Approach; Springer Science & Business Media: New York, NY, USA, 1997. [Google Scholar]
- Wang, Z.; Bovik, A.; Sheikh, H.; Simoncelli, E. Image quality assessment: From error visibility to structural similarity. IEEE Trans. Image Process. 2004, 13, 600–612. [Google Scholar] [CrossRef] [Green Version]
- Krause, E.F. Taxicab Geometry: An Adventure in Non-Euclidean Geometry; Courier Corporation: Chelmsford, MA, USA, 1986. [Google Scholar]
- Bonnici, V.; Manca, V. Informational laws of genome structures. Sci. Rep. 2016, 6, 28840. [Google Scholar] [CrossRef] [Green Version]
- Forsdyke, D.R.; Mortimer, J.R. Chargaff’s legacy. Gene 2000, 261, 127–137. [Google Scholar] [CrossRef]
- Forsdyke, D. Different Biological Species “Broadcast” Their DNAs at Different (G + C). J. Theor. Biol. 1996, 178, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Karlin, S.; Mrázek, J. What Drives Codon Choices in Human Genes? J. Mol. Biol. 1996, 262, 459–472. [Google Scholar] [CrossRef]
- Forsdyke, D.R. Relative roles of primary sequence and (G + C) hierarchy of frequencies of complementary trinucleotide pairs in DNAs of different species. J. Mol. Evol. 1995, 41, 573–581. [Google Scholar] [CrossRef]
- Hershberg, R.; Petrov, D.A. Evidence That Mutation Is Universally Biased towards AT in Bacteria. PLoS Genet. 2010, 6, e1001115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mann, S.; Chen, Y.P.P. Bacterial genomic G plus C composition-eliciting environmental adaptation. Genomics 2010, 95, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, G. Isochores and the evolutionary genomics of vertebrates. Gene 2000, 241, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Filipski, J. Evolution of DNA Sequence Contributions of Mutational Bias and Selection to the Origin of Chromosomal Compartments. In Advances in Mutagenesis Research; Springer: Berlin/Heidelberg, Germany, 1990; pp. 1–54. [Google Scholar] [CrossRef]
- Forsdyke, D.R. Success of alignment-free oligonucleotide (k-mer) analysis confirms relative importance of genomes not genes in speciation and phylogeny. Biol. J. Linn. Soc. 2019, 128, 239–250. [Google Scholar] [CrossRef]
- Hatje, K.; Kollmar, M. A phylogenetic analysis of the Brassicales clade based on an alignment-free sequence comparison method. Front. Plant Sci. 2012, 3, 192. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef] [Green Version]
- Nissen, J.; Johansen, J.; Allesøe, R.; Sønderby, C.; Armenteros, J.; Grønbech, C.; Jensen, L.; Nielsen, H.; Nordahl Petersen, T.; Winther, O.; et al. Improved metagenome binning and assembly using deep variational autoencoders. Nat. Biotechnol. 2021, 39, 555–560. [Google Scholar] [CrossRef]
- Millán Arias, P.; Alipour, F.; Hill, K.A.; Kari, L. DeLUCS: Deep learning for unsupervised clustering of DNA sequences. PLoS ONE 2022, 17, e0261531. [Google Scholar] [CrossRef] [PubMed]
- Girgis, H. MeShClust v3.0: High-quality clustering of DNA sequences using the mean shift algorithm and alignment-free identity scores. BMC Genom. 2022, 23, 423. [Google Scholar] [CrossRef]
- Lo, R.; Dougan, K.E.; Chen, Y.; Shah, S.; Bhattacharya, D.; Chan, C.X. Alignment-Free Analysis of Whole-Genome Sequences from Symbiodiniaceae Reveals Different Phylogenetic Signals in Distinct Regions. Front. Plant Sci. 2022, 13, 815714. [Google Scholar] [CrossRef] [PubMed]
- González-Pech, R.A.; Stephens, T.G.; Chen, Y.; Mohamed, A.R.; Cheng, Y.; Shah, S.; Dougan, K.E.; Fortuin, M.D.; Lagorce, R.; Burt, D.W.; et al. Comparison of 15 dinoflagellate genomes reveals extensive sequence and structural divergence in family Symbiodiniaceae and genus Symbiodinium. BMC Biol. 2021, 19, 73. [Google Scholar] [CrossRef] [PubMed]
- Bashir, A.; Ye, C.; Price, A.L.; Bafna, V. Orthologous repeats and mammalian phylogenetic inference. Genome Res. 2005, 15, 998–1006. [Google Scholar] [CrossRef] [Green Version]
- Touati, R.; Haddad-Boubaker, S.; Ferchichi, I.; Messaoudi, I.; Ouesleti, A.E.; Triki, H.; Lachiri, Z.; Kharrat, M. Comparative genomic signature representations of the emerging COVID-19 coronavirus and other coronaviruses: High identity and possible recombination between Bat and Pangolin coronaviruses. Genomics 2020, 112, 4189–4202. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Keyword | Frequency | Total Link Strength |
|---|---|---|
| Gene expression | 141 | 570 |
| Genome signature | 125 | 530 |
| cancer | 106 | 468 |
| sequence | 81 | 317 |
| evolution | 72 | 316 |
| identification | 52 | 219 |
| genes | 45 | 194 |
| genome | 45 | 170 |
| cells | 40 | 150 |
| diversity | 33 | 117 |
| survival | 31 | 135 |
| mutation | 29 | 106 |
| bacteria | 27 | 125 |
| chaos game representation | 26 | 117 |
| chemotherapy | 26 | 123 |
| DNA | 26 | 106 |
| Escherichia coli | 25 | 123 |
| prognosis | 24 | 122 |
| differentiation | 22 | 85 |
| microarray | 22 | 105 |
| adaptation | 21 | 90 |
| selection | 21 | 81 |
| biomarker | 20 | 90 |
| phylogenetic analysis | 20 | 94 |
| Scientific Field | Cluster | Num. Keywords |
|---|---|---|
| Health | C1 | 41 |
| C2 | 34 | |
| Molecular biology | C3 | 24 |
| Comparative genomics | C4 | 36 |
| Evolutionary biology | C5 | 38 |
| 1/2 | 0 | 0 | 1/2 | 0 | 0 | |
| 1/2 | 0 | 0 | 1/2 | 1/2 | 0 | |
| 1/2 | 0 | 0 | 1/2 | 1/4 | 1/2 |
| 1/2 | 0 | 0 | 1/2 | 0 | 0 | 1/4 | |
| 1/2 | 0 | 0 | 1/2 | 0 | 1/2 | 1/4 | |
| 1/2 | 0 | 0 | 1/2 | 1/2 | 1/2 | 1/4 | |
| 1/2 | 0 | 0 | 1/2 | 1/2 | 0 | 1/4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de la Fuente, R.; Díaz-Villanueva, W.; Arnau, V.; Moya, A. Genomic Signature in Evolutionary Biology: A Review. Biology 2023, 12, 322. https://doi.org/10.3390/biology12020322
de la Fuente R, Díaz-Villanueva W, Arnau V, Moya A. Genomic Signature in Evolutionary Biology: A Review. Biology. 2023; 12(2):322. https://doi.org/10.3390/biology12020322
Chicago/Turabian Stylede la Fuente, Rebeca, Wladimiro Díaz-Villanueva, Vicente Arnau, and Andrés Moya. 2023. "Genomic Signature in Evolutionary Biology: A Review" Biology 12, no. 2: 322. https://doi.org/10.3390/biology12020322