

Failure to Thrive: Impaired BDNF Transport along the Cortical–Striatal Axis in Mouse Q140 Neurons of Huntington’s Disease
 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Culture and Treatments
2.3. btBDNF Cloning
2.4. Biotinylated Mature BDNF (btBDNF) Production
2.5. btBDNF Protein Purification
2.6. Microfluidic Chambers
2.7. Live Imaging of QD Transport
2.8. Antibodies
2.9. Immunoblot
2.10. Immunostaining
2.11. Microscopy
3. Results
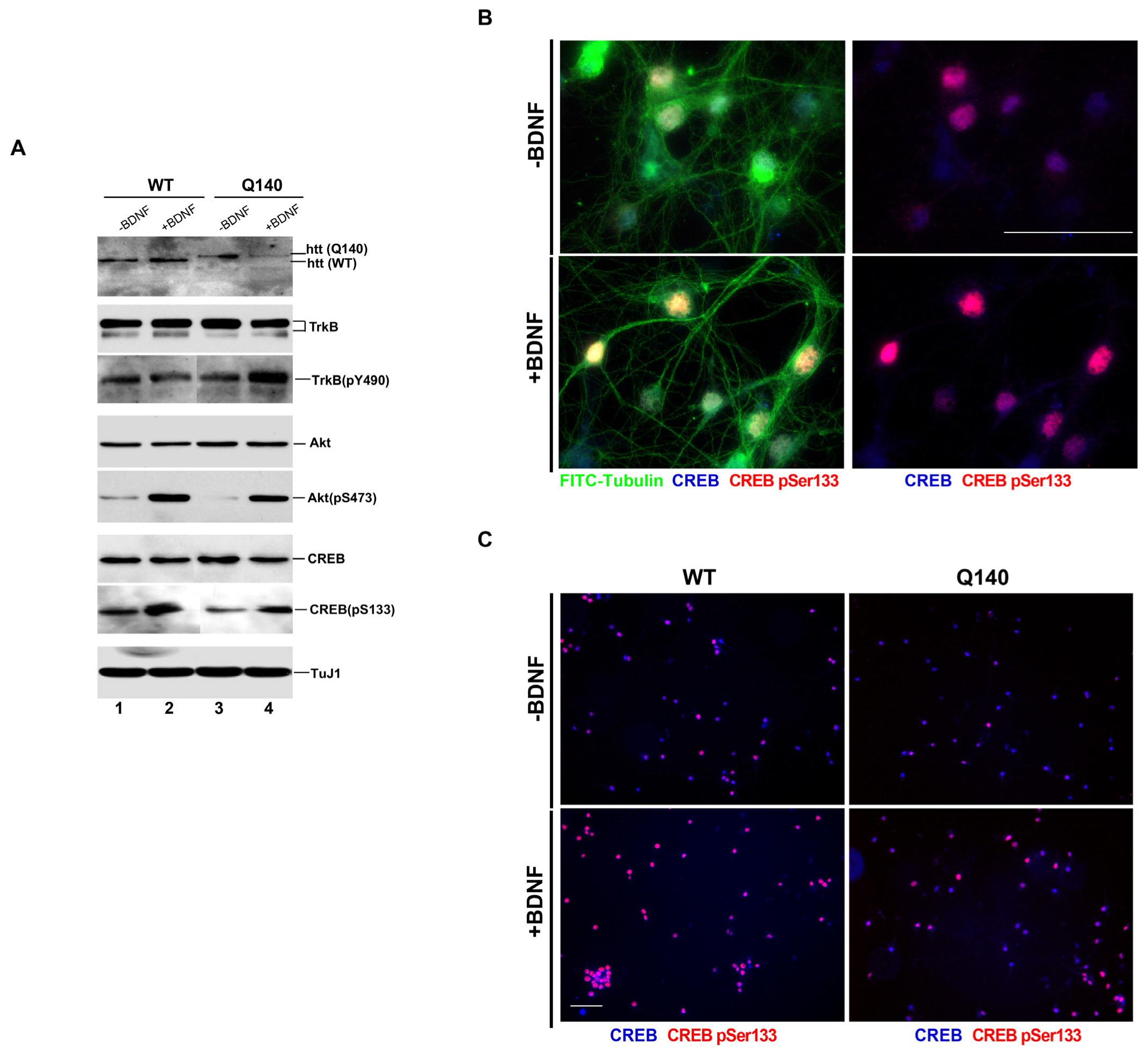
3.1. Effect of BDNF on the BDNF/TrkB Signaling Pathway in Q140 Striatal Neurons
3.2. QD625-Streptavidin Is Conjugated with BDNF
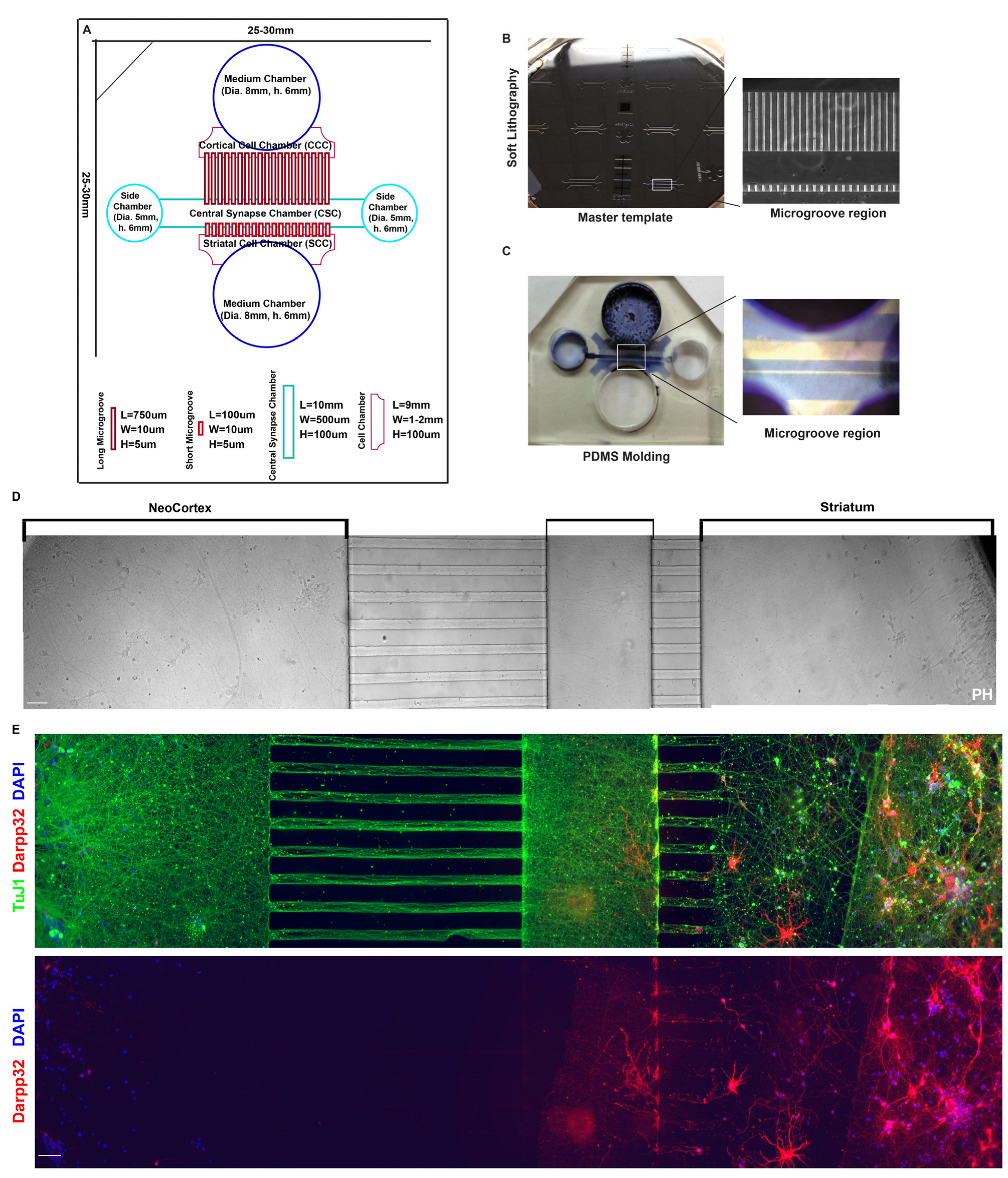
3.3. Cortical–Striatal Neuronal Microfluidic Chamber Was Designed and Fabricated for the Study
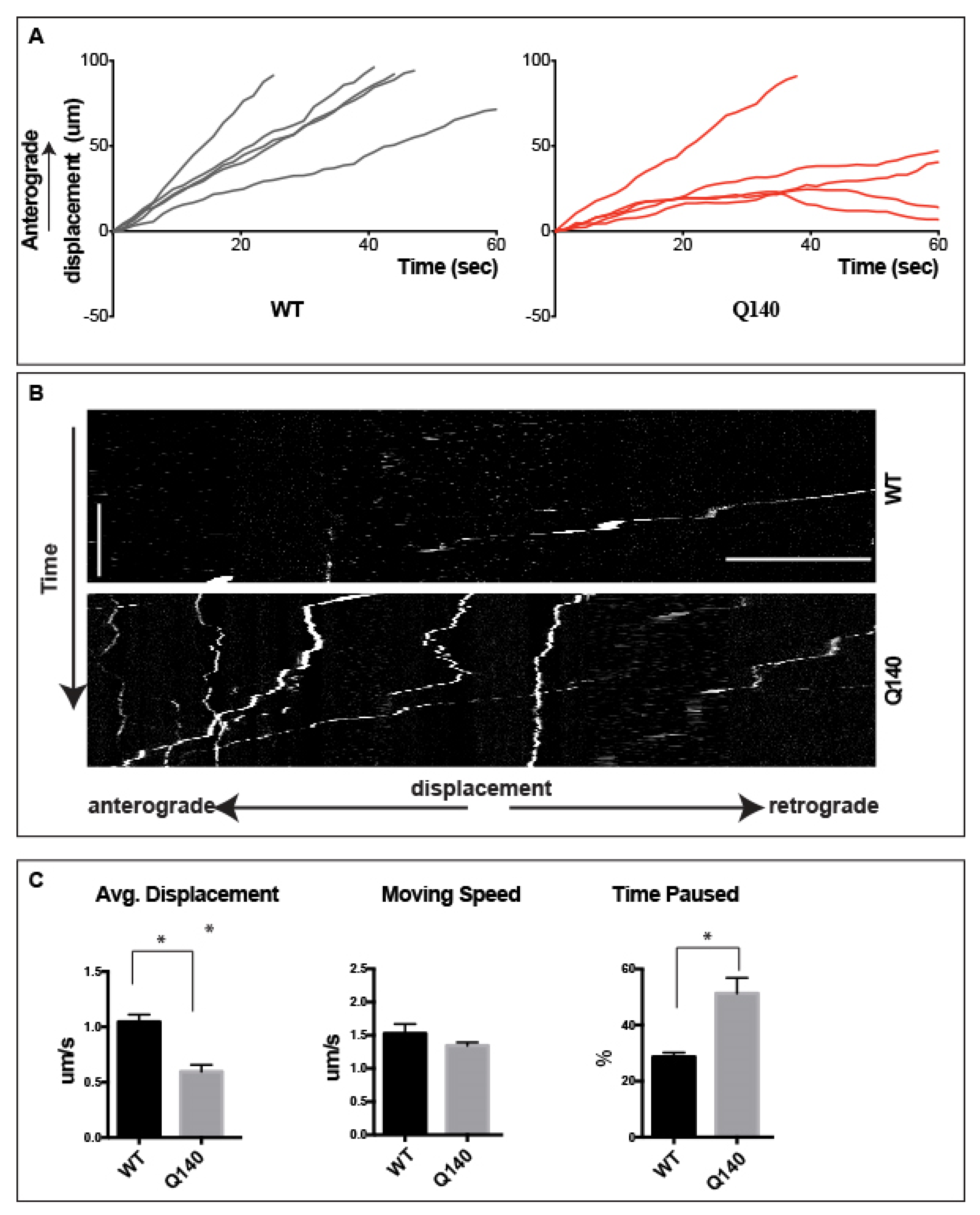
3.4. Anterograde QD-BDNF Transport Is Hindered in the Q140 Cortical Axon
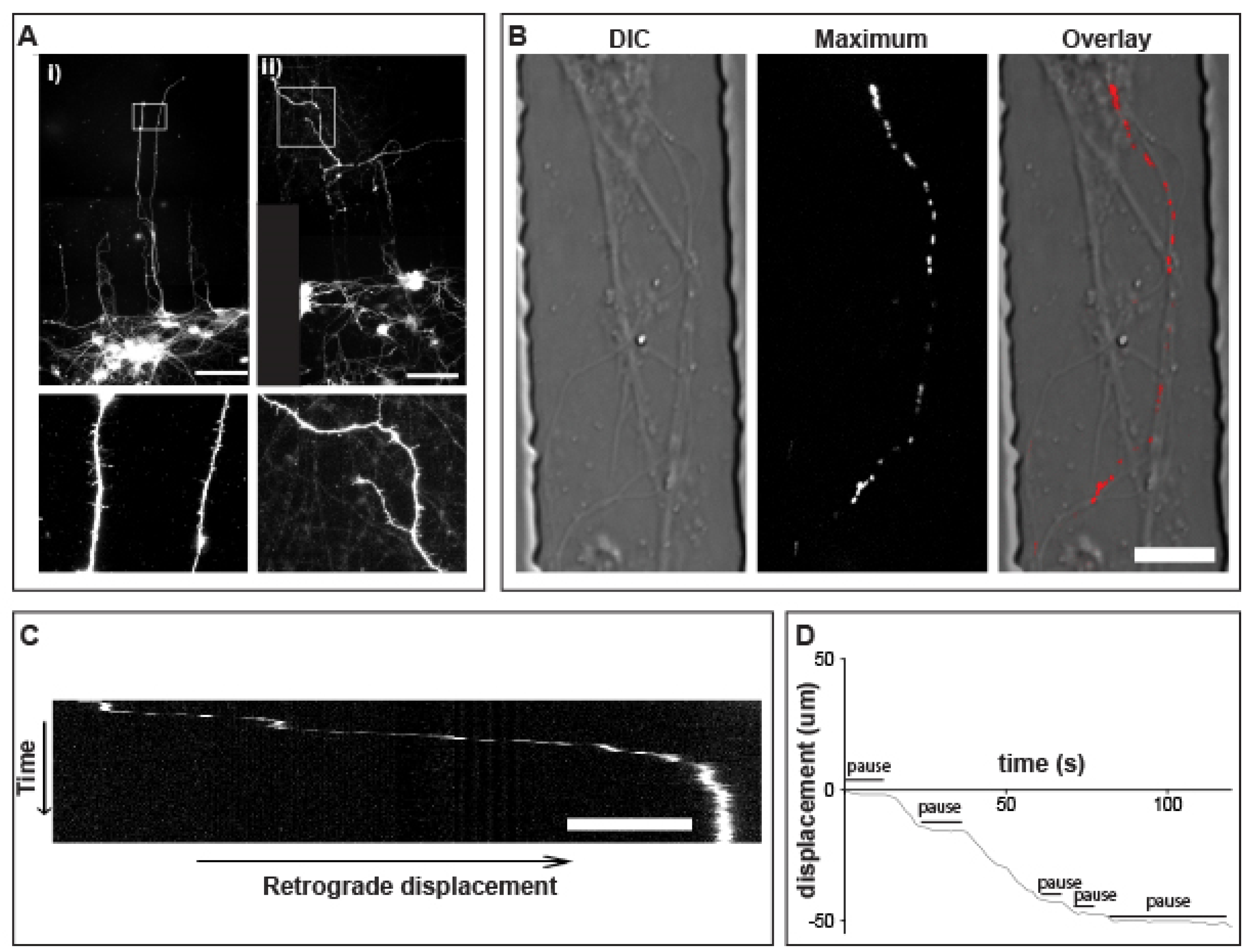
3.5. Prolonged pauses dominate the QD-BDNF Transport in MSN Striatal Dendrites
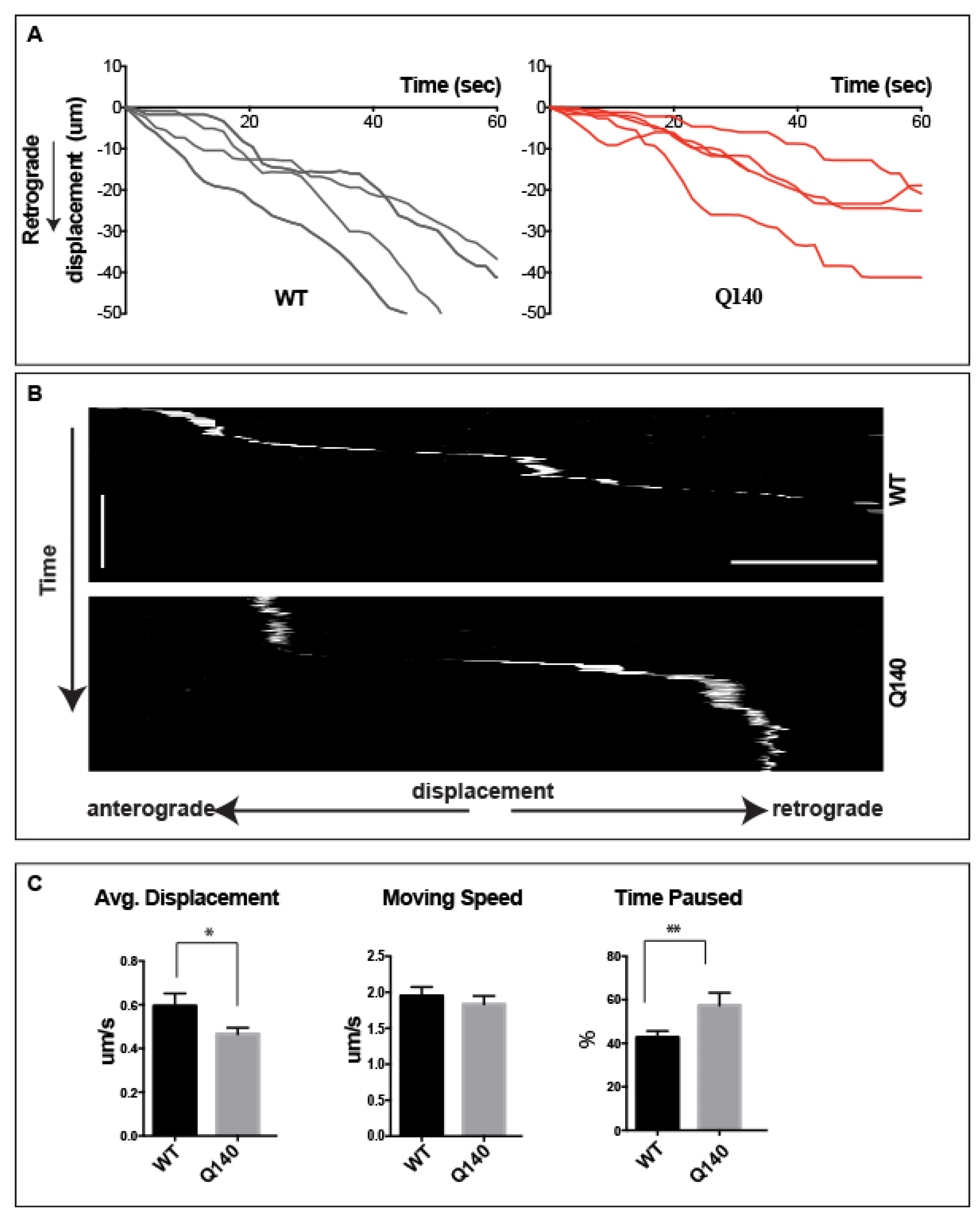
3.6. Retrograde QD-BDNF Transport Is Detained in the Q140 Striatal Dendrites
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Irfan, Z.; Khanam, S.; Karmakar, V.; Firdous, S.M.; Samih, B.; Abou, I.; Khier, E.; Khan, I.; Rehman, M.U.; Khan, A. Pathogenesis of Huntington’ s Disease: An Emphasis on Molecular Pathways and Prevention by Natural Remedies. Brain Sci. 2022, 12, 1389. [Google Scholar] [CrossRef] [PubMed]
- Nopoulos, P.C. Huntington Disease: A Single-Gene Degenerative Disorder of the Striatum. Dialogues Clin. Neurosci. 2016, 18, 91–98. [Google Scholar] [CrossRef]
- Garcia Jareño, P.; Bartley, O.J.M.; Precious, S.V.; Rosser, A.E.; Lelos, M.J. Challenges in Progressing Cell Therapies to the Clinic for Huntington’s Disease: A Review of the Progress Made with Pluripotent Stem Cell Derived Medium Spiny Neurons. Int. Rev. Neurobiol. 2022, 166, 1–48. [Google Scholar] [CrossRef]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington Disease: New Insights into Molecular Pathogenesis and Therapeutic Opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Nasir, J.; Floresco, S.B.; O’Kusky, J.R.; Diewert, V.M.; Richman, J.M.; Zeisler, J.; Borowski, A.; Marth, J.D.; Phillips, A.G.; Hayden, M.R. Targeted Disruption of the Huntington’s Disease Gene Results in Embryonic Lethality and Behavioral and Morphological Changes in Heterozygotes. Cell 1995, 81, 811–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langbehn, D.R. Longer CAG Repeat Length Is Associated with Shorter Survival after Disease Onset in Huntington Disease. Am. J. Hum. Genet. 2022, 109, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Thomaz, A.; Jaeger, M.; Brunetto, A.L.; Brunetto, A.T.; Gregianin, L.; de Farias, C.B.; Ramaswamy, V.; Nör, C.; Taylor, M.D.; Roesler, R. Neurotrophin Signaling in Medulloblastoma. Cancers 2020, 12, 2542. [Google Scholar] [CrossRef]
- Colucci-D’amato, L.; Speranza, L.; Volpicelli, F. Neurotrophic Factor Bdnf, Physiological Functions and Therapeutic Potential in Depression, Neurodegeneration and Brain Cancer. Int. J. Mol. Sci. 2020, 21, 7777. [Google Scholar] [CrossRef]
- Cappoli, N.; Tabolacci, E.; Aceto, P.; Dello Russo, C. The Emerging Role of the BDNF-TrkB Signaling Pathway in the Modulation of Pain Perception. J. Neuroimmunol. 2020, 349, 577406. [Google Scholar] [CrossRef]
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF Function and Intracellular Signaling in Neurons. Histol. Histopathol. 2010, 25, 237–258. [Google Scholar] [PubMed]
- Wu, C.H.; Hung, T.H.; Chen, C.C.; Ke, C.H.; Lee, C.Y.; Wang, P.Y.; Chen, S.F. Post-Injury Treatment with 7,8-Dihydroxyflavone, a TrkB Receptor Agonist, Protects against Experimental Traumatic Brain Injury via PI3K/Akt Signaling. PLoS ONE 2014, 9, e113397. [Google Scholar] [CrossRef] [PubMed]
- Palasz, E.; Wysocka, A.; Gasiorowska, A.; Chalimoniuk, M.; Niewiadomski, W.; Niewiadomska, G. BDNF as a Promising Therapeutic Agent in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giampà, C.; Patassini, S.; Borreca, A.; Laurenti, D.; Marullo, F.; Bernardi, G.; Menniti, F.S.; Fusco, F.R. Phosphodiesterase 10 Inhibition Reduces Striatal Excitotoxicity in the Quinolinic Acid Model of Huntington’s Disease. Neurobiol. Dis. 2009, 34, 450–456. [Google Scholar] [CrossRef] [PubMed]
- DeMarch, Z.; Giampà, C.; Patassini, S.; Martorana, A.; Bernardi, G.; Fusco, F.R. Beneficial Effects of Rolipram in a Quinolinic Acid Model of Striatal Excitotoxicity. Neurobiol. Dis. 2007, 25, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Li, C.H.; Chen, S.; Yoo, H.; Qin, X.; Park, H. Decreased BDNF Release in Cortical Neurons of a Knock-in Mouse Model of Huntington’s Disease. Sci. Rep. 2018, 8, 16976. [Google Scholar] [CrossRef]
- Vitet, H.; Brandt, V.; Saudou, F. Traffic Signaling: New Functions of Huntingtin and Axonal Transport in Neurological Disease. Curr. Opin. Neurobiol. 2020, 63, 122–130. [Google Scholar] [CrossRef]
- Azman, K.F.; Zakaria, R. Recent Advances on the Role of Brain-Derived Neurotrophic Factor (BDNF) in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 6827. [Google Scholar] [CrossRef]
- Zuccato, C.; Ciammola, A.; Rigamonti, D.; Leavitt, B.R.; Goffredo, D.; Conti, L.; MacDonald, M.E.; Friedlander, R.M.; Silani, V.; Hayden, M.R.; et al. Loss of Huntingtin-Mediated BDNF Gene Transcription in Huntington’s Disease. Science 2001, 293, 493–498. [Google Scholar] [CrossRef] [Green Version]
- Fusco, F.R.; Zuccato, C.; Tartari, M.; Martorana, A.; De March, Z.; Giampà, C.; Cattaneo, E.; Bernardi, G. Co-Localization of Brain-Derived Neurotrophic Factor (BDNF) and Wild-Type Huntingtin in Normal and Quinolinic Acid-Lesioned Rat Brain. Eur. J. Neurosci. 2003, 18, 1093–1102. [Google Scholar] [CrossRef]
- Conner, J.M.; Lauterborn, J.C.; Yan, Q.; Gall, C.M.; Varon, S. Distribution of Brain-Derived Neurotrophic Factor (BDNF) Protein and MRNA in the Normal Adult Rat CNS: Evidence for Anterograde Axonal Transport. J. Neurosci. 1997, 17, 2295–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anthony Altar, C.; Cai, N.; Bliven, T.; Juhasz, M.; Conner, J.M.; Acheson, A.L.; Lindsay, R.M.; Wiegand, S.J. Anterograde Transport of Brain-Derived Neurotrophic Factor and Its Role in the Brain. Nature 1997, 389, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Salin, P.; Tseng, G.F.; Hoffman, S.; Parada, I.; Prince, D.A. Axonal Sprouting in Layer V Pyramidal Neurons of Chronically Injured Cerebral Cortex. J. Neurosci. 1995, 15, 8234–8245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, Y.; Zhao, T.; Li, X.J.; Li, S. Mutant Huntingtin Impairs BDNF Release from Astrocytes by Disrupting Conversion of Rab3a-GTP into Rab3a-GDP. J. Neurosci. 2016, 36, 8790–8801. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhong, S.; Zhang, R.; Kang, K.; Zhang, X.; Xu, Y.; Zhao, C.; Zhao, M. Functional Analysis of Brain Derived Neurotrophic Factor (BDNF) in Huntington’s Disease. Aging (Albany NY) 2021, 13, 6103–6114. [Google Scholar] [CrossRef] [PubMed]
- Veldman, M.B.; Yang, X.W. Molecular Insights into Cortico-Striatal Miscommunications in Huntington’s Disease. Curr. Opin. Neurobiol. 2018, 48, 79–89. [Google Scholar] [CrossRef]
- Jodeiri Farshbaf, M.; Ghaedi, K. Huntington’s Disease and Mitochondria. Neurotox. Res. 2017, 32, 518–529. [Google Scholar] [CrossRef]
- Terenzio, M.; Schiavo, G.; Fainzilber, M. Compartmentalized Signaling in Neurons: From Cell Biology to Neuroscience. Neuron 2017, 96, 667–679. [Google Scholar] [CrossRef]
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L.F. Axonal Transport: Cargo-Specific Mechanisms of Motility and Regulation. Neuron 2014, 84, 292–309. [Google Scholar] [CrossRef] [Green Version]
- Tesco, G.; Lomoio, S. Pathophysiology of Neurodegenerative Diseases: An Interplay among Axonal Transport Failure, Oxidative Stress, and Inflammation? Semin. Immunol. 2022, 59, 101628. [Google Scholar] [CrossRef]
- Nagano, S.; Araki, T. Axonal Transport and Local Translation of MRNA in Neurodegenerative Diseases. Front. Mol. Neurosci. 2021, 14, 697973. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.S.; Orth, C.B.; Kim, H.J.; Jeon, N.L.; Jaffrey, S.R. Neurotrophin-Mediated Dendrite-to-Nucleus Signaling Revealed by Microfluidic Compartmentalization of Dendrites. Proc. Natl. Acad. Sci. USA 2011, 108, 11246–11251. [Google Scholar] [CrossRef] [Green Version]
- Bruyère, J.; Abada, Y.S.; Vitet, H.; Fontaine, G.; Deloulme, J.C.; Cès, A.; Denarier, E.; Pernet-Gallay, K.; Andrieux, A.; Humbert, S.; et al. Presynaptic APP Levels and Synaptic Homeostasis Are Regulated by Akt Phosphorylation of Huntingtin. eLife 2020, 9, e56371. [Google Scholar] [CrossRef] [PubMed]
- Koppers, M.; Farías, G.G. Organelle Distribution in Neurons: Logistics behind Polarized Transport. Curr. Opin. Cell Biol. 2021, 71, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Quach, T.T.; Stratton, H.J.; Khanna, R.; Kolattukudy, P.E.; Honnorat, J.; Meyer, K.; Duchemin, A.M. Intellectual Disability: Dendritic Anomalies and Emerging Genetic Perspectives. Acta Neuropathol. 2021, 141, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.; Yong, X.L.H.; Roche, K.W.; Anggono, V. Regulation of NMDA Glutamate Receptor Functions by the GluN2 Subunits. J. Neurochem. 2020, 154, 121–143. [Google Scholar] [CrossRef] [Green Version]
- Sidibé, H.; Dubinski, A.; Vande Velde, C. The Multi-Functional RNA-Binding Protein G3BP1 and Its Potential Implication in Neurodegenerative Disease. J. Neurochem. 2021, 157, 944–962. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewska, A.; Janowski, M. Microfluidic Systems in CNS Studies. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2020; Volume 1230. [Google Scholar]
- Taylor, A.M.; Jeon, N.L. Micro-Scale and Microfluidic Devices for Neurobiology. Curr. Opin. Neurobiol. 2010, 20, 640–647. [Google Scholar] [CrossRef]
- Maoz, B.M. Brain-on-a-Chip: Characterizing the next Generation of Advanced in Vitro Platforms for Modeling the Central Nervous System. APL Bioeng. 2021, 5, 030902. [Google Scholar] [CrossRef]
- Menalled, L.B.; Sison, J.D.; Dragatsis, I.; Zeitlin, S.; Chesselet, M.F. Time Course of Early Motor and Neuropathological Anomalies in a Knock-in Mouse Model of Huntington’s Disease with 140 CAG Repeats. J. Comp. Neurol. 2003, 465, 11–26. [Google Scholar] [CrossRef]
- Minamide, L.S.; Striegl, A.M.; Boyle, J.A.; Meberg, P.J.; Bamburg, J.R. Neurodegenerative Stimuli Induce Persistent ADF/Cofilin-Actin Rods That Disrupt Distal Neurite Function. Nat. Cell Biol. 2000, 2, 628–636. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Kater, S.B. Isolated Hippocampal Neurons in Cryopreserved Long-Term Cultures: Development of Neuroarchitecture and Sensitivity to NMDA. Int. J. Dev. Neurosci. 1988, 6, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, W.P.; Banker, G.A. An Electron Microscopic Study of the Development of Axons and Dendrites by Hippocampal Neurons in Culture. I. Cells Which Develop without Intercellular Contacts. J. Neurosci. 1984, 4, 1944–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schatz, P.J. Use of Peptide Libraries to Map the Substrate Specificity of a Peptide-Modifying Enzyme: A 13 Residue Consensus Peptide Specifies Biotinylation in Escherichia coli. Bio/Technology 1993, 11, 1138–1143. [Google Scholar] [CrossRef] [PubMed]
- Niers, J.M.; Chen, J.W.; Lewandrowski, G.; Kerami, M.; Garanger, E.; Wojtkiewicz, G.; Waterman, P.; Keliher, E.; Weissleder, R.; Tannous, B.A. Single Reporter for Targeted Multimodal In Vivo Imaging. J. Am. Chem. Soc. 2012, 134, 5149–5156. [Google Scholar] [CrossRef] [Green Version]
- Howarth, M.; Takao, K.; Hayashi, Y.; Ting, A.Y. Targeting Quantum Dots to Surface Proteins in Living Cells with Biotin Ligase. Proc. Natl. Acad. Sci. USA 2005, 102, 7583–7588. [Google Scholar] [CrossRef] [Green Version]
- Kapur, M.; Maloney, M.T.; Wang, W.; Chen, X.; Millan, I.; Mooney, T.; Yang, J.; Yang, Y. A SxIP Motif Interaction at the Microtubule plus End Is Important for Processive Retrograde Axonal Transport. Cell. Mol. Life Sci. 2014, 71, 4043–4054. [Google Scholar] [CrossRef]
- Ahmed, S.; Kwatra, M.; Gawali, B.; Panda, S.R.; Naidu, V.G.M. Potential Role of TrkB Agonist in Neuronal Survival by Promoting CREB/BDNF and PI3K/Akt Signaling in Vitro and in Vivo Model of 3-Nitropropionic Acid (3-NP)-Induced Neuronal Death. Apoptosis 2021, 26, 52–70. [Google Scholar] [CrossRef]
- Wang, G.; Lei, C.; Tian, Y.; Wang, Y.; Zhang, L.; Zhang, R. Rb1, the Primary Active Ingredient in Panax Ginseng C.A. Meyer, Exerts Antidepressant-like Effects via the BDNF-TrkB-CREB Pathway. Front. Pharmacol. 2019, 10, 1034. [Google Scholar] [CrossRef]
- Ginés, S.; Paoletti, P.; Alberch, J. Impaired TrkB-Mediated ERK1/2 Activation in Huntington Disease Knock-in Striatal Cells Involves Reduced P52/P46 Shc Expression. J. Biol. Chem. 2010, 285, 21537–21548. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, X.Q.; Han, E.; Hu, Y.; Paik, P.; Ding, Z.; Overman, J.; Lau, A.L.; Shahmoradian, S.H.; Chiu, W.; et al. TRiC Subunits Enhance BDNF Axonal Transport and Rescue Striatal Atrophy in Huntington’s Disease. Proc. Natl. Acad. Sci. USA 2016, 113, E5655–E5664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginés, S.; Bosch, M.; Marco, S.; Gavaldà, N.; Díaz-Hernández, M.; Lucas, J.J.; Canals, J.M.; Alberch, J. Reduced Expression of the TrkB Receptor in Huntington’s Disease Mouse Models and in Human Brain. Eur. J. Neurosci. 2006, 23, 649–658. [Google Scholar] [CrossRef]
- Zuccato, C.; Marullo, M.; Conforti, P.; MacDonald, M.E.; Tartari, M.; Cattaneo, E. Systematic Assessment of BDNF and Its Receptor Levels in Human Cortices Affected by Huntington’s Disease. Brain Pathol. 2008, 18, 225–238. [Google Scholar] [CrossRef]
- Zheng, J.; Shen, W.H.; Lu, T.J.; Zhou, Y.; Chen, Q.; Wang, Z.; Xiang, T.; Zhu, Y.C.; Zhang, C.; Duan, S.; et al. Clathrin-Dependent Endocytosis Is Required for TrkB-Dependent Akt-Mediated Neuronal Protection and Dendritic Growth. J. Biol. Chem. 2008, 283, 13280–13288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, V.V.; Stempel, M.H.; Anand, A.; Sidibe, D.K.; Maday, S. Retrograde Axonal Autophagy and Endocytic Pathways Are Parallel and Separate in Neurons. J. Neurosci. 2022, 42, 8524–8541. [Google Scholar] [CrossRef] [PubMed]
- Cui, B.; Wu, C.; Chen, L.; Ramirez, A.; Bearer, E.L.; Li, W.P.; Mobley, W.C.; Chu, S. One at a Time, Live Tracking of NGF Axonal Transport Using Quantum Dots. Proc. Natl. Acad. Sci. USA 2007, 104, 13666–13671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, M.; Shenoy, R.R. Emphasizing Roles of BDNF Promoters and Inducers in Alzheimer’s Disease for Improving Impaired Cognition and Memory. J. Basic Clin. Physiol. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Zhao, X.; Zhou, Y.; Weissmiller, A.M.; Pearn, M.L.; Mobley, W.C.; Wu, C. Real-Time Imaging of Axonal Transport of Quantum Dot-Labeled BDNF in Primary Neurons. J. Vis. Exp. 2014, 15, 51899. [Google Scholar] [CrossRef] [Green Version]
- Virlogeux, A.; Moutaux, E.; Christaller, W.; Genoux, A.; Bruyère, J.; Fino, E.; Charlot, B.; Cazorla, M.; Saudou, F. Reconstituting Corticostriatal Network On-a-Chip Reveals the Contribution of the Presynaptic Compartment to Huntington’s Disease. Cell Rep. 2018, 22, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Ivkovic, S.; Ehrlich, M.E. Expression of the Striatal DARPP-32/ARPP-21 Phenotype in GABAergic Neurons Requires Neurotrophins In Vivo and In Vitro. J. Neurosci. 1999, 19, 5409–5419. [Google Scholar] [CrossRef]
- Plotkin, J.L.; Day, M.; Peterson, J.D.; Xie, Z.; Kress, G.J.; Rafalovich, I.; Kondapalli, J.; Gertler, T.S.; Flajolet, M.; Greengard, P.; et al. Impaired TrkB Receptor Signaling Underlies Corticostriatal Dysfunction in Huntington’s Disease. Neuron 2014, 83, 178–188. [Google Scholar] [CrossRef] [Green Version]
- Jurcau, A. Molecular Pathophysiological Mechanisms in Huntington’s Disease. Biomedicines 2022, 10, 1432. [Google Scholar] [CrossRef]
- Strand, A.D.; Baquet, Z.C.; Aragaki, A.K.; Holmans, P.; Yang, L.; Cleren, C.; Beal, M.F.; Jones, L.; Kooperberg, C.; Olson, J.M.; et al. Expression Profiling of Huntington’s Disease Models Suggests That Brain-Derived Neurotrophic Factor Depletion Plays a Major Role in Striatal Degeneration. J. Neurosci. 2007, 27, 11758–11768. [Google Scholar] [CrossRef] [Green Version]
- Chao, M.V. Neurotrophins and Their Receptors: A Convergence Point for Many Signalling Pathways. Nat. Rev. Neurosci. 2003, 4, 299–309. [Google Scholar] [CrossRef]
- Huang, E.J.; Reichardt, L.F. Trk Receptors: Roles in Neuronal Signal Transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [Green Version]
- Chiang, M.C.; Lee, Y.C.; Huang, C.L.; Chern, Y. CAMP-Response Element-Binding Protein Contributes to Suppression of the A2A Adenosine Receptor Promoter by Mutant Huntingtin with Expanded Polyglutamine Residues. J. Biol. Chem. 2005, 280, 14331–14340. [Google Scholar] [CrossRef] [Green Version]
- Lonze, B.E.; Ginty, D.D. Function and Regulation of CREB Family Transcription Factors in the Nervous System. Neuron 2002, 35, 605–623. [Google Scholar] [CrossRef] [Green Version]
- Lonze, B.E.; Riccio, A.; Cohen, S.; Ginty, D.D. Apoptosis, Axonal Growth Defects, and Degeneration of Peripheral Neurons in Mice Lacking CREB. Neuron 2002, 34, 371–385. [Google Scholar] [CrossRef] [Green Version]
- Giampà, C.; DeMarch, Z.; D’Angelo, V.; Morello, M.; Martorana, A.; Sancesario, G.; Bernardi, G.; Fusco, F.R. Striatal Modulation of CAMP-Response-Element-Binding Protein (CREB) after Excitotoxic Lesions: Implications with Neuronal Vulnerability in Huntington’s Disease. Eur. J. Neurosci. 2006, 23, 11–20. [Google Scholar] [CrossRef]
- Biffi, E. Microfluidic and Compartmentalized Platforms for Neurobiological Research; Humana Press: New York, NY, USA, 2015; ISBN 9781493925100. [Google Scholar]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef]
- Osaki, T.; Shin, Y.; Sivathanu, V.; Campisi, M.; Kamm, R.D. In Vitro Microfluidic Models for Neurodegenerative Disorders. Adv. Healthc. Mater. 2018, 7, 1700489. [Google Scholar] [CrossRef] [Green Version]
- Millet, L.J.; Gillette, M.U. New Perspectives on Neuronal Development via Microfluidic Environments. Trends Neurosci. 2012, 35, 752–761. [Google Scholar] [CrossRef] [Green Version]
- Parlato, S.; De Ninno, A.; Molfetta, R.; Toschi, E.; Salerno, D.; Mencattini, A.; Romagnoli, G.; Fragale, A.; Roccazzello, L.; Buoncervello, M.; et al. 3D Microfluidic Model for Evaluating Immunotherapy Efficacy by Tracking Dendritic Cell Behaviour toward Tumor Cells. Sci. Rep. 2017, 7, 1093. [Google Scholar] [CrossRef] [Green Version]
- Gidwani, B.; Sahu, V.; Shukla, S.S.; Pandey, R.; Joshi, V.; Jain, V.K.; Vyas, A. Quantum Dots: Prospectives, Toxicity, Advances and Applications. J. Drug Deliv. Sci. Technol. 2021, 61, 102308. [Google Scholar] [CrossRef]
- Badıllı, U.; Mollarasouli, F.; Bakirhan, N.K.; Ozkan, Y.; Ozkan, S.A. Role of Quantum Dots in Pharmaceutical and Biomedical Analysis, and Its Application in Drug Delivery. TrAC—Trends Anal. Chem. 2020, 131, 116013. [Google Scholar] [CrossRef]
- Sung, K.; Maloney, M.T.; Yang, J.; Wu, C. A Novel Method for Producing Mono-Biotinylated, Biologically Active Neurotrophic Factors: An Essential Reagent for Single Molecule Study of Axonal Transport. J. Neurosci. Methods 2011, 200, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Sleigh, J.N. Axonal Transport: The Delivery System Keeping Nerve Cells Alive. Front. Young Minds 2020, 8. [Google Scholar] [CrossRef] [Green Version]
- Colin, E.; Zala, D.; Liot, G.; Rangone, H.; Borrell-Pagès, M.; Li, X.J.; Saudou, F.; Humbert, S. Huntingtin Phosphorylation Acts as a Molecular Switch for Anterograde/Retrograde Transport in Neurons. EMBO J. 2008, 27, 2124–2134. [Google Scholar] [CrossRef] [Green Version]
- Morfini, G.A.; You, Y.M.; Pollema, S.L.; Kaminska, A.; Liu, K.; Yoshioka, K.; Björkblom, B.; Coffey, E.T.; Bagnato, C.; Han, D.; et al. Pathogenic Huntingtin Inhibits Fast Axonal Transport by Activating JNK3 and Phosphorylating Kinesin. Nat. Neurosci. 2009, 12, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Resch-Genger, U.; Grabolle, M.; Cavaliere-Jaricot, S.; Nitschke, R.; Nann, T. Quantum Dots versus Organic Dyes as Fluorescent Labels. Nat. Methods 2008, 5, 763–775. [Google Scholar] [CrossRef]
- Bruno, J.G. Advantages and Disadvantages of Using Quantum Dots in Lateral Flow and Other Biological Assay Formats. In Application of Quantum Dots in Biology and Medicine; Springer: Berlin/Heidelberg, Germany, 2022; pp. 91–102. [Google Scholar]
- Buentello, D.C.; García-Corral, M.; Santiago, G.T.; Alvarez, M.M. Neuron(s)-on-a-Chip: A Review of the Design and Use of Microfluidic Systems for Neural Tissue Culture. IEEE Rev. Biomed. Eng. 2022, 1–24. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagents (Antibodies) | Source | Cat# | Used in Manuscript |
|---|---|---|---|
| Primary antibodies | |||
| Rabbit anti-p44/42 ERK1/2 (137FS) | Cell Signaling Technology, Inc., Danvers, MA, USA | 4695S | Figure S3 (WB, 1:1000) |
| Rabbit anti-phospho-p44/42 ERK1/2 (T202&Y204) | Cell Signaling Technology, Inc., Danvers, MA, USA | 4370S | Figure S3 (WB, 1:1000) |
| Rabbit anti-Akt (11E7) | Cell Signaling Technology, Inc., Danvers, MA, USA | 4685S | Figure 1 and Figure S3 (WB, 1:1000) |
| Rabbit anti-phospho-Akt (S473) (D9E) | Cell Signaling Technology, Inc., Danvers, MA, USA | 4060S | Figure 1 and Figure S3 (WB, 1:1000) |
| Mouse anti-CREB (86B10) | Cell Signaling Technology, Inc., Danvers, MA, USA | 9104S | Figure 1 (WB, 1:1000, IF, 1:100) |
| Rabbit anti-phospho-CREB(S133) | Cell Signaling Technology, Inc., Danvers, MA, USA | 9198S | Figure 1 (WB, 1:1000, IF, 1:100) |
| Rabbit anti-TrkB | EMD Millipore Corp, Burlington, MA, USA | 07-225 | Figure 1 (WB, 1:1000) and Figure S3 (IF, 1:100) |
| Rabbit anti-phospho-TrkB(Y490) | Thermo Fisher Scientific, Waltham, MA, USA. | CHDI900520 | Figure 1 (WB, 1:1000) |
| Mouse anti-β-Tubulin iso3 (TuJ1) | AbCam, Cambridge, UK | Ab78078 | Figure 2 (IF, 1:250) |
| Rabbit anti- β-Tubulin iso3 (TuJ1) | AbCam, Cambridge, UK | Ab18027 | Figure 1 (WB, 1:1000–2000) |
| Rabbit anti-Darpp32 (19A3) | Cell Signaling Technology, Inc., Danvers, MA, USA | 2306S | Figure 2 and Figure 3 (IF, 1:250) |
| Mouse anti-huntingtin | EMD Millipore Corp, Burlington, MA, USA | MABN2540 | Figure 1 (WB, 1:1000) |
| Rabbit anti-Tau | Agilent. Santa Clara, CA, USA | A0024 | Figure S2 (WB, 1:5000) |
| Mouse anti-BDNF (N-20) | Santa Cruz Bio, Santa Cruz, CA, USA | SC-546 | Figure S2 (WB, 1:1000) |
| Rabbit anti-MAP2 | Covance, Princeton, NJ, USA | PBB-547C | Figure 3 (IF, 1:250) |
| Rabbit anti-Biotin (D5A7) | Cell Signaling Technology, Inc., Danvers, MA, USA | 5597S | Sup.Figure 2 (WB, 1:1000) |
| Mouse anti-beta Actin (C4) | Santa Cruz Bio, Santa Cruz, CA, USA | SC-47778 | Figure S3 (WB, 1:1000) |
| Alexa Flur488-alpha Tubulin Mono Antibody (DM1A) | eBioscience, San Diego, CA, USA | 53-4502-82 | Figure 1 (IF, 1:100) |
| Secondary Antibodies | |||
| HRP-Conjugated Goat anti-Rabbit | Jackson ImmunoResearch, West Grove, PA, USA | 111-035-144 | For WB (1:10,000) |
| HRP-Conjugated Goat anti-Mouse | Jackson ImmunoResearch, West Grove, PA, USA | 111-035-146 | For WB (1:10,000) |
| Alexa Flur488-conjugated Goat anti-Mouse | Jackson ImmunoResearch, West Grove, PA, USA | 115-545-003 | For IF (1:1000) |
| Alexa Flur488-conjugated Goat anti-Rabbit | Jackson ImmunoResearch, West Grove, PA, USA | 115-545-062 | For IF (1:1000) |
| Alexa Flur594-conjugated Goat anti-Rabbit | Jackson ImmunoResearch, West Grove, PA, USA | 115-585-062 | For IF (1:1000) |
| Alexa Flur647-conjugated Goat anti-Mouse | Jackson ImmunoResearch, West Grove, PA, USA | 115-605-003 | For IF (1:1000) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maloney, M.T.; Wang, W.; Bhowmick, S.; Millan, I.; Kapur, M.; Herrera, N.; Frost, E.; Zhang, E.Y.; Song, S.; Wang, M.; et al. Failure to Thrive: Impaired BDNF Transport along the Cortical–Striatal Axis in Mouse Q140 Neurons of Huntington’s Disease. Biology 2023, 12, 157. https://doi.org/10.3390/biology12020157
Maloney MT, Wang W, Bhowmick S, Millan I, Kapur M, Herrera N, Frost E, Zhang EY, Song S, Wang M, et al. Failure to Thrive: Impaired BDNF Transport along the Cortical–Striatal Axis in Mouse Q140 Neurons of Huntington’s Disease. Biology. 2023; 12(2):157. https://doi.org/10.3390/biology12020157
Chicago/Turabian StyleMaloney, Michael T., Wei Wang, Sumana Bhowmick, Ivan Millan, Mridu Kapur, Nicolas Herrera, Everett Frost, Elena Y. Zhang, Scott Song, Melissa Wang, and et al. 2023. "Failure to Thrive: Impaired BDNF Transport along the Cortical–Striatal Axis in Mouse Q140 Neurons of Huntington’s Disease" Biology 12, no. 2: 157. https://doi.org/10.3390/biology12020157