
Immune Escape in Glioblastoma: Mechanisms of Action and Implications for Immune Checkpoint Inhibitors and CAR T-Cell Therapy
 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Overview of Immunotherapy
3. Glioblastoma Achieves Resistance to Immune Checkpoint Inhibitors with Unique Immune Escape Mechanisms
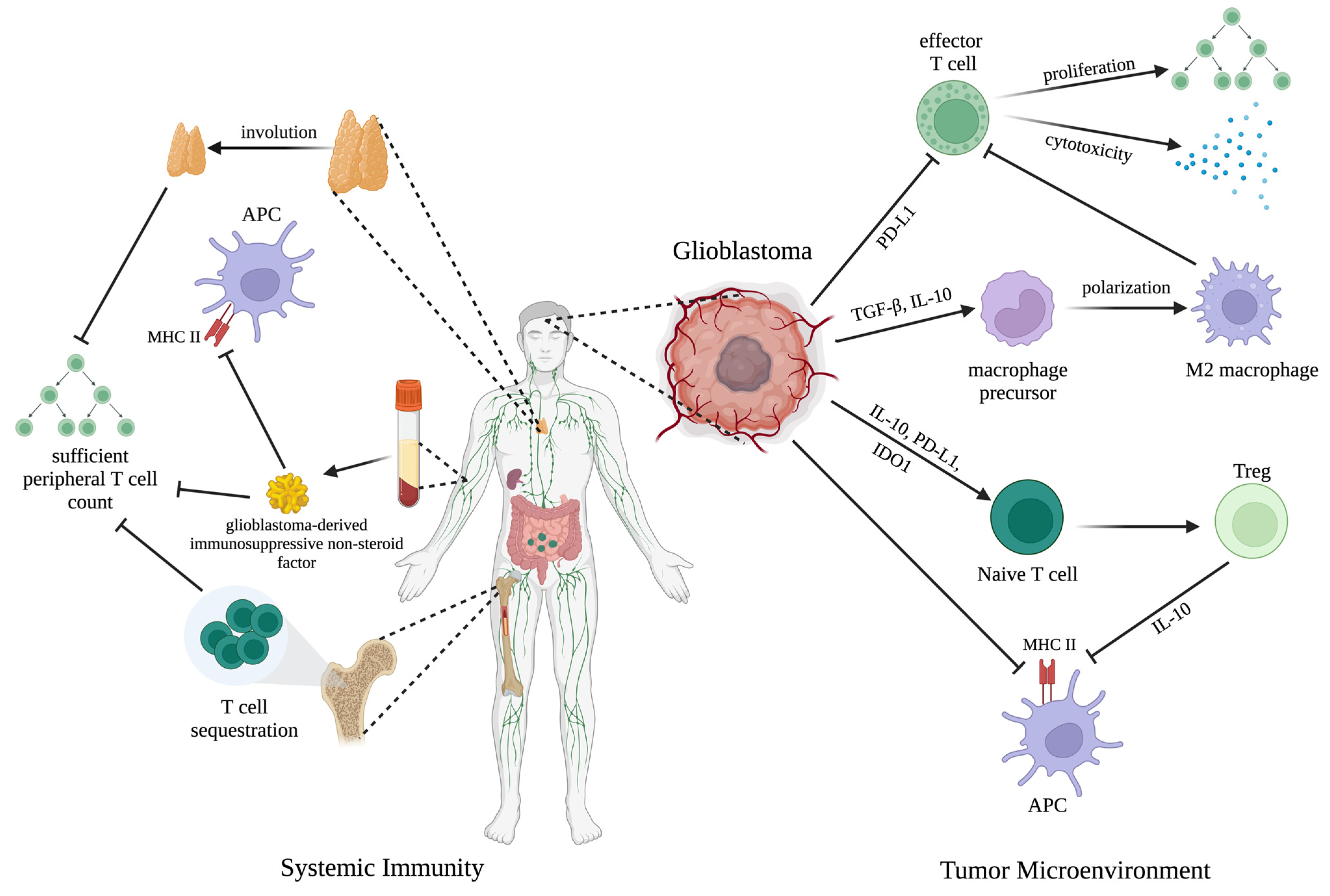
3.1. ICI Efficacy Is Predicated on Functional Innate and Adaptive Immunity
3.2. Glioblastoma Achieves Immune Escape in the Local Tumor Microenvironment
3.3. Glioblastoma Induces Widespread Systemic Immunosuppression
3.4. Potential Combined Approaches to Overcome the Limitations of ICIs in Glioblastoma
4. Glioblastoma Demonstrates Multiple Mechanisms of Resistance to CAR T-Cell Therapy
4.1. Glioblastoma-Induced Immunosuppression and Tumor Heterogeneity Limit the Efficacy of CAR-T Therapy
4.2. Physical Parameters and Features of Glioblastoma Present Additional Challenges to Effective CAR-T Therapy
4.3. Potential Combined Approaches to Overcome the Limitations of CAR T-Cell Therapy in Glioblastoma
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schaff, L.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults. JAMA 2023, 329, 574–587. [Google Scholar] [CrossRef]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaïh, A.; Ahluwalia, M.; Fink, K.; et al. Effect of Tumor-Treating Fields plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Bélanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; Curschmann, J.; et al. Mirimanoff. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Weber, J.S.; Mandalà, M.; Del Vecchio, M.; Gogas, H.; Arance, A.; Cowey, C.L.; Dalle, S.; Schenker, M.; Chiarion-Sileni, V.; Márquez-Rodas, I.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III or IV Melanoma. N. Engl. J. Med. 2017, 377, 1824–1835. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.M.; Kalinka, E.; et al. Nivolumab in Previously Untreated Melanoma without BRAF Mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.-J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and Tumor Responses with Lambrolizumab (Anti–PD-1) in Melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Lee, J.-S.; Ciuleanu, T.-E.; Caro, R.B.; Nishio, M.; Urbán, L.; Audigier-Valette, C.; Lupinacci, L.; Sangha, R.; Płużański, A.; et al. Five-Year Survival Outcomes with Nivolumab plus Ipilimumab versus Chemotherapy as First-Line Treatment for Metastatic Non–Small-Cell Lung Cancer in CheckMate 227. J. Clin. Oncol. 2023, 41, 1200–1212. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.-H.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.-C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor–Transduced T Cells Targeting EGFRvIII in Patients with Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.G.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Lim, M.; Weller, M.; Idbaïh, A.; Steinbach, J.P.; Finocchiaro, G.; Raval, R.R.; Ansstas, G.; Baehring, J.M.; Taylor, J.; Honnorat, J.; et al. Phase III Trial of Chemoradiotherapy with Temozolomide plus Nivolumab or Placebo for Newly Diagnosed Glioblastoma with Methylated MGMT Promoter. Neuro-Oncology 2022, 24, 1935–1949. [Google Scholar] [CrossRef]
- Omuro, A.; Brandes, A.A.; Carpentier, A.F.; Idbaïh, A.; Reardon, D.A.; Cloughesy, T.F.; Sumrall, A.; Baehring, J.M.; van den Bent, M.; Bähr, O.; et al. Radiotherapy Combined with Nivolumab or Temozolomide for Newly Diagnosed Glioblastoma with Unmethylated MGMT Promoter: An International Randomized Phase III Trial. Neuro-Oncology 2022, 25, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Brandes, A.A.; Omuro, A.; Mulholland, P.J.; Lim, M.; Wick, A.; Baehring, J.M.; Ahluwalia, M.; Roth, P.; Bähr, O.; et al. Effect of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma. JAMA Oncol. 2020, 6, 1003–1010. [Google Scholar] [CrossRef]
- Sindhu, K.K.; Nehlsen, A.D.; Lehrer, E.J.; Rowley, J.P.; Stock, R.G.; Galsky, M.D.; Buckstein, M. Oligoprogression of Solid Tumors on Immune Checkpoint Inhibitors: The Impact of Local Ablative Radiation Therapy. Biomedicines 2022, 10, 2481. [Google Scholar] [CrossRef] [PubMed]
- Sindhu, K.K.; Leiter, A.; Moshier, E.; Lin, J.; Carroll, E.; Brooks, D.; Shimol, J.B.; Eisenberg, E.; Gallagher, E.J.; Stock, R.G.; et al. Durable Disease Control with Local Treatment for Oligoprogression of Metastatic Solid Tumors Treated with Immune Checkpoint Blockade. Cancer Treat. Res. Commun. 2020, 25, 100216. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Menon, H.; Verma, V.; Guo, C.; Ramapriyan, R.; Barsoumian, H.B.; Younes, A.; Hu, Y.; Wasley, M.; Cortez, M.A.; et al. Response and Outcomes after Anti-CTLA4 versus Anti-PD1 Combined with Stereotactic Body Radiation Therapy for Metastatic Non-Small Cell Lung Cancer: Retrospective Analysis of Two Single-Institution Prospective Trials. J. ImmunoTherapy Cancer 2020, 8, e000492. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.; McDermott, D.F.; Weber, R.; Sosman, J.A.; Haanen, J.B.; González, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, R.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef]
- Herbst, R.S.; Giaccone, G.; de Marinis, F.; Reinmuth, N.; Vergnenègre, A.; Barrios, C.H.; Morise, M.; Felip, E.; Andrić, Z.; Geater, S.; et al. Atezolizumab for First-Line Treatment of PD-L1–Selected Patients with NSCLC. N. Engl. J. Med. 2020, 383, 1328–1339. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T Cell Immunotherapy for Human Cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.J.; Rivière, I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.G.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.S.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene Maraleucel for Patients with Relapsed or Refractory Large B-Cell Lymphomas (TRANSCEND NHL 001): A Multicentre Seamless Design Study. Lancet 2020, 396, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.M.; et al. Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T-Cell Therapy in Patients with Relapsed or Refractory Multiple Myeloma (CARTITUDE-1): A Phase 1b/2 Open-Label Study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Muñoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Zimmer, N.; Kim, E.; Sprang, B.; Leukel, P.; Khafaji, F.; Ringel, F.; Sommer, C. Jochen Tuettenberg; Tuettenberg, A. GARP as an Immune Regulatory Molecule in the Tumor Microenvironment of Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 3676. [Google Scholar] [CrossRef]
- Kennedy, B.C.; Showers, C.; Anderson, D.E.; Anderson, L.; Canoll, P.; Bruce, J.N.; Anderson, R.C.E. Tumor-Associated Macrophages in Glioma: Friend or Foe? J. Oncol. 2013, 2013, 1–11. [Google Scholar] [CrossRef]
- Abbott, M.; Ustoyev, Y. Cancer and the Immune System: The History and Background of Immunotherapy. Semin. Oncol. Nurs. 2019, 35, 150923. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Chang, R.B.; Beatty, G.L. The Interplay between Innate and Adaptive Immunity in Cancer Shapes the Productivity of Cancer Immunosurveillance. J. Leukoc. Biol. 2020, 108, 363–376. [Google Scholar] [CrossRef]
- Hilligan, K.L.; Ronchese, F. Antigen Presentation by Dendritic Cells and Their Instruction of CD4+ T Helper Cell Responses. Cell. Mol. Immunol. 2020, 17, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.; Simon, C.M. The Tumor Microenvironment. Curr. Biol. 2020, 30, R921–R925. [Google Scholar] [CrossRef]
- Elhanani, O.; Ben-Uri, R.; Keren, L. Spatial Profiling Technologies Illuminate the Tumor Microenvironment. Cancer Cell 2023, 41, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to Treat Immune Hot, Altered and Cold Tumours with Combination Immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Srikrishnan, R.; Labadie, B.; Argulian, A.; Patnaik, A. Targeting Innate Immunity in Cancer Therapy. Vaccines 2021, 9, 138. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.; Kersten, K.; Chan, V.; Fearon, D.F.; Mérad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the Tumor Immune Microenvironment (TIME) for Effective Therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Ochoa, M.; Rodrigo, B.N.; Zimmermann, S.; Coukos, G. Turning up the Heat on Non-Immunoreactive Tumours: Opportunities for Clinical Development. Lancet Oncol. 2020, 21, e419–e430. [Google Scholar] [CrossRef]
- Spitzer, M.H.; Carmi, Y.; Reticker-Flynn, N.E.; Kwek, S.S.; Madhireddy, D.; Martins, M.S.; Gherardini, P.F.; Prestwood, T.R.; Chabon, J.; Bendall, S.C.; et al. Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 2017, 168, 487–502.e15. [Google Scholar] [CrossRef]
- Mathios, D.; Kim, J.E.; Mangraviti, A.; Phallen, J.; Park, S.J.; Jackson, C.M.; Garzón-Muvdi, T.; Kim, E.; Theodros, D.; Polanczyk, M.J.; et al. Anti–PD-1 Antitumor Immunity Is Enhanced by Local and Abrogated by Systemic Chemotherapy in GBM. Sci. Transl. Med. 2016, 8, 370ra180. [Google Scholar] [CrossRef]
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial Activation Chemicals Synergize with Surface Receptor PD-1 Blockade for T Cell-Dependent Antitumor Activity. Proc. Natl. Acad. Sci. USA 2017, 114, E761–E770. [Google Scholar] [CrossRef]
- Paul, S.; Lal, G. The Molecular Mechanism of Natural Killer Cells Function and Its Importance in Cancer Immunotherapy. Front. Immunol. 2017, 8, 1124. [Google Scholar] [CrossRef] [PubMed]
- Himes, B.; Geiger, P.; Ayasoufi, K.; Bhargav, A.G.; Brown, D.; Parney, I.F. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef]
- Qian, J.; Luo, F.; Jiao, Y.; Li, J.; Wang, L.; Wang, C.; Deng, Y.; Zhou, L.; Wang, Y.; Wang, J.; et al. TLR2 Promotes Glioma Immune Evasion by Downregulating MHC Class II Molecules in Microglia. Cancer Immunol. Res. 2018, 6, 1220–1233. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The Role of Microglia and Macrophages in Glioma Maintenance and Progression. Nat. Neurosci. 2015, 19, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hambardzumyan, D. Immune Microenvironment in Glioblastoma Subtypes. Front. Immunol. 2018, 9, 1004. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, J.; Fu, H.; Liu, C.; Yu, Z.; Sun, Y.; She, X.; Li, P.; Chun-hua, Z.; Liu, Y.; et al. Coagulation Factor X Regulated by CASC2c Recruited Macrophages and Induced M2 Polarization in Glioblastoma Multiforme. Front. Immunol. 2018, 9, 1557. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage Plasticity and Interaction with Lymphocyte Subsets: Cancer as a Paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Chen, H.; Li, M.; Guo, Y.; Zhong, Y.; He, Z.; Xu, Y.; Zou, J. Immune Response in Glioma’s Microenvironment. Innov. Surg. Sci. 2020, 5, 115–125. [Google Scholar] [CrossRef]
- Himes, B.; Peterson, T.E.; de Mooij, T.; Milbeth, L.; Jung, M.; Uhm, S.; Yan, D.Z.; Tyson, J.; Jin-Lee, H.J.; Parney, D.; et al. The Role of Extracellular Vesicles and PD-L1 in Glioblastoma-Mediated Immunosuppressive Monocyte Induction. Neuro-Oncology 2020, 22, 967–978. [Google Scholar] [CrossRef]
- Ricklefs, F.; Alayo, Q.A.; Krenzlin, H.; Mahmoud, A.B.; Speranza, M.C.; Nakashima, H.; Hayes, J.; Lee, K.; Balaj, L.; Passaro, C.; et al. Immune Evasion Mediated by PD-L1 on Glioblastoma-Derived Extracellular Vesicles. Sci. Adv. 2018, 4, 2766. [Google Scholar] [CrossRef]
- Liu, T.; Zhu, C.; Chen, X.; Guan, G.; Zou, C.; Shen, S.; Wu, J.; Wang, Y.; Lin, Z.; Chen, L.; et al. Ferroptosis, as the Most Enriched Programmed Cell Death Process in Glioma, Induces Immunosuppression and Immunotherapy Resistance. Neuro-Oncology 2022, 24, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zhang, D.; Jiang, H.; Ye, J.; Zhang, L.; Bagley, S.; Winkler, J.; Gong, Y.; Fan, Y. Small-Molecule Toosendanin Reverses Macrophage-Mediated Immunosuppression to Overcome Glioblastoma Resistance to Immunotherapy. Sci. Transl. Med. 2023, 15, 3558. [Google Scholar] [CrossRef]
- Chen, X.; Fan, X.; Zhao, C.; Zhao, Z.; Hu, L.; Wang, D.; Wang, R.; Fang, Z. Molecular Subtyping of Glioblastoma Based on Immune-Related Genes for Prognosis. Sci. Rep. 2020, 10, 15495. [Google Scholar] [CrossRef] [PubMed]
- Nduom, E.K.; Wei, J.; Yaghi, N.K.; Huang, N.; Kong, L.Y.; Gabrusiewicz, K.; Ling, X.; Zhou, S.; Ivan, C.; Chen, J.Q.; et al. PD-L1 Expression and Prognostic Impact in Glioblastoma. Neuro-Oncology 2015, 18, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Han, S.I.; Ma, E.; Wang, X.; Yu, C.; Dong, T.; Zhan, W.Z.; Wei, X.; Liang, G.; Feng, S. Rescuing Defective Tumor-Infiltrating T-Cell Proliferation in Glioblastoma Patients. Oncol. Lett. 2016, 12, 2924–2929. [Google Scholar] [CrossRef]
- Mathewson, N.D.; Ashenberg, O.; Tirosh, I.; Gritsch, S.; Perez, E.M.; Marx, S.; Jerby-Arnon, L.; Chanoch-Myers, R.; Hara, T.; Richman, A.; et al. Inhibitory CD161 Receptor Identified in Glioma-Infiltrating T Cells by Single-Cell Analysis. Cell 2021, 184, 1281–1298.e26. [Google Scholar] [CrossRef]
- Tang, H.; Wang, Y.; Chlewicki, L.K.; Zhang, Y.; Guo, J.; Liang, W.; Wang, J.; Wang, X.; Fu, Y. Facilitating T Cell Infiltration in Tumor Microenvironment Overcomes Resistance to PD-L1 Blockade. Cancer Cell 2016, 29, 285–296. [Google Scholar] [CrossRef]
- Gustafson, M.P.; Lin, Y.; New, K.C.; Bulur, P.A.; O’Neill, B.P.; Gastineau, D.A.; Dietz, A.B. Systemic Immune Suppression in Glioblastoma: The Interplay between CD14+HLA-DRlo/Neg Monocytes, Tumor Factors, and Dexamethasone. Neuro-Oncology 2010, 12, 631–644. [Google Scholar] [CrossRef]
- Chongsathidkiet, P.; Jackson, C.; Koyama, S.; Loebel, F.; Cui, X.; Farber, H.S.; Woroniecka, K.; Elsamadicy, A.A.; Dechant, C.; Kemeny, H.; et al. Sequestration of T Cells in Bone Marrow in the Setting of Glioblastoma and Other Intracranial Tumors. Nat. Med. 2018, 24, 1459–1468. [Google Scholar] [CrossRef]
- Fecci, P.E.; Mitchell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E.; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased Regulatory T-Cell Fraction amidst a Diminished CD4 Compartment Explains Cellular Immune Defects in Patients with Malignant Glioma. Cancer Res. 2006, 66, 3294–3302. [Google Scholar] [CrossRef] [PubMed]
- Schietinger, A.; Greenberg, P.D. Tolerance and Exhaustion: Defining Mechanisms of T Cell Dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Katayoun, A.; Pfaller, C.K.; Evgin, L.; Khadka, R.H.; Tritz, Z.; Goddery, E.; Fain, C.E.; Yokanovich, L.T.; Himes, B.; Jin, F.; et al. Brain Cancer Induces Systemic Immunosuppression through Release of Non-Steroid Soluble Mediators. Brain 2020, 143, 3629–3652. [Google Scholar] [CrossRef]
- Chae, M.; Peterson, T.E.; Balgeman, A.J.; Chen, S.; Zhang, L.; Renner, D.N.; Johnson, A.J.; Parney, I.F. Increasing Glioma-Associated Monocytes Leads to Increased Intratumoral and Systemic Myeloid-Derived Suppressor Cells in a Murine Model. Neuro-Oncology 2014, 17, 978–991. [Google Scholar] [CrossRef] [PubMed]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic Immunity in Cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Huang, Y.; Chen, Y.; Liu, Y.; Xie, L.; You, Y.; Tong, S.; Xu, J.; Jiang, G.; Song, Q.; et al. Reprogramming Systemic and Local Immune Function to Empower Immunotherapy against Glioblastoma. Nat. Commun. 2023, 14, 435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, A.M.; Hu, G.; Huang, M.; Fan, Y.; Zhang, L.; Wellen, K.E.; Xu, X.; Conn, C.S.; Zou, W.; et al. PHGDH-Mediated Endothelial Metabolism Drives Glioblastoma Resistance to Chimeric Antigen Receptor T Cell Immunotherapy. Cell Metab. 2023, 35, 517–534.e8. [Google Scholar] [CrossRef]
- Meir, V. Human Glioblastoma Cells Release Interleukin 6 In Vivo and In Vitro. Cancer Res. 2023, 50, 6683–6688. [Google Scholar]
- Lohr, J.; Ratliff, T.; Huppertz, A.; Ge, Y.; Dictus, C.; Ahmadi, R.; Grau, S.; Hiraoka, N.; Eckstein, V.; Ecker, R.; et al. Effector T-Cell Infiltration Positively Impacts Survival of Glioblastoma Patients and Is Impaired by Tumor-Derived TGF-β. Clin. Cancer Res. 2011, 17, 4296–4308. [Google Scholar] [CrossRef]
- Skaga, E.; Kulesskiy, E.; Fayzullin, A.; Sandberg, C.; Potdar, S.; Kyttälä, A.; Langmoen, I.A.; Laakso, A.; Gaál-Paavola, E.; Perola, M.; et al. Intertumoral Heterogeneity in Patient-Specific Drug Sensitivities in Treatment-Naïve Glioblastoma. BMC Cancer 2019, 19, 628. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Grazia, S.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor Heterogeneity in Human Glioblastoma Reflects Cancer Evolutionary Dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.; Silverbush, D.; Shaw, M.; Hebert, C.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef] [PubMed]
- Hara, T.; Chanoch-Myers, R.; Mathewson, N.D.; Myskiw, C.; Atta, L.; Bussema, L.; Eichhorn, S.W.; Greenwald, A.C.; Gabriela Kinker, S.; Rodman, C.; et al. Interactions between Cancer Cells and Immune Cells Drive Transitions to Mesenchymal-like States in Glioblastoma. Cancer Cell 2021, 39, 779–792.e11. [Google Scholar] [CrossRef] [PubMed]
- Pant, A.; Lim, M. CAR-T Therapy in GBM: Current Challenges and Avenues for Improvement. Cancers 2023, 15, 1249. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, H.; Soldati, S.; Mossu, A.; Rosito, M.; Rudolph, H.; Müller, W.A.; Latorre, D.; Sallusto, F.; Sospedra, M.; Martin, R.; et al. Human CD4+ T Cell Subsets Differ in Their Abilities to Cross Endothelial and Epithelial Brain Barriers In Vitro. Fluids Barriers CNS 2020, 17, 3. [Google Scholar] [CrossRef]
- Mosteiro, A.; Pedrosa, L.; Ferrés, A.; Diao, D.; Sierra, À.; González, J.J. The Vascular Microenvironment in Glioblastoma: A Comprehensive Review. Biomedicines 2022, 10, 1285. [Google Scholar] [CrossRef]
- Hira, V.V.V.; Ploegmakers, K.; Grevers, F.; Verbovšek, U.; Silvestre-Roig, C.; Aronica, E.; Tigchelaar, W.; Turnšek, T.L.; Molenaar, R.J.; van Noorden, C. CD133+ and Nestin+ Glioma Stem-like Cells Reside around CD31+ Arterioles in Niches That Express SDF-1α, CXCR4, Osteopontin and Cathepsin K. J. Histochem. Cytochem. 2015, 63, 481–493. [Google Scholar] [CrossRef]
- Hira, V.V.V.; Aderetti, D.A.; Noorden, V. Glioma Stem Cell Niches in Human Glioblastoma Are Periarteriolar. J. Histochem. Cytochem. 2018, 66, 349–358. [Google Scholar] [CrossRef]
- Pober, J.S.; Tellides, G. Participation of Blood Vessel Cells in Human Adaptive Immune Responses. Trends Immunol. 2012, 33, 49–57. [Google Scholar] [CrossRef]
- Chen, J.; Liu, G.; Wang, X.; Hong, H.; Li, T.; Li, L.; Wang, H.; Xie, J.; Li, B.; Li, T.; et al. Glioblastoma Stem Cell-Specific Histamine Secretion Drives Pro-Angiogenic Tumor Microenvironment Remodeling. Cell Stem Cell 2022, 29, 1531–1546.e7. [Google Scholar] [CrossRef]
- Jiang, Y.; Han, S.; Cheng, W.; Wang, Z.; Wu, A. NFAT1-Regulated IL6 Signalling Contributes to Aggressive Phenotypes of Glioma. Cell Commun. Signal. 2017, 15, 54. [Google Scholar] [CrossRef]
- Pillay, J.; Tak, T.; Kamp, V.; Koenderman, L. Immune Suppression by Neutrophils and Granulocytic Myeloid-Derived Suppressor Cells: Similarities and Differences. Cell. Mol. Life Sci. 2013, 70, 3813–3827. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-Infiltrated Innate Immune Cells Resemble M0 Macrophage Phenotype. JCI Insight 2016, 1, e85841. [Google Scholar] [CrossRef] [PubMed]
- Orrego, E.; Castañeda, C.A.; Castillo, M.; Bernabe, L.A.; Casavilca, S.; Chakravarti, A.; Wang, M.; García-Corrochano, P.; Villa-Robles, M.R.; Zevallos, R.; et al. Distribution of Tumor-Infiltrating Immune Cells in Glioblastoma. CNS Oncol. 2018, 7, CNS21. [Google Scholar] [CrossRef] [PubMed]
- Fossati, G.; Ricevuti, G.; Edwards, S.W.; Walker, C.; Dalton, A.; Rossi, M. Neutrophil Infiltration into Human Gliomas. Acta Neuropathol. 1999, 98, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Maas, R.R.; Soukup, K.; Fournier, N.; Massara, M.; Galland, S.; Kornete, M.; Wischnewski, V.; Lourenço, J.; Croci, D.; Álvarez-Prado, Á.F.; et al. The Local Microenvironment Drives Activation of Neutrophils in Human Brain Tumors. Cell 2023, 186, 4546–4566.e27. [Google Scholar] [CrossRef] [PubMed]
- Luksik, A.S.; Yazigi, E.; Shah, P.; Jackson, C.M. CAR T Cell Therapy in Glioblastoma: Overcoming Challenges Related to Antigen Expression. Cancers 2023, 15, 1414. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.C.; Kann, M.C.; Bailey, S.R.; Haradhvala, N.J.; Montero-Llopis, P.; Bouffard, A.A.; Scarfò, I.; Leick, M.B.; Grauwet, K.; Berger, T.R.; et al. CAR T Cell Killing Requires the IFNγR Pathway in Solid but Not Liquid Tumours. Nature 2022, 604, 563–570. [Google Scholar] [CrossRef]
- Chen, Y.; Abila, B.; Kamel, Y.M. CAR-T: What Is Next? Cancers 2023, 15, 663. [Google Scholar] [CrossRef]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142.e17. [Google Scholar] [CrossRef]
- Gu, T.; Hu, K.; Si, X.; Hu, Y.; Huang, H. Mechanisms of Immune Effector Cell-Associated Neurotoxicity Syndrome after CAR-T Treatment. Wires Mech. Dis. 2022, 14, 1576. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.; Parney, I.F.; Pafundi, D.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the Blood–Brain Barrier Really Disrupted in All Glioblastomas? A Critical Assessment of Existing Clinical Data. Neuro-Oncology 2017, 20, 184–191. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.R.; Wright, C.J.; et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, Y.; Gu, L.; Chen, R.; Zhu, H.; Zhang, X.; Zhang, Y.; Shi, F.; Sheng, Q.; Jian, Z.; et al. Gene Targets of CAR-T Cell Therapy for Glioblastoma. Cancers 2023, 15, 2351. [Google Scholar] [CrossRef] [PubMed]
- Wykosky, J.; Gibo, D.M.; Stanton, C.A.; Debinski, W. Interleukin-13 Receptor α2, EphA2, and Fos-Related Antigen 1 as Molecular Denominators of High-Grade Astrocytomas and Specific Targets for Combinatorial Therapy. Clin. Cancer Res. 2008, 14, 199–208. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Adjunctive Treatment | Mechanism of Adjunctive Treatment | Results |
|---|---|---|
| Ferroptosis inhibitor (Ferrostatin-1) | ↑ M2 to M1 polarization ↑ T-cell activity | Combined treatment with a ferroptosis inhibitor and PD-1/PD-L1 blockade reversed the immunosuppressive phenotype in glioblastoma-bearing mice, resulting in prolonged survival time and reduced tumor size compared to treatment with PD-1/PD-L1 blockade alone or ferroptosis inhibition alone [52]. |
| Small-molecule toosendanin | ↑ M2 to M1 polarization ↑ T-cell infiltration and activation ↓ T-cell exhaustion | a. Combined treatment with toosendanin and ICIs (anti-PD-1 and anti-CTLA-4 antibodies) delayed tumor growth and enhanced mouse survival compared to treatment with toosendanin or ICIs alone [53]. b. Combined treatment with toosendanin and CAR T-cell therapy significantly enhanced mouse survival compared to treatment with toosendanin or CAR T-cell therapy alone [53]. |
| Nanostructure Nano-reshaper | ↑ number of systemic T cells ↑ local T-cell recruitment ↑ APC activity ↑ normalization of blood vessels ↑ M2 to M1 polarization | Combined treatment with Nano-reshaper and PD-1 blockade improved long-term survival in glioblastoma-bearing mice and generated immunological memory to prevent recurrence [66]. |
| PHGDH inhibition | ↓ aberrant vessel sprouting ↓ intratumoral hypoxia ↑ T-cell infiltration and activity | Combined treatment with PHGDH inhibition and CAR T-cell therapy improved overall survival and delayed tumor growth compared to treatment with PHGDH inhibition or CAR T-cell therapy alone [67]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, C.; Hsieh, K.; Cherry, D.R.; Nehlsen, A.D.; Resende Salgado, L.; Lazarev, S.; Sindhu, K.K. Immune Escape in Glioblastoma: Mechanisms of Action and Implications for Immune Checkpoint Inhibitors and CAR T-Cell Therapy. Biology 2023, 12, 1528. https://doi.org/10.3390/biology12121528
Yu C, Hsieh K, Cherry DR, Nehlsen AD, Resende Salgado L, Lazarev S, Sindhu KK. Immune Escape in Glioblastoma: Mechanisms of Action and Implications for Immune Checkpoint Inhibitors and CAR T-Cell Therapy. Biology. 2023; 12(12):1528. https://doi.org/10.3390/biology12121528
Chicago/Turabian StyleYu, Catherine, Kristin Hsieh, Daniel R. Cherry, Anthony D. Nehlsen, Lucas Resende Salgado, Stanislav Lazarev, and Kunal K. Sindhu. 2023. "Immune Escape in Glioblastoma: Mechanisms of Action and Implications for Immune Checkpoint Inhibitors and CAR T-Cell Therapy" Biology 12, no. 12: 1528. https://doi.org/10.3390/biology12121528