
Targets of Immune Escape Mechanisms in Cancer: Basis for Development and Evolution of Cancer Immune Checkpoint Inhibitors
 ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Overview of Immune Checkpoint Inhibitors
2.1. PD-1/PD-L1 Axis as an Immune Checkpoint Target
2.1.1. PD-1/PD-L1: Structure, Function, and Overview of Pathway
2.1.2. Relationship among Signaling Pathways and PD-1/PD-L1 in Cancer
2.1.3. Treatments Targeted at PD-1/PD-L1 Pathway: Role in Cancer Immunotherapy
2.2. Cytotoxic T Lymphocyte-Associated Antigen (CTLA-4) as an Immune Checkpoint Target
2.2.1. Structure and Basic Role of CTLA-4 in Immune Checkpoint
2.2.2. Negative Co-Stimulation Mediated by CTLA-4
2.2.3. Therapeutic Potential of CTLA-4 Blockade Therapy in Cancer
2.3. HLA-E/NKG2A Axis as an Immune Checkpoint Target
2.3.1. Relationship between Tumor Microenvironment and HLA-E
2.3.2. Interaction of HLA-E with Immune Cells
2.3.3. HLA-E/NKG2A and Its Role in Cancer Immunotherapy
2.4. NKG2D as an Immune Checkpoint Target
2.4.1. Relationship between Tumor Microenvironment and NKG2D
2.4.2. NKG2D: Its Overall Function and Structural Configuration
2.4.3. NKG2D and Its Role in Cancer Immunotherapy
2.5. A2AR and A2BR as Immune Checkpoint Targets
2.5.1. Relationship between Tumor Microenvironment and A2AR
2.5.2. A2AR: Its Overall Function and Structural Configuration
2.5.3. A2AR and Its Role in Cancer Immunotherapy
2.5.4. Relationship between Tumor Microenvironment and A2BR
2.5.5. A2BR: Its Overall Function and Structural Configuration
2.5.6. A2BR and Its Role in Cancer Immunotherapy
2.6. SIRPα/CD47 as an Immune Checkpoint Target
2.6.1. SIRPα/CD47 Functions
2.6.2. SIRPα/CD47 Role in Cancer Immunosuppression
2.7. TIM-3 as an Immune Checkpoint Target
2.7.1. Role of TIM-3
2.7.2. TIM-3 in Cancer Immune Escape and Clinical Applications of TIM-3 Antibodies
2.8. LAG-3 as an Immune Checkpoint Target
2.8.1. Function of LAG-3
2.8.2. Clinical Application of LAG-3 Blocking Antibodies
2.9. B7-H3 as Immune Checkpoint Target in Cancer
2.9.1. Role of B7-H3/CD276 in Immune Response
2.9.2. Role of B7-H3/CD276 in Immune Suppression in Tumors
2.10. PARPs as Promising Immune Checkpoint Targets in Cancer
2.10.1. Functions of PARPs in DNA Damage Response
2.10.2. Antitumor Role of PARP Inhibitors (PARPi) in Cancer in the Context of the Tumor Microenvironment (TME)
2.11. TIGIT as a Promising Immune Checkpoint Target in Cancer
2.11.1. Structure and Functions of TIGIT in Immune Cells
2.11.2. Role of TIGIT in Immune Suppression in the TME
2.12. VISTA as a Target of Cancer Immunotherapy
2.12.1. VISTA’s Structure and Functions
2.12.2. VISTA, a Potential Target for Cancer Immunotherapy
2.13. mARTs as Promising Immune Checkpoint Targets in Cancer
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| A2AR | adenosine A2A receptor |
| ADCC | antibody-dependent cellular cytotoxicity |
| AML | acute myeloid leukemia |
| APC | antigen-presenting cells |
| ART1 | mono-ADP-ribosyl transferase 1 |
| ATC | activated T cells |
| ATP | adenosine triphosphate |
| (CAR)-T | chimeric antigen receptor T cell |
| CD | cluster of differentiation |
| CRC | colorectal cancer |
| CREB | cAMP response element-binding protein |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| CTL | cytotoxic T lymphocytes |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| DC | dendritic cells |
| DSB | double-strand break |
| EMT | epithelial-to-mesenchymal transition |
| FGL1 | fibrinogen-like protein 1 |
| Gal-9 | galectin-9 |
| GITR | glucocorticoid-induced tumor necrosis factor receptor-related protein |
| GPCR | G-protein-coupled receptor |
| HAVCR2 | hepatitis A virus cellular receptor 2 |
| HCC | hepatocellular carcinoma |
| HDAC | histone deacetylase |
| HIF | hypoxia-inducible factor |
| HLA | human leukocyte antigen |
| HMGB1 | High Mobility Group Box 1 |
| HNSCC | head and neck squamous cell carcinoma |
| IAP | integrin-related protein |
| ICB | immune checkpoint blockade |
| ICI | immune checkpoint inhibitor |
| ICOS | inducible T cell COStimulator |
| IFN γ | interferon-γ |
| IG | immunoglobulin |
| IL-10 | interleukin-10 |
| IL-15R | interleukin15 receptor |
| IL-2 | interleukin-2 |
| IL-4 | interleukin-4 |
| ITIM | immune receptor tyrosine-based inhibitory motif |
| ITSM | immune receptor tyrosine-based switch motif |
| LAG-3 | lymphocyte activation gene-3 |
| MAR | mono-ADP-ribosyl |
| mART | mono-ADP-ribosyl transferase |
| MDSC | myeloid-derived suppressor cells |
| MHC | major histocompatibility complex |
| miRNA | micro-RNA |
| mRNA | messenger RNA |
| MMP | matrix metalloproteinases |
| mAb | Monoclonal antibody |
| NECA | 5′-(N-ethylcarboxamido) adenosine |
| NHL | non-Hodgkin’s lymphoma |
| NICD | NAD+-induced cell death |
| NK | natural killer |
| NKAE | activated and expanded NK cells |
| NKG2D/A | natural killer group 2D/A |
| NKG2DL | NKG2D ligand |
| NSCLC | non-small-cell lung cancer |
| ORR | objective response rate |
| MAR | poly-ADP-ribosyl |
| PARP | poly-ADP-ribose polymerase |
| PD-1 | programmed cell death protein 1 |
| PD-L1/2 | programmed cell death ligand 1/2 |
| PI3K | phosphatidylinositol 3-kinase |
| PKD2 | protein kinase D isoform 2 |
| PRR | pattern recognition receptor |
| PSGL-1 | P-selectin glycoprotein ligand 1 |
| Ptdser | phosphatidylserine |
| PTEN | phosphatase and TENsin homolog |
| PTPase | protein tyrosine phosphatases |
| SCLC | small cell lung cancer |
| SIRPα | signal regulatory protein α |
| STING | stimulator of interferon genes |
| TAA | tumor-associated antigen |
| TCR | T cell receptor |
| TIGIT | T cell immunoreceptor with immunoglobulin and ITIM domain |
| TIL | tumor-infiltrating lymphocyte |
| TIM-3 | T cell immunoglobulin and mucin domain 3 |
| TLR7 | toll-like receptor 7 |
| TME | tumor microenvironment |
| TNBC | triple-negative breast cancer |
| TNF α | tumor necrosis factor-α |
| Treg | regulatory T cell |
| TRIM | T cell interacting molecule |
| TSA | tumor-specific antigen |
| VEGF | vascular endothelial growth factor |
| VISTA | V-domain Ig suppressor of T cell activation |
| WNT | wingless-related integration site |
References
- Beatty, G.L.; Gladney, W.L. Immune Escape Mechanisms as a Guide for Cancer ImmunotherapyTailoring Cancer Immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnet, F. The concept of immunological surveillance. Immunol. Asp. Neoplasia 1970, 13, 1–27. [Google Scholar]
- Thomas, L. On immunosurveillance in human cancer. Yale J. Biol. Med. 1982, 55, 329. [Google Scholar] [PubMed]
- Schreiber, P.D.; Old, L.J.; Smyth, M.J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardis, E.R. Neoantigens and genome instability: Impact on immunogenomic phenotypes and immunotherapy response. Genome Med. 2019, 11, 71. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.; Sad, S. The Expanding Universe of T-cell subsets: Th1, Th2 and More. Immunol. Today 1996, 17, 138. [Google Scholar] [CrossRef]
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274. [Google Scholar] [CrossRef]
- Ferrone, C.; Dranoff, G. Dual roles for immunity in gastrointestinal cancers. J. Clin. Oncol. 2010, 28, 4045. [Google Scholar] [CrossRef] [Green Version]
- Bader, L.; Solberg, S.; Kaada, S.; Bolstad, N.; Warren, D.; Gavasso, S.; Gjesdal, C.; Vedeler, C. Assays for infliximab drug levels and antibodies: A matter of scales and categories. Scand. J. Immunol. 2017, 86, 165–170. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 pathways: Similarities, differences, and implications of their inhibition. Am. J. Clin. Oncol. 2016, 39, 98. [Google Scholar] [CrossRef] [Green Version]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer–immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Okazaki, T.; Honjo, T. PD-1 and PD-1 ligands: From discovery to clinical application. Int. Immunol. 2007, 19, 813–824. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Ni, Y.; Liang, X.; Lin, Y.; An, B.; He, X.; Zhao, X. Mechanisms of tumor resistance to immune checkpoint blockade and combination strategies to overcome resistance. Front. Immunol. 2022, 13, 915094. [Google Scholar] [CrossRef]
- Le Dréan, E.; Vély, F.; Olcese, L.; Cambiaggi, A.; Guia, S.; Krystal, G.; Gervois, N.; Moretta, A.; Jotereau, F.; Vivier, E. Inhibition of antigen-induced T cell response and antibody-induced NK cell cytotoxicity by NKG2A: Association of NKG2A with SHP-1 and SHP-2 protein-tyrosine phosphatases. Eur. J. Immunol. 1998, 28, 264–276. [Google Scholar] [CrossRef]
- Haanen, J.B.; Cerundolo, V. NKG2A, a New Kid on the Immune Checkpoint Block. Cell 2018, 175, 1720–1722. [Google Scholar] [CrossRef] [Green Version]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727. [Google Scholar]
- Ceeraz, S.; Nowak, E.C.; Noelle, R.J. B7 family checkpoint regulators in immune regulation and disease. Trends Immunol. 2013, 34, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Neel, B.G.; Gu, H.; Pao, L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 2003, 28, 284–293. [Google Scholar] [CrossRef]
- Riley, J.L. PD-1 signaling in primary T cells. Immunol. Rev. 2009, 229, 114–125. [Google Scholar] [CrossRef]
- Polesso, F.; Weinberg, A.D.; Moran, A.E. Late-Stage Tumor Regression after PD-L1 Blockade Plus a Concurrent OX40 AgonistLarge Tumors Regress with Anti–PD-L1 and Anti-OX40 Treatment. Cancer Immunol. Res. 2019, 7, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Salinas, V.H.; Brown, K.E.; Vanguri, V.K.; Freeman, G.J.; Kuchroo, V.K.; Sharpe, A.H. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J. Exp. Med. 2009, 206, 3015–3029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Spaapen, R.M.; Zha, Y.; Williams, J.; Meng, Y.; Ha, T.T.; Gajewski, T.F. Up-regulation of PD-L1, IDO, and Tregs in the melanoma tumor microenvironment is driven by CD8+ T cells. Sci. Transl. Med. 2013, 5, 200ra116. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.H.; Gillett, M.D.; Cheville, J.C.; Lohse, C.M.; Dong, H.; Webster, W.S.; Chen, L.; Zincke, H.; Blute, M.L.; Leibovich, B.C. Costimulatory molecule B7-H1 in primary and metastatic clear cell renal cell carcinoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2005, 104, 2084–2091. [Google Scholar] [CrossRef]
- Bernstein, M.B.; Garnett, C.T.; Zhang, H.; Velcich, A.; Wattenberg, M.M.; Gameiro, S.R.; Kalnicki, S.; Hodge, J.W.; Guha, C. Radiation-induced modulation of costimulatory and coinhibitory T-cell signaling molecules on human prostate carcinoma cells promotes productive antitumor immune interactions. Cancer Biother. Radiopharm. 2014, 29, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Taube, J.M.; Anders, R.A.; Young, G.D.; Xu, H.; Sharma, R.; McMiller, T.L.; Chen, S.; Klein, A.P.; Pardoll, D.M.; Topalian, S.L. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci. Transl. Med. 2012, 4, 127ra37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Bonam, S.R.; Chauvin, C.; Mathew, M.J.; Bayry, J. IFN-γ Induces PD-L1 Expression in Primed Human Basophils. Cells 2022, 11, 801. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A.; Yamashita, T.; Tamura, H.; Zhao, W.; Tsuji, T.; Shimizu, M.; Shinya, E.; Takahashi, H.; Tamada, K.; Chen, L. Interferon-γ and tumor necrosis factor-α induce an immunoinhibitory molecule, B7-H1, via nuclear factor-κB activation in blasts in myelodysplastic syndromes. Blood J. Am. Soc. Hematol. 2010, 116, 1124–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sznol, M.; Chen, L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clin. Cancer Res. 2013, 19, 1021–1034. [Google Scholar] [CrossRef] [Green Version]
- Parsa, A.T.; Waldron, J.S.; Panner, A.; Crane, C.A.; Parney, I.F.; Barry, J.J.; Cachola, K.E.; Murray, J.C.; Tihan, T.; Jensen, M.C. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat. Med. 2007, 13, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Watari, H. Tumor-Intrinsic PD-L1 Signaling in Cancer Initiation, Development and Treatment: Beyond Immune Evasion. Front. Oncol. 2018, 8, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kammerer-Jacquet, S.-F.; Deleuze, A.; Saout, J.; Mathieu, R.; Laguerre, B.; Verhoest, G.; Dugay, F.; Belaud-Rotureau, M.-A.; Bensalah, K.; Rioux-Leclercq, N. Targeting the PD-1/PD-L1 pathway in renal cell carcinoma. Int. J. Mol. Sci. 2019, 20, 1692. [Google Scholar] [CrossRef] [Green Version]
- Nunes-Xavier, C.E.; Angulo, J.C.; Pulido, R.; Lopez, J.I. A Critical Insight into the Clinical Translation of PD-1/PD-L1 Blockade Therapy in Clear Cell Renal Cell Carcinoma. Curr. Urol. Rep. 2019, 20, 1. [Google Scholar] [CrossRef]
- Ramsay, A.G. Immune checkpoint blockade immunotherapy to activate anti-tumour T-cell immunity. Br. J. Haematol. 2013, 162, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jie, H.B.; Lei, Y.; Gildener-Leapman, N.; Trivedi, S.; Green, T.; Kane, L.P.; Ferris, R.L. PD-1/SHP-2 inhibits Tc1/Th1 phenotypic responses and the activation of T cells in the tumor microenvironment. Cancer Res. 2015, 75, 508–518. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.R.; Gupta, G.K.; Sharma, A.K.; Batra, N.; Sharma, D.K.; Joshi, A.; Sharma, A.K. PI3K/Akt/mTOR Intracellular Pathway and Breast Cancer: Factors, Mechanism and Regulation. Curr. Pharm. Des. 2017, 23, 1633–1638. [Google Scholar] [CrossRef]
- O’Donnell, J.S.; Massi, D.; Teng, M.W.; Mandala, M. PI3K-AKT-mTOR inhibition in cancer immunotherapy, redux. Semin. Cancer Biol. 2018, 48, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.; Song, Y.; Wang, Y.; Huang, Y.; Li, Z.; Cui, Y.; Yi, M.; Xia, L.; Zhuang, W.; Wu, X. PD-1/PD-L1 blockade rescue exhausted CD8+ T cells in gastrointestinal stromal tumours via the PI3K/Akt/mTOR signalling pathway. Cell Prolif. 2019, 52, e12571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wang, D.; Xie, G.; Yin, Y.; Zhao, E.; Tao, K.; Li, R. MicroRNA-152 regulates immune response via targeting B7-H1 in gastric carcinoma. Oncotarget 2017, 8, 28125–28134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stutvoet, T.S.; Kol, A.; de Vries, E.G.; de Bruyn, M.; Fehrmann, R.S.; Terwisscha van Scheltinga, A.G.; de Jong, S. MAPK pathway activity plays a key role in PD-L1 expression of lung adenocarcinoma cells. J. Pathol. 2019, 249, 52–64. [Google Scholar] [CrossRef]
- Jahangir, M.; Yazdani, O.; Kahrizi, M.S.; Soltanzadeh, S.; Javididashtbayaz, H.; Mivefroshan, A.; Ilkhani, S.; Esbati, R. Clinical potential of PD-1/PD-L1 blockade therapy for renal cell carcinoma (RCC): A rapidly evolving strategy. Cancer Cell Int. 2022, 22, 401. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Okayama, T.; Oka, K.; Mizushima, K.; Yasuda, T.; Sakamoto, N.; Katada, K.; Kamada, K.; Uchiyama, K.; Handa, O. The JAK/STAT pathway is involved in the upregulation of PD-L1 expression in pancreatic cancer cell lines. Oncol. Rep. 2017, 37, 1545–1554. [Google Scholar]
- Galluzzi, L.; Spranger, S.; Fuchs, E.; López-Soto, A. WNT signaling in cancer immunosurveillance. Trends Cell Biol. 2019, 29, 44–65. [Google Scholar] [CrossRef]
- Castagnoli, L.; Cancila, V.; Cordoba-Romero, S.L.; Faraci, S.; Talarico, G.; Belmonte, B.; Iorio, M.V.; Milani, M.; Volpari, T.; Chiodoni, C. WNT signaling modulates PD-L1 expression in the stem cell compartment of triple-negative breast cancer. Oncogene 2019, 38, 4047–4060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, X.-w.; Wang, H.; Zhang, W.-w.; Wang, J.-h.; Liu, W.-j.; Xia, Z.-j.; Huang, H.-q.; Jiang, W.-q.; Zhang, Y.-j.; Wang, L. PD-L1 is upregulated by EBV-driven LMP1 through NF-κB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J. Hematol. Oncol. 2016, 9, 109. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Hamanishi, J.; Matsumura, N.; Abiko, K.; Murat, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Hosoe, Y.; Murphy, S.K. Chemotherapy induces programmed cell death-ligand 1 overexpression via the nuclear factor-κB to foster an immunosuppressive tumor microenvironment in ovarian cancer. Cancer Res. 2015, 75, 5034–5045. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, J.; Holokai, L.; Syu, L.; Steele, N.G.; Chang, J.; Wang, J.; Ahmed, S.; Dlugosz, A.; Zavros, Y. Hedgehog signaling induces PD-L1 expression and tumor cell proliferation in gastric cancer. Oncotarget 2018, 9, 37439. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.; Huang, J.; Gao, X.; Du, J.; Wang, Y.; Zhao, L. CircPCBP2 promotes the stemness and chemoresistance of DLBCL via targeting miR-33a/b to disinhibit PD-L1. Cancer Sci. 2022, 113, 2888. [Google Scholar] [CrossRef]
- Boldrini, L.; Giordano, M.; Niccoli, C.; Melfi, F.; Lucchi, M.; Mussi, A.; Fontanini, G. Role of microRNA-33a in regulating the expression of PD-1 in lung adenocarcinoma. Cancer Cell Int. 2017, 17, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Johnston, N.; Zheng, X.; Wang, H.; Zhang, X.; Gao, D.; Min, W. miR-28 modulates exhaustive differentiation of T cells through silencing programmed cell death-1 and regulating cytokine secretion. Oncotarget 2016, 7, 53735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Nduom, E.K.; Kong, L.-Y.; Hashimoto, Y.; Xu, S.; Gabrusiewicz, K.; Ling, X.; Huang, N.; Qiao, W.; Zhou, S. MiR-138 exerts anti-glioma efficacy by targeting immune checkpoints. Neuro-Oncology 2016, 18, 639–648. [Google Scholar] [CrossRef]
- Kao, S.C.; Cheng, Y.Y.; Williams, M.; Kirschner, M.B.; Madore, J.; Lum, T.; Sarun, K.H.; Linton, A.; McCaughan, B.; Klebe, S. Tumor suppressor microRNAs contribute to the regulation of PD-L1 expression in malignant pleural mesothelioma. J. Thorac. Oncol. 2017, 12, 1421–1433. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, J.; Dong, K.; Lin, F.; Long, M.; Ouyang, Y.; Wei, J.; Chen, X.; Weng, Y.; He, T. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell. Signal. 2015, 27, 443–452. [Google Scholar] [CrossRef]
- Cioffi, M.; Trabulo, S.M.; Vallespinos, M.; Raj, D.; Kheir, T.B.; Lin, M.-L.; Begum, J.; Baker, A.-M.; Amgheib, A.; Saif, J. The miR-25-93-106b cluster regulates tumor metastasis and immune evasion via modulation of CXCL12 and PD-L1. Oncotarget 2017, 8, 21609. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Yu, H.; Yi, S.; Peng, X.; Su, P.; Xiao, Z.; Liu, R.; Tang, A.; Li, X.; Liu, F. The tumor suppressor miR-138-5p targets PD-L1 in colorectal cancer. Oncotarget 2016, 7, 45370. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Xi, Q.; Wang, H.; Zhang, Z.; Liu, H.; Cheng, Y.; Guo, X.; Zhang, J.; Zhang, Q.; Zhang, L. miR-142-5p regulates tumor cell PD-L1 expression and enhances anti-tumor immunity. Biochem. Biophys. Res. Commun. 2017, 488, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Mastroianni, J.; Stickel, N.; Andrlova, H.; Hanke, K.; Melchinger, W.; Duquesne, S.; Schmidt, D.; Falk, M.; Andrieux, G.; Pfeifer, D. miR-146a controls immune response in the melanoma microenvironment. Cancer Res. 2019, 79, 183–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.-H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Tao, Z.; Hai, B.; Liang, H.; Shi, Y.; Wang, T.; Song, W.; Chen, Y.; OuYang, J.; Chen, J. miR-424 (322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat. Commun. 2016, 7, 11406. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.-M.; Lian, G.-Y.; Song, Y.; Huang, Y.-F.; Gong, Y. LncRNA MALAT1 promotes tumorigenesis and immune escape of diffuse large B cell lymphoma by sponging miR-195. Life Sci. 2019, 231, 116335. [Google Scholar] [CrossRef]
- Fan, F.; Chen, K.; Lu, X.; Li, A.; Liu, C.; Wu, B. Dual targeting of PD-L1 and PD-L2 by PCED1B-AS1 via sponging hsa-miR-194-5p induces immunosuppression in hepatocellular carcinoma. Hepatol. Int. 2021, 15, 444–458. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, Y.; Zhang, J.; Liu, Y.; Qi, Q. LncRNA SNHG14/miR-5590-3p/ZEB1 positive feedback loop promoted diffuse large B cell lymphoma progression and immune evasion through regulating PD-1/PD-L1 checkpoint. Cell Death Dis. 2019, 10, 731. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Liu, Y.; Li, X.; Chen, S.; Xie, R.; Chen, D.; Gao, H.; Wang, G.; Cai, B.; Yang, X. Long noncoding RNA CASC11 promotes hepatocarcinogenesis and HCC progression through EIF4A3-mediated E2F1 activation. Clin. Transl. Med. 2020, 10, e220. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, C.; He, Z.; Chen, S.; Li, L.; Sun, C. LncRNA PSMB8-AS1 contributes to pancreatic cancer progression via modulating miR-382-3p/STAT1/PD-L1 axis. J. Exp. Clin. Cancer Res. 2020, 39, 179. [Google Scholar] [CrossRef]
- Zhu, F.; Niu, R.; Shao, X.; Shao, X. FGD5-AS1 promotes cisplatin resistance of human lung adenocarcinoma cell via the miR-142-5p/PD-L1 axis. Int. J. Mol. Med. 2021, 47, 523–532. [Google Scholar] [CrossRef]
- Haderk, F.; Schulz, R.; Iskar, M.; Cid, L.L.; Worst, T.; Willmund, K.V.; Schulz, A.; Warnken, U.; Seiler, J.; Benner, A. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci. Immunol. 2017, 2, eaah5509. [Google Scholar] [CrossRef] [Green Version]
- Gabrusiewicz, K.; Li, X.; Wei, J.; Hashimoto, Y.; Marisetty, A.L.; Ott, M.; Wang, F.; Hawke, D.; Yu, J.; Healy, L.M. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology 2018, 7, e1412909. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Kathuria, H.; Millien, G.; McNally, L.; Gower, A.C.; Tagne, J.-B.; Cao, Y.; Ramirez, M.I. NKX2-1-AS1 negatively regulates CD274/PD-L1, cell-cell interaction genes, and limits human lung carcinoma cell migration. Sci. Rep. 2018, 8, 14418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Du, Y.; Yang, X.; Mo, Y.; Fan, C.; Xiong, F.; Ren, D.; Ye, X.; Li, C.; Wang, Y. Circular RNAs function as ceRNAs to regulate and control human cancer progression. Mol. Cancer 2018, 17, 79. [Google Scholar] [CrossRef] [PubMed]
- Patop, I.L.; Kadener, S. circRNAs in Cancer. Curr. Opin. Genet. Dev. 2018, 48, 121–127. [Google Scholar] [CrossRef]
- He, R.; Liu, P.; Xie, X.; Zhou, Y.; Liao, Q.; Xiong, W.; Li, X.; Li, G.; Zeng, Z.; Tang, H. circGFRA1 and GFRA1 act as ceRNAs in triple negative breast cancer by regulating miR-34a. J. Exp. Clin. Cancer Res. 2017, 36, 145. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Mo, Y.; Gong, Z.; Yang, X.; Yang, M.; Zhang, S.; Xiong, F.; Xiang, B.; Zhou, M.; Liao, Q. Circular RNAs in human cancer. Mol. Cancer 2017, 16, 25. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.l.; Xu, L.l.; Wang, F. Hsa_circ_0020397 regulates colorectal cancer cell viability, apoptosis and invasion by promoting the expression of the miR-138 targets TERT and PD-L1. Cell Biol. Int. 2017, 41, 1056–1064. [Google Scholar] [CrossRef]
- Zhen, S.; Lu, J.; Chen, W.; Zhao, L.; Li, X. Synergistic antitumor effect on bladder cancer by rational combination of programmed cell death 1 blockade and CRISPR-Cas9-mediated long non-coding RNA urothelial carcinoma associated 1 knockout. Hum. Gene Ther. 2018, 29, 1352–1363. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in cancer immunotherapy: Clinical implications and future considerations. Hum. Vaccines Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Chen, L. A paradigm shift in cancer immunotherapy: From enhancement to normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Armand, P.; Engert, A.; Younes, A.; Fanale, M.; Santoro, A.; Zinzani, P.L.; Timmerman, J.M.; Collins, G.P.; Ramchandren, R.; Cohen, J.B. Nivolumab for relapsed/refractory classic Hodgkin lymphoma after failure of autologous hematopoietic cell transplantation: Extended follow-up of the multicohort single-arm phase II CheckMate 205 trial. J. Clin. Oncol. 2018, 36, 1428. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H., 3rd. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.X.; Ou, W.; Diede, S.J.; Whitman, E.D. Real-world experience with pembrolizumab in patients with advanced melanoma: A large retrospective observational study. Medicine 2019, 98, e16542. [Google Scholar] [CrossRef] [PubMed]
- Armand, P.; Shipp, M.A.; Ribrag, V.; Michot, J.-M.; Zinzani, P.L.; Kuruvilla, J.; Snyder, E.S.; Ricart, A.D.; Balakumaran, A.; Rose, S. Programmed death-1 blockade with pembrolizumab in patients with classical Hodgkin lymphoma after brentuximab vedotin failure. J. Clin. Oncol. 2016, 34, 3733. [Google Scholar] [CrossRef]
- Tahara, M.; Muro, K.; Hasegawa, Y.; Chung, H.C.; Lin, C.C.; Keam, B.; Takahashi, K.; Cheng, J.D.; Bang, Y.J. Pembrolizumab in Asia-Pacific patients with advanced head and neck squamous cell carcinoma: Analyses from KEYNOTE-012. Cancer Sci. 2018, 109, 771–776. [Google Scholar] [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S. PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [Green Version]
- McDermott, D.F.; Sosman, J.A.; Sznol, M.; Massard, C.; Gordon, M.S.; Hamid, O.; Powderly, J.D.; Infante, J.R.; Fassò, M.; Wang, Y.V. Atezolizumab, an anti-programmed death-ligand 1 antibody, in metastatic renal cell carcinoma: Long-term safety, clinical activity, and immune correlates from a phase Ia study. J. Clin. Oncol. 2016, 34, 833–842. [Google Scholar] [CrossRef]
- Bernard-Tessier, A.; Bonnet, C.; Lavaud, P.; Gizzi, M.; Loriot, Y.; Massard, C. Atézolizumab (Tecentriq®): Activité, indication et modalités d’utilisation dans les carcinomes urothéliaux localement avancés ou métastatiques. Bull. Du Cancer 2018, 105, 140–145. [Google Scholar] [CrossRef]
- Atezolizumab Extends Survival for Breast Cancer. Cancer Discov. 2017, 7, OF10. [CrossRef] [Green Version]
- D’Angelo, S.P.; Russell, J.; Lebbé, C.; Chmielowski, B.; Gambichler, T.; Grob, J.-J.; Kiecker, F.; Rabinowits, G.; Terheyden, P.; Zwiener, I. Efficacy and safety of first-line avelumab treatment in patients with stage IV metastatic Merkel cell carcinoma: A preplanned interim analysis of a clinical trial. JAMA Oncol. 2018, 4, e180077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, L.L.; Even, C.; Mesía, R.; Remenar, E.; Daste, A.; Delord, J.-P.; Krauss, J.; Saba, N.F.; Nabell, L.; Ready, N.E. Safety and efficacy of durvalumab with or without tremelimumab in patients with PD-L1–low/negative recurrent or metastatic HNSCC: The phase 2 CONDOR randomized clinical trial. JAMA Oncol. 2019, 5, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Khan, F.; Qari, H.A.; Upadhyay, T.K.; Alkhateeb, A.F.; Oves, M. Revolutionization in Cancer Therapeutics via Targeting Major Immune Checkpoints PD-1, PD-L1 and CTLA-4. Pharmaceuticals 2022, 15, 335. [Google Scholar] [CrossRef]
- Perkins, D.; Wang, Z.; Donovan, C.; He, H.; Mark, D.; Guan, G.; Wang, Y.; Walunas, T.; Bluestone, J.; Listman, J.; et al. Regulation of CTLA-4 expression during T cell activation. J. Immunol. 1996, 156, 4154–4159. [Google Scholar] [CrossRef] [PubMed]
- Alegre, M.-L.; Noel, P.J.; Eisfelder, B.J.; Chuang, E.; Clark, M.R.; Reiner, S.L.; Thompson, C.B. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. J. Immunol. 1996, 157, 4762–4770. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudd, C.E.; Taylor, A.; Schneider, H. CD28 and CTLA-4 coreceptor expression and signal transduction. Immunol. Rev. 2009, 229, 12–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [Green Version]
- Chambers, C.A.; Kuhns, M.S.; Egen, J.G.; Allison, J.P. CTLA-4-mediated inhibition in regulation of T cell responses: Mechanisms and manipulation in tumor immunotherapy. Annu. Rev. Immunol. 2001, 19, 565. [Google Scholar] [CrossRef] [Green Version]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef]
- Fife, B.T.; Bluestone, J.A. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol. Rev. 2008, 224, 166–182. [Google Scholar] [CrossRef] [PubMed]
- Stamper, C.C.; Zhang, Y.; Tobin, J.F.; Erbe, D.V.; Ikemizu, S.; Davis, S.J.; Stahl, M.L.; Seehra, J.; Somers, W.S.; Mosyak, L. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature 2001, 410, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Wing, K.; Onishi, Y.; Prieto-Martin, P.; Yamaguchi, T.; Miyara, M.; Fehervari, Z.; Nomura, T.; Sakaguchi, S. CTLA-4 control over Foxp3+ regulatory T cell function. Science 2008, 322, 271–275. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Thompson, C.B. At the bench: Preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J. Leukoc. Biol. 2013, 94, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Chikuma, S.; Abbas, A.K.; Bluestone, J.A. B7-independent inhibition of T cells by CTLA-4. J. Immunol. 2005, 175, 177–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, H.; Valk, E.; Leung, R.; Rudd, C.E. CTLA-4 activation of phosphatidylinositol 3-kinase (PI 3-K) and protein kinase B (PKB/AKT) sustains T-cell anergy without cell death. PLoS ONE 2008, 3, e3842. [Google Scholar] [CrossRef] [Green Version]
- Fraser, J.H.; Rincón, M.; McCoy, K.D.; Le Gros, G. CTLA4 ligation attenuates AP-1, NFAT and NF-κB activity in activated T cells. Eur. J. Immunol. 1999, 29, 838–844. [Google Scholar] [CrossRef]
- Bhandaru, M.; Rotte, A. Monoclonal antibodies for the treatment of melanoma: Present and future strategies. Hum. Monoclon. Antibodies 2019, 1904, 83–108. [Google Scholar]
- Chambers, C.A.; Cado, D.; Truong, T.; Allison, J.P. Thymocyte development is normal in CTLA-4-deficient mice. Proc. Natl. Acad. Sci. USA 1997, 94, 9296–9301. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar] [CrossRef]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, M.F.; Köhler, G.; Ecabert, B.; Mak, T.W.; Kopf, M. Cutting edge: Lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J. Immunol. 1999, 163, 1128–1131. [Google Scholar] [CrossRef] [PubMed]
- Friedline, R.H.; Brown, D.S.; Nguyen, H.; Kornfeld, H.; Lee, J.; Zhang, Y.; Appleby, M.; Der, S.D.; Kang, J.; Chambers, C.A. CD4+ regulatory T cells require CTLA-4 for the maintenance of systemic tolerance. J. Exp. Med. 2009, 206, 421–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, S.; Greenwald, R.; Izcue, A.; Robinson, N.; Mandelbrot, D.; Francisco, L.; Sharpe, A.H.; Powrie, F. Blockade of CTLA-4 on CD4+ CD25+ regulatory T cells abrogates their function in vivo. J. Immunol. 2006, 177, 4376–4383. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, O.S.; Zheng, Y.; Nakamura, K.; Attridge, K.; Manzotti, C.; Schmidt, E.M.; Baker, J.; Jeffery, L.E.; Kaur, S.; Briggs, Z. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science 2011, 332, 600–603. [Google Scholar] [CrossRef] [Green Version]
- Paterson, A.M.; Lovitch, S.B.; Sage, P.T.; Juneja, V.R.; Lee, Y.; Trombley, J.D.; Arancibia-Cárcamo, C.V.; Sobel, R.A.; Rudensky, A.Y.; Kuchroo, V.K. Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J. Exp. Med. 2015, 212, 1603–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simeone, E.; Ascierto, P.A. Immunomodulating antibodies in the treatment of metastatic melanoma: The experience with anti-CTLA-4, anti-CD137, and anti-PD1. J. Immunotoxicol. 2012, 9, 241–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [Green Version]
- Sansom, D. CD28, CTLA-4 and their ligands: Who does what and to whom? Immunology 2000, 101, 169. [Google Scholar] [CrossRef]
- Lipson, E.J.; Drake, C.G. Ipilimumab: An Anti-CTLA-4 Antibody for Metastatic MelanomaIpilimumab for Metastatic Melanoma. Clin. Cancer Res. 2011, 17, 6958–6962. [Google Scholar] [CrossRef] [Green Version]
- Klein, O.; Kee, D.; Markman, B.; Carlino, M.S.; Underhill, C.; Palmer, J.; Power, D.; Cebon, J.; Behren, A. Evaluation of TMB as a predictive biomarker in patients with solid cancers treated with anti-PD-1/CTLA-4 combination immunotherapy. Cancer Cell 2021, 39, 592–593. [Google Scholar] [CrossRef] [PubMed]
- Coillie, S.V.; Wiernicki, B.; Xu, J. Molecular and cellular functions of CTLA-4. Regul. Cancer Immune Checkp. 2020, 1248, 7–32. [Google Scholar]
- Robert, L.; Tsoi, J.; Wang, X.; Emerson, R.; Homet, B.; Chodon, T.; Mok, S.; Huang, R.R.; Cochran, A.J.; Comin-Anduix, B.; et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin. Cancer Res. 2014, 20, 2424–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, E.; Klinger, M.; Hou, Y.; Cummings, C.; Ribas, A.; Faham, M.; Fong, L. Improved survival with T cell clonotype stability after anti–CTLA-4 treatment in cancer patients. Sci. Transl. Med. 2014, 6, 238ra70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Liu, Z.; Wang, H.; Zhang, G. HLA-E Binding Peptide as a Potential Therapeutic Candidate for High-Risk Multiple Myeloma. Front. Oncol 2021, 11, 670673. [Google Scholar] [CrossRef]
- Robinson, J.; Halliwell, J.A.; McWilliam, H.; Lopez, R.; Marsh, S.G. IPD—The Immuno Polymorphism Database. Nucleic Acids Res. 2013, 41, D1234–D1240. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, M.; Wiley, D.C. Importance of peptide amino and carboxyl termini to the stability of MHC class I molecules. Science 1994, 265, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Lo Monaco, E.; Tremante, E.; Cerboni, C.; Melucci, E.; Sibilio, L.; Zingoni, A.; Nicotra, M.R.; Natali, P.G.; Giacomini, P. Human leukocyte antigen E contributes to protect tumor cells from lysis by natural killer cells. Neoplasia 2011, 13, 822–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrido, C.; Paco, L.; Romero, I.; Berruguilla, E.; Stefansky, J.; Collado, A.; Algarra, I.; Garrido, F.; Garcia-Lora, A.M. MHC class I molecules act as tumor suppressor genes regulating the cell cycle gene expression, invasion and intrinsic tumorigenicity of melanoma cells. Carcinogenesis 2012, 33, 687–693. [Google Scholar] [CrossRef] [Green Version]
- Mendez, R.; Aptsiauri, N.; Del Campo, A.; Maleno, I.; Cabrera, T.; Ruiz-Cabello, F.; Garrido, F.; Garcia-Lora, A. HLA and melanoma: Multiple alterations in HLA class I and II expression in human melanoma cell lines from ESTDAB cell bank. Cancer Immunol. Immunother. 2009, 58, 1507–1515. [Google Scholar] [CrossRef]
- de Kruijf, E.M.; Sajet, A.; van Nes, J.G.; Natanov, R.; Putter, H.; Smit, V.T.; Liefers, G.J.; van den Elsen, P.J.; van de Velde, C.J.; Kuppen, P.J. HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J. Immunol. 2010, 185, 7452–7459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, E.M.; Bianchini, M.; Von Euw, E.M.; Barrio, M.M.; Bravo, A.I.; Furman, D.; Domenichini, E.; Macagno, C.; Pinsky, V.; Zucchini, C. Human leukocyte antigen-E protein is overexpressed in primary human colorectal cancer. Int. J. Oncol. 2008, 32, 633–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeestraten, E.; Reimers, M.; Saadatmand, S.; Dekker, J.T.; Liefers, G.; Van Den Elsen, P.; Van De Velde, C.; Kuppen, P. Combined analysis of HLA class I, HLA-E and HLA-G predicts prognosis in colon cancer patients. Br. J. Cancer 2014, 110, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Eugène, J.; Jouand, N.; Ducoin, K.; Dansette, D.; Oger, R.; Deleine, C.; Leveque, E.; Meurette, G.; Podevin, J.; Matysiak, T. The inhibitory receptor CD94/NKG2A on CD8+ tumor-infiltrating lymphocytes in colorectal cancer: A promising new druggable immune checkpoint in the context of HLAE/β2m overexpression. Mod. Pathol. 2020, 33, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Braud, V.M.; Allan, D.S.; O’Callaghan, C.A.; Söderström, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- García, P.; Llano, M.; Heredia, A.B.d.; Willberg, C.B.; Caparrós, E.; Aparicio, P.; Braud, V.M.; López-Botet, M. Human T cell receptor-mediated recognition of HLA-E. Eur. J. Immunol. 2002, 32, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Pietra, G.; Romagnani, C.; Falco, M.; Vitale, M.; Castriconi, R.; Pende, D.; Millo, E.; Anfossi, S.; Biassoni, R.; Moretta, L. The analysis of the natural killer-like activity of human cytolytic T lymphocytes revealed HLA-E as a novel target for TCR α/β-mediated recognition. Eur. J. Immunol. 2001, 31, 3687–3693. [Google Scholar] [CrossRef]
- Moretta, L.; Romagnani, C.; Pietra, G.; Moretta, A.; Mingari, M.C. NK-CTLs, a novel HLA-E-restricted T-cell subset. Trends Immunol. 2003, 24, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Houchins, J.P.; Yabe, T.; McSherry, C.; Bach, F.H. DNA sequence analysis of NKG2, a family of related cDNA clones encoding type II integral membrane proteins on human natural killer cells. J. Exp. Med. 1991, 173, 1017–1020. [Google Scholar] [CrossRef] [Green Version]
- Pegram, H.J.; Andrews, D.M.; Smyth, M.J.; Darcy, P.K.; Kershaw, M.H. Activating and inhibitory receptors of natural killer cells. Immunol. Cell Biol. 2011, 89, 216–224. [Google Scholar] [CrossRef]
- Sheu, B.-C.; Chiou, S.-H.; Lin, H.-H.; Chow, S.-N.; Huang, S.-C.; Ho, H.-N.; Hsu, S.-M. Up-regulation of inhibitory natural killer receptors CD94/NKG2A with suppressed intracellular perforin expression of tumor-infiltrating CD8+ T lymphocytes in human cervical carcinoma. Cancer Res. 2005, 65, 2921–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanier, L.L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef]
- Petrie, E.J.; Clements, C.S.; Lin, J.; Sullivan, L.C.; Johnson, D.; Huyton, T.; Heroux, A.; Hoare, H.L.; Beddoe, T.; Reid, H.H. CD94-NKG2A recognition of human leukocyte antigen (HLA)-E bound to an HLA class I leader sequence. J. Exp. Med. 2008, 205, 725–735. [Google Scholar] [CrossRef] [Green Version]
- Marín, R.; Ruiz-Cabello, F.; Pedrinaci, S.; Méndez, R.; Jiménez, P.; Geraghty, D.E.; Garrido, F. Analysis of HLA-E expression in human tumors. Immunogenetics 2003, 54, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Kren, L.; Slaby, O.; Muckova, K.; Lzicarova, E.; Sova, M.; Vybihal, V.; Svoboda, T.; Fadrus, P.; Lakomy, R.; Vanhara, P. Expression of immune-modulatory molecules HLA-G and HLA-E by tumor cells in glioblastomas: An unexpected prognostic significance? Neuropathology 2011, 31, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Mandruzzato, S.; Callegaro, A.; Turcatel, G.; Francescato, S.; Montesco, M.C.; Chiarion-Sileni, V.; Mocellin, S.; Rossi, C.R.; Bicciato, S.; Wang, E. A gene expression signature associated with survival in metastatic melanoma. J. Transl. Med. 2006, 4, 50. [Google Scholar] [CrossRef] [Green Version]
- Silva, T.G.; Crispim, J.C.; Miranda, F.A.; Hassumi, M.K.; de Mello, J.M.; Simões, R.T.; Souto, F.; Soares, E.G.; Donadi, E.A.; Soares, C.P. Expression of the nonclassical HLA-G and HLA-E molecules in laryngeal lesions as biomarkers of tumor invasiveness. Histol. Histopathol. 2011, 26, 1487–1497. [Google Scholar] [PubMed]
- Yazdi, M.T.; van Riet, S.; van Schadewijk, A.; Fiocco, M.; van Hall, T.; Taube, C.; Hiemstra, P.S. The positive prognostic effect of stromal CD8+ tumor-infiltrating T cells is restrained by the expression of HLA-E in non-small cell lung carcinoma. Oncotarget 2016, 7, 3477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, E.; Poschke, I.; Villabona, L.; Carlson, J.W.; Lundqvist, A.; Kiessling, R.; Seliger, B.; Masucci, G.V. Non-classical HLA-class I expression in serous ovarian carcinoma: Correlation with the HLA-genotype, tumor infiltrating immune cells and prognosis. Oncoimmunology 2016, 5, e1052213. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.-Y.; Lv, Y.-G.; Wang, L.; Shi, S.-J.; Yang, F.; Zheng, G.-X.; Wen, W.-H.; Yang, A.-G. Predictive value of HLA-G and HLA-E in the prognosis of colorectal cancer patients. Cell. Immunol. 2015, 293, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Z.-J.; Ling, J.-Y.; Cai, Y.; Luo, W.-B.; He, Y.-J. Impact of HLA-E gene polymorphism on HLA-E expression in tumor cells and prognosis in patients with stage III colorectal cancer. Med. Oncol. 2013, 30, 482. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, S.; Beziat, V.; Dhedin, N.; Kuentz, M.; Vernant, J.; Debre, P.; Vieillard, V. HLA-E upregulation on IFN-γ-activated AML blasts impairs CD94/NKG2A-dependent NK cytolysis after haplo-mismatched hematopoietic SCT. Bone Marrow Transplant. 2009, 43, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Montfoort, N.; Borst, L.; Korrer, M.J.; Sluijter, M.; Marijt, K.A.; Santegoets, S.J.; van Ham, V.J.; Ehsan, I.; Charoentong, P.; André, P. NKG2A blockade potentiates CD8 T cell immunity induced by cancer vaccines. Cell 2018, 175, 1744–1755.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ClinicalTrials.gov. Combination Study of IPH2201 (Monalizumab) with Ibrutinib in Relapsed, Refractory or Previously Untreated CLL; NIH: Bethesda, MD, USA, 2019.
- ClinicalTrials.gov. Monalizumab and Trastuzumab in Metastatic HER2-pOSitive breAst Cancer: MIMOSA-Trial (MIMOSA); NIH: Bethesda, MD, USA, 2022.
- ClinicalTrials.gov. Study of Monalizumab and Cetuximab in Patients with Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck; NIH: Bethesda, MD, USA, 2021.
- van Hall, T.; Andre, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; van der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the novel immune checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef] [PubMed]
- Raulet, D.H.; Gasser, S.; Gowen, B.G.; Deng, W.; Jung, H. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 2013, 31, 413–441. [Google Scholar] [CrossRef] [PubMed]
- Spear, P.; Wu, M.R.; Sentman, M.L.; Sentman, C.L. NKG2D ligands as therapeutic targets. Cancer Immun. 2013, 13, 8. [Google Scholar] [PubMed]
- Cerboni, C.; Fionda, C.; Soriani, A.; Zingoni, A.; Doria, M.; Cippitelli, M.; Santoni, A. The DNA damage response: A common pathway in the regulation of NKG2D and DNAM-1 ligand expression in normal, infected, and cancer cells. Front. Immunol. 2014, 4, 508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamieson, A.M.; Diefenbach, A.; McMahon, C.W.; Xiong, N.; Carlyle, J.R.; Raulet, D.H. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity 2002, 17, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Wensveen, F.M.; Jelencic, V.; Polic, B. NKG2D: A Master Regulator of Immune Cell Responsiveness. Front. Immunol. 2018, 9, 441. [Google Scholar] [CrossRef]
- Lopez-Soto, A.; Gonzalez, S.; Galluzzi, L. Soluble NKG2D ligands limit the efficacy of immune checkpoint blockade. Oncoimmunology 2017, 6, e1346766. [Google Scholar] [CrossRef]
- Sagiv, A.; Burton, D.G.; Moshayev, Z.; Vadai, E.; Wensveen, F.; Ben-Dor, S.; Golani, O.; Polic, B.; Krizhanovsky, V. NKG2D ligands mediate immunosurveillance of senescent cells. Aging 2016, 8, 328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sagiv, A.; Biran, A.; Yon, M.; Simon, J.; Lowe, S.; Krizhanovsky, V. Granule exocytosis mediates immune surveillance of senescent cells. Oncogene 2013, 32, 1971–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Santo, J.P. Natural killer cell developmental pathways: A question of balance. Annu. Rev. Immunol. 2006, 24, 257–286. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef] [PubMed]
- Curio, S.; Jonsson, G.; Marinović, S. A summary of current NKG2D-based CAR clinical trials. Immunother. Adv. 2021, 1, ltab018. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; Dai, C.; Ge, X.; Tang, W.; Lin, Y.; Wang, Y.; Li, J. Prognostic significance and functional implication of immune activating receptor NKG2D in gastric cancer. Biochem. Biophys. Res. Commun. 2017, 487, 619–624. [Google Scholar] [CrossRef]
- Liu, X.; Sun, M.; Yu, S.; Liu, K.; Li, X.; Shi, H. Potential therapeutic strategy for gastric cancer peritoneal metastasis by NKG2D ligands-specific T cells. OncoTargets Ther. 2015, 8, 3095. [Google Scholar]
- Han, Y.; Xie, W.; Song, D.-G.; Powell, D.J. Control of triple-negative breast cancer using ex vivo self-enriched, costimulated NKG2D CAR T cells. J. Hematol. Oncol. 2018, 11, 92. [Google Scholar] [CrossRef]
- Siemaszko, J.; Marzec-Przyszlak, A.; Bogunia-Kubik, K. NKG2D Natural Killer Cell Receptor-A Short Description and Potential Clinical Applications. Cells 2021, 10, 1420. [Google Scholar] [CrossRef]
- Fernández-Sánchez, A.; Baragaño Raneros, A.; Carvajal Palao, R.; Sanz, A.B.; Ortiz, A.; Ortega, F.; Suárez-Álvarez, B.; López-Larrea, C. DNA demethylation and histone H3K9 acetylation determine the active transcription of the NKG2D gene in human CD8+ T and NK cells. Epigenetics 2013, 8, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Raneros, A.B.; Minguela, A.; Rodriguez, R.M.; Colado, E.; Bernal, T.; Anguita, E.; Mogorron, A.V.; Gil, A.C.; Vidal-Castiñeira, J.R.; Márquez-Kisinousky, L.; et al. Increasing TIMP3 expression by hypomethylating agents diminishes soluble MICA, MICB and ULBP2 shedding in acute myeloid leukemia, facilitating NK cell-mediated immune recognition. Oncotarget 2017, 8, 31959–31976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baragaño Raneros, A.; Martin-Palanco, V.; Fernandez, A.; Rodriguez, R.; Fraga, M.; Lopez-Larrea, C.; Suarez-Alvarez, B. Methylation of NKG2D ligands contributes to immune system evasion in acute myeloid leukemia. Genes Immun. 2015, 16, 71–82. [Google Scholar] [CrossRef]
- Leivas, A.; Valeri, A.; Cordoba, L.; Garcia-Ortiz, A.; Ortiz, A.; Sanchez-Vega, L.; Grana-Castro, O.; Fernandez, L.; Carreno-Tarragona, G.; Perez, M.; et al. NKG2D-CAR-transduced natural killer cells efficiently target multiple myeloma. Blood Cancer J. 2021, 11, 146. [Google Scholar] [CrossRef]
- Basher, F.; Jeng, E.K.; Wong, H.; Wu, J. Cooperative therapeutic anti-tumor effect of IL-15 agonist ALT-803 and co-targeting soluble NKG2D ligand sMIC. Oncotarget 2016, 7, 814. [Google Scholar] [CrossRef] [Green Version]
- Raeeszadeh-Sarmazdeh, M.; Do, L.D.; Hritz, B.G. Metalloproteinases and their inhibitors: Potential for the development of new therapeutics. Cells 2020, 9, 1313. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, M.B.; Domaica, C.I.; Zwirner, N.W. Leveraging NKG2D Ligands in Immuno-Oncology. Front. Immunol. 2021, 12, 713158. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Sampath, P.; Yan, X.; Thorne, S.H. Potential for enhanced therapeutic activity of biological cancer therapies with doxycycline combination. Gene 2013, 20, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-F.; Lee, C.-F.; Chern, Y. Chapter One—Adenosine Receptor Neurobiology: Overview. Int. Rev. Neurobiol. 2014, 119, 1–49. [Google Scholar]
- Ohta, A.; Sitkovsky, M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001, 414, 916–920. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Lo, Y.-C.; Powell, J.D. A2aR antagonists: Next generation checkpoint blockade for cancer immunotherapy. Comput. Struct. Biotechnol. J. 2015, 13, 265–272. [Google Scholar] [CrossRef] [Green Version]
- Olah, M.E.; Stiles, G.L. Adenosine receptor subtypes: Characterization and therapeutic regulation. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 581–606. [Google Scholar] [CrossRef] [PubMed]
- Pelleg, A.; Porter, R.S. The pharmacology of adenosine. Pharmacother. J. Hum. Pharmacol. Drug Ther. 1990, 10, 157–174. [Google Scholar]
- Robeva, A.S.; Woodard, R.L.; Jin, X.; Gao, Z.; Bhattacharya, S.; Taylor, H.E.; Rosin, D.L.; Linden, J. Molecular characterization of recombinant human adenosine receptors. Drug Dev. Res. 1996, 39, 243–252. [Google Scholar] [CrossRef]
- Muller-Haegele, S.; Muller, L.; Whiteside, T.L. Immunoregulatory activity of adenosine and its role in human cancer progression. Expert Rev. Clin. Immunol. 2014, 10, 897–914. [Google Scholar] [CrossRef]
- Sun, C.; Wang, B.; Hao, S. Adenosine-A2A Receptor Pathway in Cancer Immunotherapy. Front. Immunol. 2022, 13, 837230. [Google Scholar] [CrossRef] [PubMed]
- Slaats, J.; Wagena, E.; Smits, D.; Berends, A.A.; Peters, E.; Bakker, G.-J.; Erp, M.; Weigelin, B.; Adema, G.J.; Friedl, P. Adenosine A2a receptor antagonism restores additive cytotoxicity by cytotoxic T cells in metabolically perturbed tumors. Cancer Immunol. Res. 2022, 10, 1462–1474. [Google Scholar] [CrossRef]
- Young, A.; Ngiow, S.F.; Gao, Y.; Patch, A.-M.; Barkauskas, D.S.; Messaoudene, M.; Lin, G.; Coudert, J.D.; Stannard, K.A.; Zitvogel, L. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor MicroenvironmentAdenosine Impairs Proliferation of Terminal NK Cells. Cancer Res. 2018, 78, 1003–1016. [Google Scholar] [CrossRef] [Green Version]
- Halpin-Veszeleiova, K.; Hatfield, S.M. Oxygenation and A2AR blockade to eliminate hypoxia/HIF-1α-adenosinergic immunosuppressive axis and improve cancer immunotherapy. Curr. Opin. Pharmacol. 2020, 53, 84–90. [Google Scholar] [CrossRef]
- Ohta, A.; Sitkovsky, M. The adenosinergic immunomodulatory drugs. Curr. Opin. Pharmacol. 2009, 9, 501–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Apasov, S.; Koshiba, M.; Sitkovsky, M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood J. Am. Soc. Hematol. 1997, 90, 1600–1610. [Google Scholar]
- Decking, U.K.; Schlieper, G.; Kroll, K.; Schrader, J.r. Hypoxia-induced inhibition of adenosine kinase potentiates cardiac adenosine release. Circ. Res. 1997, 81, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandapathil, M.; Szczepanski, M.J.; Szajnik, M.; Ren, J.; Lenzner, D.E.; Jackson, E.K.; Gorelik, E.; Lang, S.; Johnson, J.T.; Whiteside, T.L. Increased ectonucleotidase expression and activity in regulatory T cells of patients with head and neck cancer. Clin. Cancer Res. 2009, 15, 6348–6357. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Vijayan, D.; Li, X.Y.; Robson, S.C.; Geetha, N.; Teng, M.W.L.; Smyth, M.J. The role of NK cells and CD39 in the immunological control of tumor metastases. Oncoimmunology 2019, 8, e1593809. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.W.; Hoshi, T.; Wu, Y.; Sun, X.; Enjyoji, K.; Cszimadia, E.; Sundberg, C.; Robson, S.C. Disordered purinergic signaling inhibits pathological angiogenesis in cd39/Entpd1-null mice. Am. J. Pathol. 2007, 171, 1395–1404. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Wu, Y.; Gao, W.; Enjyoji, K.; Csizmadia, E.; Müller, C.E.; Murakami, T.; Robson, S.C. CD39/ENTPD1 expression by CD4+ Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology 2010, 139, 1030–1040. [Google Scholar] [CrossRef] [Green Version]
- Roh, M.; Wainwright, D.A.; Wu, J.D.; Wan, Y.; Zhang, B. Targeting CD73 to augment cancer immunotherapy. Curr. Opin. Pharm. 2020, 53, 66–76. [Google Scholar] [CrossRef]
- Wennerberg, E.; Spada, S.; Rudqvist, N.P.; Lhuillier, C.; Gruber, S.; Chen, Q.; Zhang, F.; Zhou, X.K.; Gross, S.S.; Formenti, S.C.; et al. CD73 Blockade Promotes Dendritic Cell Infiltration of Irradiated Tumors and Tumor Rejection. Cancer Immunol. Res. 2020, 8, 465–478. [Google Scholar] [CrossRef]
- Jin, D.; Fan, J.; Wang, L.; Thompson, L.F.; Liu, A.; Daniel, B.J.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 on Tumor Cells Impairs Antitumor T-Cell Responses: A Novel Mechanism of Tumor-Induced Immune SuppressionTumor CD73 Impairs Antitumor T-Cell Responses. Cancer Res. 2010, 70, 2245–2255. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Divisekera, U.; McLaughlin, N.; Sharkey, J.; Pommey, S.; Denoyer, D.; Dwyer, K.M.; Smyth, M.J. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1547–1552. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, J.M.; Chen, J.F.; Schwarzschild, M.A.; Apasov, S.; Smith, P.T.; Caldwell, C.; Chen, P.; Figler, H.; Sullivan, G.; Fink, S. Gene dose effect reveals no Gs-coupled A2A adenosine receptor reserve in murine T-lymphocytes: Studies of cells from A2A-receptor-gene-deficient mice. Biochem. J. 2001, 354, 123–130. [Google Scholar] [CrossRef]
- Sitkovsky, M.V. Lessons from the A2A adenosine receptor antagonist–enabled tumor regression and survival in patients with treatment-refractory renal cell cancer. Cancer Discov. 2020, 10, 16–19. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.R.; Deng, W.W.; Liu, J.F.; Mao, L.; Yu, G.T.; Bu, L.L.; Kulkarni, A.B.; Zhang, W.F.; Sun, Z.J. Blockade of adenosine A2A receptor enhances CD8(+) T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol. Cancer 2017, 16, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenzen, S.; Siu, L.; Burris, H.; Le, D.; Hollebecque, A.; Steeghs, N.; Delord, J.; Hilton, J.; Barnhart, B.; Sega, E. Preliminary phase 1 profile of BMS-986179, an anti-CD73 antibody, in combination with nivolumab in patients with advanced solid tumors. Cancer Res. 2018, 78, CT180. [Google Scholar]
- Luke, J.; Merchan, J.; Harshman, L.; Marron, T.; Powderly, J.; Barve, M.; LoRusso, P.; Johnson, M.; Hotson, A.; Gittelman, R. Immunobiology and clinical activity of CPI-006, an anti-CD73 antibody with immunomodulating properties in a phase 1/1b trial in advanced cancers. J. Immunother. Cancer 2019, 37, 2505. [Google Scholar]
- Chiappori, A.; Williams, C.; Creelan, B.; Tanvetyanon, T.; Gray, J.; Haura, E.; Chen, D.; Thapa, R.; Beg, A.; Boyle, T. P1. 04-32 Phase I/II Study of the A2AR Antagonist NIR178 (PBF-509), an Oral Immunotherapy, in Patients (pts) with Advanced NSCLC. J. Thorac. Oncol. 2018, 13, S538. [Google Scholar] [CrossRef] [Green Version]
- Buisseret, L.; Rottey, S.; De Bono, J.S.; Migeotte, A.; Delafontaine, B.; Manickavasagar, T.; Martinoli, C.; Wald, N.; Rossetti, M.; Gangolli, E.A. Phase 1 trial of the adenosine A2A receptor antagonist inupadenant (EOS-850): Update on tolerability, and antitumor activity potentially associated with the expression of the A2A receptor within the tumor. J. Clin. Oncol. 2021, 39, 2562. [Google Scholar] [CrossRef]
- Berges, B.M.; Cambareri, C.; Takvorian, S.U.; Serpa, M.; Shulman, L.N.; Bekelman, J.E.; Rendle, K.A.; Argon, J.; Rosin, R. Leveraging a conversational agent to support adherence to oral anticancer agents: A usability study. J. Clin. Oncol. 2019, 37, 6534. [Google Scholar] [CrossRef]
- Fong, L.; Hotson, A.; Powderly, J.D.; Sznol, M.; Heist, R.S.; Choueiri, T.K.; George, S.; Hughes, B.G.; Hellmann, M.D.; Shepard, D.R. Adenosine 2A Receptor Blockade as an Immunotherapy for Treatment-Refractory Renal Cell CancerAdenosine 2A Receptor Blockade as a Cancer Immunotherapy. Cancer Discov. 2020, 10, 40–53. [Google Scholar] [CrossRef] [Green Version]
- Seitz, L.; Jin, L.; Leleti, M.; Ashok, D.; Jeffrey, J.; Rieger, A.; Tiessen, R.G.; Arold, G.; Tan, J.B.; Powers, J.P. Safety, tolerability, and pharmacology of AB928, a novel dual adenosine receptor antagonist, in a randomized, phase 1 study in healthy volunteers. Investig. New Drugs 2019, 37, 711–721. [Google Scholar] [CrossRef]
- Powderly, J.; Spira, A.; Gutierrez, R.; DiRenzo, D.; Udyavar, A.; Karakunnel, J.; Rieger, A.; Colabella, J.; Lai, D.; de Souza, P. Phase I evaluation of AB928, a novel dual adenosine receptor antagonist, combined with chemotherapy or AB122 (anti-PD-1) in patients (pts) with advanced malignancies. Ann. Oncol. 2019, 30, v493. [Google Scholar] [CrossRef]
- Willingham, S.B.; Hotson, A.N.; Miller, R.A. Targeting the A2AR in cancer; early lessons from the clinic. Curr. Opin Pharm. 2020, 53, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.; Bauer, T.; Patel, M.; Falchook, G.; Karlix, J.L.; Lim, E.; Mugundu, G.; Mitchell, P.D.; Pouliot, G.P.; Moorthy, G. Abstract CT026: Evidence of immune activation in the first-in-human Phase Ia dose escalation study of the adenosine 2a receptor antagonist, AZD4635, in patients with advanced solid tumors. Cancer Res. 2019, 79, CT026. [Google Scholar] [CrossRef]
- Powderly, J.D.; de Souza, P.L.; Gutierrez, R.; Horvath, L.; Seitz, L.; Ashok, D.; Park, A.; Walters, M.J.; Karakunnel, J.J.; Berry, W. AB928, a novel dual adenosine receptor antagonist, combined with chemotherapy or AB122 (anti-PD-1) in patients (pts) with advanced tumors: Preliminary results from ongoing phase I studies. J. Clin. Oncol. 2019, 37, 2604. [Google Scholar] [CrossRef]
- Houthuys, E.; Basilico, P.; Brouwer, M.; Deregnaucourt, T.; Detheux, M.; Driessens, G.; Gomes, B.; Leroy, X.; Marchante, J.; Marillier, R. EOS100850, a non-brain penetrant highly selective A2Areceptor antagonist, uniquely maintains high potency within the adenosine rich tumor microenvironment. Cancer Res. 2019, 79, 3261. [Google Scholar] [CrossRef]
- Buisseret, L.; Rottey, S.; de Bono, J.; Mossakowski, M.; Delafontaine, B.; Manickavasagar, T.; Kotecki, N.; Martinoli, C.; Schneider, M.; De Henau, O. Abstract CT152: First in human study with EOS100850, a novel potent A2A antagonist, shows excellent tolerance and clinical benefit in immune resistant advanced cancers. Cancer Res. 2020, 80, CT152. [Google Scholar] [CrossRef]
- Wang, Z.; Che, P.-L.; Du, J.; Ha, B.; Yarema, K.J. Static magnetic field exposure reproduces cellular effects of the Parkinson’s disease drug candidate ZM241385. PLoS ONE 2010, 5, e13883. [Google Scholar] [CrossRef] [PubMed]
- Poucher, S.; Keddie, J.; Singh, P.; Stoggall, S.; Caulkett, P.; Jones, G.; Collis, M. The in vitro pharmacology of ZM 241385, a potent, non-xanthine, A2a selective adenosine receptor antagonist. Br. J. Pharmacol. 1995, 115, 1096–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.F.; Xu, K.; Petzer, J.P.; Staal, R.; Xu, Y.H.; Beilstein, M.; Sonsalla, P.K.; Castagnoli, K.; Castagnoli, N., Jr.; Schwarzschild, M.A. Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson’s disease. J. Neurosci. 2001, 21, Rc143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zocchi, C.; Ongini, E.; Ferrara, S.; Baraldi, P.G.; Dionisotti, S. Binding of the radioligand [3H]-SCH 58261, a new non-xanthine A2A adenosine receptor antagonist, to rat striatal membranes. Br. J. Pharmacol. 1996, 117, 1381–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, J.; McIntosh, R.; Shen, X.; Lee, S.; Chanoit, G.; Criswell, H.; Zvara, D.A.; Xu, Z. Adenosine A2A and A2B receptors work in concert to induce a strong protection against reperfusion injury in rat hearts. J. Mol. Cell. Cardiol. 2009, 47, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Young, A.; Stannard, K.; Yong, M.; Teng, M.W.; Allard, B.; Stagg, J.; Smyth, M.J. Antimetastatic effects of blocking PD-1 and the adenosine A2A receptor. Cancer Res. 2014, 74, 3652–3658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beavis, P.A.; Divisekera, U.; Paget, C.; Chow, M.T.; John, L.B.; Devaud, C.; Dwyer, K.; Stagg, J.; Smyth, M.J.; Darcy, P.K. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 14711–14716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgson, R.A.; Bertorelli, R.; Varty, G.B.; Lachowicz, J.E.; Forlani, A.; Fredduzzi, S.; Cohen-Williams, M.E.; Higgins, G.A.; Impagnatiello, F.; Nicolussi, E. Characterization of the potent and highly selective A2A receptor antagonists preladenant and SCH 412348 [7-[2-[4-2, 4-difluorophenyl]-1-piperazinyl] ethyl]-2-(2-furanyl)-7H-pyrazolo [4, 3-e][1, 2, 4] triazolo [1, 5-c] pyrimidin-5-amine] in rodent models of movement disorders and depression. J. Pharmacol. Exp. Ther. 2009, 330, 294–303. [Google Scholar] [PubMed] [Green Version]
- Beavis, P.A.; Milenkovski, N.; Henderson, M.A.; John, L.B.; Allard, B.; Loi, S.; Kershaw, M.H.; Stagg, J.; Darcy, P.K. Adenosine receptor 2A blockade increases the efficacy of anti–PD-1 through enhanced antitumor T-cell responses. Cancer Immunol. Res. 2015, 3, 506–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cekic, C.; Linden, J. Adenosine A2A Receptors Intrinsically Regulate CD8+ T Cells in the Tumor MicroenvironmentAdenosine Maintains CD8+ T Cells in Solid Tumors. Cancer Res. 2014, 74, 7239–7249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, S.P.H. A-2B Adenosine Receptor. In xPharm: The Comprehensive Pharmacology Reference; Enna, S.J., Bylund, D.B., Eds.; Elsevier: New York, NY, USA, 2007; pp. 1–18. [Google Scholar] [CrossRef]
- Gessi, S.; Merighi, S.; Sacchetto, V.; Simioni, C.; Borea, P.A. Adenosine receptors and cancer. Biochim. Biophys. Acta BBA-Biomembr. 2011, 1808, 1400–1412. [Google Scholar] [CrossRef] [Green Version]
- Kazemi, M.H.; Raoofi Mohseni, S.; Hojjat-Farsangi, M.; Anvari, E.; Ghalamfarsa, G.; Mohammadi, H.; Jadidi-Niaragh, F. Adenosine and adenosine receptors in the immunopathogenesis and treatment of cancer. J. Cell. Physiol. 2018, 233, 2032–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredholm, B.B.; Irenius, E.; Kull, B.; Schulte, G. Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells. Biochem. Pharmacol. 2001, 61, 443–448. [Google Scholar] [CrossRef]
- Aherne, C.M.; Kewley, E.M.; Eltzschig, H.K. The resurgence of A2B adenosine receptor signaling. Biochim. Et Biophys. Acta BBA-Biomembr. 2011, 1808, 1329–1339. [Google Scholar] [CrossRef]
- Hajiahmadi, S.; Panjehpour, M.; Aghaei, M.; Shabani, M. Activation of A2b adenosine receptor regulates ovarian cancer cell growth: Involvement of Bax/Bcl-2 and caspase-3. Biochem. Cell Biol. 2015, 93, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Sinha, D.; Barkauskas, D.; Young, A.; Kalimutho, M.; Stannard, K.; Caramia, F.; Haibe-Kains, B.; Stagg, J.; Khanna, K.K.; et al. Adenosine 2B Receptor Expression on Cancer Cells Promotes Metastasis. Cancer Res. 2016, 76, 4372–4382. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Guo, X.; Zong, W.; Song, B.; Liu, G.; He, S. MicroRNA-128b suppresses tumor growth and promotes apoptosis by targeting A2bR in gastric cancer. Biochem. Biophys. Res. Commun. 2015, 467, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda, C.; Palomo, I.; Fuentes, E. Role of adenosine A2b receptor overexpression in tumor progression. Life Sci. 2016, 166, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.; Zhou, Y.; Chu, X.; Zheng, X.; Fei, D.; Lei, J.; Qi, H.; Dai, Y. Blockade of adenosine A2b receptor reduces tumor growth and migration in renal cell carcinoma. J. Cancer 2020, 11, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousavi, S.; Panjehpour, M.; Izadpanahi, M.H.; Aghaei, M. Expression of adenosine receptor subclasses in malignant and adjacent normal human prostate tissues. Prostate 2015, 75, 735–747. [Google Scholar] [CrossRef]
- Kasama, H.; Sakamoto, Y.; Kasamatsu, A.; Okamoto, A.; Koyama, T.; Minakawa, Y.; Ogawara, K.; Yokoe, H.; Shiiba, M.; Tanzawa, H.; et al. Adenosine A2b receptor promotes progression of human oral cancer. BMC Cancer 2015, 15, 563. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.G.; Jacobson, K.A. A2B Adenosine Receptor and Cancer. Int. J. Mol. Sci. 2019, 20, 5139. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. A Phase II Study to Test the Efficacy of AB928 (Dual Adenosine Receptor Antagonist) and AB122 (a PD1 Checkpoint Inhibitor) in Combination with Short Course Radiotherapy and Consolidation Chemotherapy for Rectal Cancer. (PANTHER); NIH: Bethesda, MD, USA, 2022.
- Walters, M.J.; Piovesan, D.; Tan, J.; DiRenzo, D.; Yin, F.; Miles, D.; Leleti, M.R.; Park, T.; Soriano, F.; Sharif, E. Combining adenosine receptor inhibition with AB928 and chemotherapy results in greater immune activation and tumor control. Cancer Res. 2018, 78, 5556. [Google Scholar] [CrossRef] [Green Version]
- Lu, Q.; Chen, X.; Wang, S.; Lu, Y.; Yang, C.; Jiang, G. Potential New Cancer Immunotherapy: Anti-CD47-SIRPα Antibodies. Onco Targets 2020, 13, 9323–9331. [Google Scholar] [CrossRef]
- Wu, Z.H.; Li, N.; Mei, X.F.; Chen, J.; Wang, X.Z.; Guo, T.T.; Chen, G.; Nie, L.; Chen, Y.; Jiang, M.Z.; et al. Preclinical characterization of the novel anti-SIRPα antibody BR105 that targets the myeloid immune checkpoint. J. Immunother. Cancer 2022, 10, e004054. [Google Scholar] [CrossRef] [PubMed]
- Maute, R.; Xu, J.; Weissman, I.L. CD47-SIRPα-targeted therapeutics: Status and prospects. Immunooncol. Technol. 2022, 13, 100070. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S. Molecular functions of SIRPα and its role in cancer. Biomed. Rep. 2018, 9, 3–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatherley, D.; Harlos, K.; Dunlop, D.C.; Stuart, D.I.; Barclay, A.N. The structure of the macrophage signal regulatory protein alpha (SIRPalpha) inhibitory receptor reveals a binding face reminiscent of that used by T cell receptors. J. Biol. Chem. 2007, 282, 14567–14575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatherley, D.; Graham, S.C.; Harlos, K.; Stuart, D.I.; Barclay, A.N. Structure of signal-regulatory protein alpha: A link to antigen receptor evolution. J. Biol. Chem. 2009, 284, 26613–26619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, E.J.; Frazier, W.A. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001, 11, 130–135. [Google Scholar] [CrossRef]
- Fenalti, G.; Villanueva, N.; Griffith, M.; Pagarigan, B.; Lakkaraju, S.K.; Huang, R.Y.; Ladygina, N.; Sharma, A.; Mikolon, D.; Abbasian, M.; et al. Structure of the human marker of self 5-transmembrane receptor CD47. Nat. Commun. 2021, 12, 5218. [Google Scholar] [CrossRef]
- Matozaki, T.; Murata, Y.; Okazawa, H.; Ohnishi, H. Functions and molecular mechanisms of the CD47-SIRPalpha signalling pathway. Trends Cell Biol. 2009, 19, 72–80. [Google Scholar] [CrossRef]
- Hayat, S.M.G.; Bianconi, V.; Pirro, M.; Jaafari, M.R.; Hatamipour, M.; Sahebkar, A. CD47: Role in the immune system and application to cancer therapy. Cell Oncol. 2020, 43, 19–30. [Google Scholar] [CrossRef]
- Weiskopf, K. Cancer immunotherapy targeting the CD47/SIRPα axis. Eur. J. Cancer 2017, 76, 100–109. [Google Scholar] [CrossRef]
- Qu, T.; Li, B.; Wang, Y. Targeting CD47/SIRPα as a therapeutic strategy, where we are and where we are headed. Biomark Res. 2022, 10, 20. [Google Scholar] [CrossRef]
- Oldenborg, P.-A.; Zheleznyak, A.; Fang, Y.-F.; Lagenaur, C.F.; Gresham, H.D.; Lindberg, F.P. Role of CD47 as a marker of self on red blood cells. Science 2000, 288, 2051–2054. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Yu, G.-T.; Deng, W.-W.; Mao, L.; Yang, L.-L.; Ma, S.-R.; Bu, L.-L.; Kulkarni, A.B.; Zhang, W.-F.; Zhang, L. Anti-CD47 treatment enhances anti-tumor T-cell immunity and improves immunosuppressive environment in head and neck squamous cell carcinoma. Oncoimmunology 2018, 7, e1397248. [Google Scholar] [CrossRef] [Green Version]
- Chao, M.P.; Tang, C.; Pachynski, R.K.; Chin, R.; Majeti, R.; Weissman, I.L. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood J. Am. Soc. Hematol. 2011, 118, 4890–4901. [Google Scholar] [CrossRef]
- Zhang, M.; Hutter, G.; Kahn, S.A.; Azad, T.D.; Gholamin, S.; Xu, C.Y.; Liu, J.; Achrol, A.S.; Richard, C.; Sommerkamp, P. Anti-CD47 treatment stimulates phagocytosis of glioblastoma by M1 and M2 polarized macrophages and promotes M1 polarized macrophages in vivo. PLoS ONE 2016, 11, e0153550. [Google Scholar] [CrossRef]
- Hu, T.; Liu, H.; Liang, Z.; Wang, F.; Zhou, C.; Zheng, X.; Zhang, Y.; Song, Y.; Hu, J.; He, X. Tumor-intrinsic CD47 signal regulates glycolysis and promotes colorectal cancer cell growth and metastasis. Theranostics 2020, 10, 4056. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Ren, Z.; Tseng, K.-F.; Liu, X.; Li, H.; Lu, C.; Cai, Y.; Minna, J.D.; Fu, Y.-X. Dual targeting of CTLA-4 and CD47 on Treg cells promotes immunity against solid tumors. Sci. Transl. Med. 2021, 13, eabg8693. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Qian, P.; Wang, F.; Yu, H.; Guo, Y. Targeting CD47 enhances the efficacy of anti-PD-1 and CTLA-4 in an esophageal squamous cell cancer preclinical model. Oncol. Res. 2017, 25, 1579. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F. TIM-3, a promising target for cancer immunotherapy. OncoTargets Ther. 2018, 11, 7005–7009. [Google Scholar] [CrossRef] [PubMed]
- Acharya, N.; Sabatos-Peyton, C.; Anderson, A.C. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer 2020, 8, e000911. [Google Scholar] [CrossRef] [PubMed]
- Kandel, S.; Adhikary, P.; Li, G.; Cheng, K. The TIM3/Gal9 signaling pathway: An emerging target for cancer immunotherapy. Cancer Lett. 2021, 510, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Kashio, Y.; Nakamura, K.; Abedin, M.J.; Seki, M.; Nishi, N.; Yoshida, N.; Nakamura, T.; Hirashima, M. Galectin-9 induces apoptosis through the calcium-calpain-caspase-1 pathway. J. Immunol. 2003, 170, 3631–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Sun, L.; Li, C.-F.; Wang, Y.-H.; Yao, J.; Li, H.; Yan, M.; Chang, W.-C.; Hsu, J.-M.; Cha, J.-H. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 2021, 12, 832. [Google Scholar] [CrossRef]
- Nagahara, K.; Arikawa, T.; Oomizu, S.; Kontani, K.; Nobumoto, A.; Tateno, H.; Watanabe, K.; Niki, T.; Katoh, S.; Miyake, M. Galectin-9 increases Tim-3+ dendritic cells and CD8+ T cells and enhances antitumor immunity via galectin-9-Tim-3 interactions. J. Immunol. 2008, 181, 7660–7669. [Google Scholar] [CrossRef] [Green Version]
- Lv, K.; Zhang, Y.; Zhang, M.; Zhong, M.; Suo, Q. Galectin-9 promotes TGF-β1-dependent induction of regulatory T cells via the TGF-β/Smad signaling pathway. Mol. Med. Rep. 2013, 7, 205–210. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chen, Y.; Liu, X.; Zhang, J.; He, X.; Teng, G.; Yu, D. Tim3/Gal9 interactions between T cells and monocytes result in an immunosuppressive feedback loop that inhibits Th1 responses in osteosarcoma patients. Int. Immunopharmacol. 2017, 44, 153–159. [Google Scholar] [CrossRef]
- Gleason, M.K.; Lenvik, T.R.; McCullar, V.; Felices, M.; O’Brien, M.S.; Cooley, S.A.; Verneris, M.R.; Cichocki, F.; Holman, C.J.; Panoskaltsis-Mortari, A. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood J. Am. Soc. Hematol. 2012, 119, 3064–3072. [Google Scholar]
- Chiba, S.; Baghdadi, M.; Akiba, H.; Yoshiyama, H.; Kinoshita, I.; Dosaka-Akita, H.; Fujioka, Y.; Ohba, Y.; Gorman, J.V.; Colgan, J.D. Tumor-infiltrating DCs suppress nucleic acid–mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 2012, 13, 832–842. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, M.; Akiba, H.; Takeda, K.; Kojima, Y.; Hashiguchi, M.; Azuma, M.; Yagita, H.; Okumura, K. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood J. Am. Soc. Hematol. 2009, 113, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Guan, R.; Yang, H.; Zhou, Y.; Hong, W.; Ma, L.; Zhao, G.; Yu, M. Assessment of the expression of the immune checkpoint molecules PD-1, CTLA4, TIM-3 and LAG-3 across different cancers in relation to treatment response, tumor-infiltrating immune cells and survival. Int. J. Cancer 2020, 147, 423–439. [Google Scholar] [CrossRef]
- Klein, C.; Schaefer, W.; Regula, J.T.; Dumontet, C.; Brinkmann, U.; Bacac, M.; Umaña, P. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods 2019, 154, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Gelderblom, H.; Mach, N.; Doi, T.; Tai, D.; Forde, P.M.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.C.; Hodi, F.S.; et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti-TIM-3 Antibody, Alone and in Combination with Spartalizumab, an Anti-PD-1 Antibody, in Advanced Solid Tumors. Clin. Cancer Res. 2021, 27, 3620–3629. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.L.; Wang, Y.T.; Fu, W.J.; Lu, N.; Liu, Z.S. The promising immune checkpoint LAG-3 in cancer immunotherapy: From basic research to clinical application. Front. Immunol. 2022, 13, 956090. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demeure, C.E.; Wolfers, J.; Martin-Garcia, N.; Gaulard, P.; Triebel, F. T Lymphocytes infiltrating various tumour types express the MHC class II ligand lymphocyte activation gene-3 (LAG-3): Role of LAG-3/MHC class II interactions in cell-cell contacts. Eur. J. Cancer 2001, 37, 1709–1718. [Google Scholar] [CrossRef]
- Durham, N.M.; Nirschl, C.J.; Jackson, C.M.; Elias, J.; Kochel, C.M.; Anders, R.A.; Drake, C.G. Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE 2014, 9, e109080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña, J.; Jones, N.G.; Bousheri, S.; Bangsberg, D.R.; Cao, H. Lymphocyte activation gene-3 expression defines a discrete subset of HIV-specific CD8+ T cells that is associated with lower viral load. AIDS Res. Hum. Retrovir. 2014, 30, 535–541. [Google Scholar] [CrossRef] [Green Version]
- Gagliani, N.; Magnani, C.F.; Huber, S.; Gianolini, M.E.; Pala, M.; Licona-Limon, P.; Guo, B.; Herbert, D.R.; Bulfone, A.; Trentini, F.; et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat. Med. 2013, 19, 739–746. [Google Scholar] [CrossRef]
- Huang, C.T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Andreae, S.; Buisson, S.; Triebel, F. MHC class II signal transduction in human dendritic cells induced by a natural ligand, the LAG-3 protein (CD223). Blood 2003, 102, 2130–2137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workman, C.J.; Wang, Y.; El Kasmi, K.C.; Pardoll, D.M.; Murray, P.J.; Drake, C.G.; Vignali, D.A. LAG-3 regulates plasmacytoid dendritic cell homeostasis. J. Immunol. 2009, 182, 1885–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kisielow, M.; Kisielow, J.; Capoferri-Sollami, G.; Karjalainen, K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur. J. Immunol. 2005, 35, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; Mastrangeli, R.; Prigent, P.; Bruniquel, D.; Donini, S.; El-Tayar, N.; Maigret, B.; Dréano, M.; Triebel, F. Characterization of the major histocompatibility complex class II binding site on LAG-3 protein. Proc. Natl. Acad. Sci. USA 1997, 94, 5744–5749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, R.; Zhang, M.; Hu, Q.; Zheng, S.; Soh, A.; Zheng, Y.; Yuan, H. Galectin-3 as a novel biomarker for disease diagnosis and a target for therapy (Review). Int. J. Mol. Med. 2018, 41, 599–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouo, T.; Huang, L.; Pucsek, A.B.; Cao, M.; Solt, S.; Armstrong, T.; Jaffee, E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol. Res. 2015, 3, 412–423. [Google Scholar] [CrossRef] [Green Version]
- Huard, B.; Prigent, P.; Tournier, M.; Bruniquel, D.; Triebel, F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur. J. Immunol. 1995, 25, 2718–2721. [Google Scholar] [CrossRef] [PubMed]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.M.; Shimizu, K.; Maeda, T.K.; Ikubo, J.; Yoshikawa, H.; Maenaka, K.; Ishimaru, N.; Kosako, H.; et al. Binding of LAG-3 to stable peptide-MHC class II limits T cell function and suppresses autoimmunity and anti-cancer immunity. Immunity 2022, 55, 912–924.e918. [Google Scholar] [CrossRef]
- Hemon, P.; Jean-Louis, F.; Ramgolam, K.; Brignone, C.; Viguier, M.; Bachelez, H.; Triebel, F.; Charron, D.; Aoudjit, F.; Al-Daccak, R.; et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J. Immunol. 2011, 186, 5173–5183. [Google Scholar] [CrossRef] [Green Version]
- Liang, B.; Workman, C.; Lee, J.; Chew, C.; Dale, B.M.; Colonna, L.; Flores, M.; Li, N.; Schweighoffer, E.; Greenberg, S.; et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J. Immunol. 2008, 180, 5916–5926. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.; Yi, Z.; Wang, L.; Li, Z.; Niu, B.; Ren, G. The co-expression characteristics of LAG3 and PD-1 on the T cells of patients with breast cancer reveal a new therapeutic strategy. Int. Immunopharmacol. 2020, 78, 106113. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Tang, L.; Zhang, G.; Wei, H.; Cui, Y.; Guo, L.; Gou, Z.; Chen, X.; Jiang, D.; Zhu, Y.; et al. Characterization of a novel C-type lectin-like gene, LSECtin: Demonstration of carbohydrate binding and expression in sinusoidal endothelial cells of liver and lymph node. J. Biol. Chem. 2004, 279, 18748–18758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, F.; Liu, J.; Liu, D.; Liu, B.; Wang, M.; Hu, Z.; Du, X.; Tang, L.; He, F. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. 2014, 74, 3418–3428. [Google Scholar] [CrossRef] [Green Version]
- Workman, C.J.; Cauley, L.S.; Kim, I.J.; Blackman, M.A.; Woodland, D.L.; Vignali, D.A. Lymphocyte activation gene-3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J. Immunol. 2004, 172, 5450–5455. [Google Scholar] [CrossRef] [Green Version]
- Maçon-Lemaître, L.; Triebel, F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology 2005, 115, 170–178. [Google Scholar] [CrossRef]
- Previte, D.M.; Martins, C.P.; O’Connor, E.C.; Marre, M.L.; Coudriet, G.M.; Beck, N.W.; Menk, A.V.; Wright, R.H.; Tse, H.M.; Delgoffe, G.M.; et al. Lymphocyte Activation Gene-3 Maintains Mitochondrial and Metabolic Quiescence in Naive CD4. Cell Rep. 2019, 27, 129–141.e4. [Google Scholar] [CrossRef] [Green Version]
- Camisaschi, C.; Casati, C.; Rini, F.; Perego, M.; De Filippo, A.; Triebel, F.; Parmiani, G.; Belli, F.; Rivoltini, L.; Castelli, C. LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J. Immunol. 2010, 184, 6545–6551. [Google Scholar] [CrossRef] [Green Version]
- Graydon, C.G.; Mohideen, S.; Fowke, K.R. LAG3’s Enigmatic Mechanism of Action. Front. Immunol. 2020, 11, 615317. [Google Scholar] [CrossRef]
- Li, N.; Wang, Y.; Forbes, K.; Vignali, K.M.; Heale, B.S.; Saftig, P.; Hartmann, D.; Black, R.A.; Rossi, J.J.; Blobel, C.P.; et al. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 2007, 26, 494–504. [Google Scholar] [CrossRef]
- Zhai, W.; Zhou, X.; Wang, H.; Li, W.; Chen, G.; Sui, X.; Li, G.; Qi, Y.; Gao, Y. A novel cyclic peptide targeting LAG-3 for cancer immunotherapy by activating antigen-specific CD8. Acta Pharm. Sin. B 2020, 10, 1047–1060. [Google Scholar] [CrossRef]
- Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; González-Rodríguez, A.P.; Payer, Á.; González-García, E.; López-Soto, A.; Gonzalez, S. LAG-3 Blockade with Relatlimab (BMS-986016) Restores Anti-Leukemic Responses in Chronic Lymphocytic Leukemia. Cancers 2021, 13, 2112. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wei, W.; Zhao, Q. B7-H3, a checkpoint molecule, as a target for cancer immunotherapy. Int. J. Biol. Sci. 2020, 16, 1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapoval, A.I.; Ni, J.; Lau, J.S.; Wilcox, R.A.; Flies, D.B.; Liu, D.; Dong, H.; Sica, G.L.; Zhu, G.; Tamada, K. B7-H3: A costimulatory molecule for T cell activation and IFN-γ production. Nat. Immunol. 2001, 2, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Chen, L.; Wang, F.; Zhu, D.; Ge, X.; Hua, D.; Sun, J. Cancer cell-expressed B7-H3 regulates the differentiation of tumor-associated macrophages in human colorectal carcinoma. Oncol. Lett. 2017, 14, 6177–6183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Ji, J.; Zhang, G.; Fang, C.; Jiang, F.; Ma, S.; Hou, J. Expression and significance of B7-H3 and Tie-2 in the tumor vasculature of clear cell renal carcinoma. OncoTargets Ther. 2017, 10, 5417. [Google Scholar] [CrossRef] [Green Version]
- Yonesaka, K.; Haratani, K.; Takamura, S.; Sakai, H.; Kato, R.; Takegawa, N.; Takahama, T.; Tanaka, K.; Hayashi, H.; Takeda, M. B7-H3 Negatively Modulates CTL-Mediated Cancer ImmunityB7-H3 Negatively Modulates Cancer Immunity. Clin. Cancer Res. 2018, 24, 2653–2664. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Li, D.-C.; Zhu, X.-G.; Gan, W.-J.; Li, Z.; Xiong, F.; Zhang, Z.-X.; Zhang, G.-B.; Zhang, X.-G.; Zhao, H. B7-H3 overexpression in pancreatic cancer promotes tumor progression. Int. J. Mol. Med. 2013, 31, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Suh, W.-K.; Gajewska, B.U.; Okada, H.; Gronski, M.A.; Bertram, E.M.; Dawicki, W.; Duncan, G.S.; Bukczynski, J.; Plyte, S.; Elia, A. The B7 family member B7-H3 preferentially down-regulates T helper type 1–mediated immune responses. Nat. Immunol. 2003, 4, 899–906. [Google Scholar] [CrossRef]
- Chen, J.-T.; Chen, C.-H.; Ku, K.-L.; Hsiao, M.; Chiang, C.-P.; Hsu, T.-L.; Chen, M.-H.; Wong, C.-H. Glycoprotein B7-H3 overexpression and aberrant glycosylation in oral cancer and immune response. Proc. Natl. Acad. Sci. USA 2015, 112, 13057–13062. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Zhang, P.; Li, J.; Zhao, J.; Liu, C.; Yang, F.; Yang, D.; Gao, A.; Lin, W.; Ma, X. B7-H3 in combination with regulatory T cell is associated with tumor progression in primary human non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 13987. [Google Scholar] [PubMed]
- Li, M.; Zhang, G.; Zhang, X.; Lv, G.; Wei, X.; Yuan, H.; Hou, J. Overexpression of B7-H3 in CD14+ monocytes is associated with renal cell carcinoma progression. Med. Oncol. 2014, 31, 349. [Google Scholar] [CrossRef]
- Bin, Z.; Guangbo, Z.; Yan, G.; Huan, Z.; Desheng, L.; Xueguang, Z. Overexpression of B7-H3 in CD133+ colorectal cancer cells is associated with cancer progression and survival in human patients. J. Surg. Res. 2014, 188, 396–403. [Google Scholar] [CrossRef]
- Liu, C.; Liu, J.; Wang, J.; Liu, Y.; Zhang, F.; Lin, W.; Gao, A.; Sun, M.; Wang, Y.; Sun, Y. B7-H3 expression in ductal and lobular breast cancer and its association with IL-10. Mol. Med. Rep. 2013, 7, 134–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, D.; Alderson, R.F.; Chen, F.Z.; Huang, L.; Zhang, W.; Gorlatov, S.; Burke, S.; Ciccarone, V.; Li, H.; Yang, Y. Development of an Fc-Enhanced Anti–B7-H3 Monoclonal Antibody with Potent Antitumor ActivityDevelopment of Fc-Enhanced Anti–B7-H3 Monoclonal Antibody. Clin. Cancer Res. 2012, 18, 3834–3845. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Ma, P.; Zhao, C.; Xue, X.; Han, H.; Liu, C.; Tao, H.; Xiu, W.; Cai, J.; Zhang, M. B7-H3 as a promising target for cytotoxicity T cell in human cancer therapy. Oncotarget 2016, 7, 29480. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Peyraud, F.; Italiano, A. Combined PARP inhibition and immune checkpoint therapy in solid tumors. Cancers 2020, 12, 1502. [Google Scholar] [CrossRef]
- De Vos, M.; Schreiber, V.; Dantzer, F. The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem. Pharmacol. 2012, 84, 137–146. [Google Scholar] [CrossRef]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair 2018, 71, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Yu, X. Functions of PARylation in DNA damage repair pathways. Genom. Proteom. Bioinform. 2016, 14, 131–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langelier, M.-F.; Eisemann, T.; Riccio, A.A.; Pascal, J.M. PARP family enzymes: Regulation and catalysis of the poly (ADP-ribose) posttranslational modification. Curr. Opin. Struct. Biol. 2018, 53, 187–198. [Google Scholar] [CrossRef]
- Wu, Z.; Cui, P.; Tao, H.; Zhang, S.; Ma, J.; Liu, Z.; Wang, J.; Qian, Y.; Chen, S.; Huang, Z. The synergistic effect of PARP inhibitors and immune checkpoint inhibitors. Clin. Med. Insights Oncol. 2021, 15, 1179554921996288. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung CancerDDR Inhibition Enhances Antitumor Immunity in SCLC. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, C.; Xu, Y.; Ye, W.; Xie, Q.; Gao, H.; Xu, B.; Zhang, D.; Jiang, J. Expression of PD-L1 in ovarian cancer and its synergistic antitumor effect with PARP inhibitor. Gynecol. Oncol. 2020, 157, 222–233. [Google Scholar] [CrossRef]
- Huang, J.; Wang, L.; Cong, Z.; Amoozgar, Z.; Kiner, E.; Xing, D.; Orsulic, S.; Matulonis, U.; Goldberg, M.S. The PARP1 inhibitor BMN 673 exhibits immunoregulatory effects in a Brca1−/− murine model of ovarian cancer. Biochem. Biophys. Res. Commun. 2015, 463, 551–556. [Google Scholar] [CrossRef]
- Aurelius, J.; Martner, A.; Riise, R.E.; Romero, A.I.; Palmqvist, L.; Brune, M.; Hellstrand, K.; Thorén, F.B. Chronic myeloid leukemic cells trigger poly (ADP-ribose) polymerase-dependent inactivation and cell death in lymphocytes. J. Leukoc. Biol. 2013, 93, 155–160. [Google Scholar] [CrossRef]
- Fenerty, K.E.; Padget, M.; Wolfson, B.; Gameiro, S.R.; Su, Z.; Lee, J.H.; Rabizadeh, S.; Soon-Shiong, P.; Hodge, J.W. Immunotherapy utilizing the combination of natural killer–and antibody dependent cellular cytotoxicity (ADCC)–mediating agents with poly (ADP-ribose) polymerase (PARP) inhibition. J. ImmunoTherapy Cancer 2018, 6, 133. [Google Scholar] [CrossRef]
- Kaye, S.B.; Lubinski, J.; Matulonis, U.; Ang, J.E.; Gourley, C.; Karlan, B.Y.; Amnon, A.; Bell-McGuinn, K.M.; Chen, L.-M.; Friedlander, M. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly (ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J. Clin. Oncol. 2012, 30, 372–379. [Google Scholar] [CrossRef]
- Lin, K.Y.; Kraus, W.L. PARP inhibitors for cancer therapy. Cell 2017, 169, 183. [Google Scholar] [CrossRef] [PubMed]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Tinker, A.V.; Oaknin, A.; Shapira-Frommer, R.; McNeish, I.A.; Swisher, E.M.; Ray-Coquard, I.; Bell-McGuinn, K.; Coleman, R.L.; O’Malley, D.M. Antitumor activity and safety of the PARP inhibitor rucaparib in patients with high-grade ovarian carcinoma and a germline or somatic BRCA1 or BRCA2 mutation: Integrated analysis of data from Study 10 and ARIEL2. Gynecol. Oncol. 2017, 147, 267–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAnessPARP1 Inhibitors Trigger Antitumor Immunity. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, K.; Xiao, Y.; Feng, B.; Mikule, K.; Ma, X.; Feng, N.; Vellano, C.P.; Federico, L.; Marszalek, J.R. Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci. Rep. 2019, 9, 1853. [Google Scholar] [CrossRef] [Green Version]
- Vikas, P.; Borcherding, N.; Chennamadhavuni, A.; Garje, R. Therapeutic potential of combining PARP inhibitor and immunotherapy in solid tumors. Front. Oncol. 2020, 10, 570. [Google Scholar] [CrossRef]
- Chauvin, J.M.; Zarour, H.M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef]
- Boles, K.S.; Vermi, W.; Facchetti, F.; Fuchs, A.; Wilson, T.J.; Diacovo, T.G.; Cella, M.; Colonna, M. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur. J. Immunol 2009, 39, 695–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Levin, S.D.; Taft, D.W.; Brandt, C.S.; Bucher, C.; Howard, E.D.; Chadwick, E.M.; Johnston, J.; Hammond, A.; Bontadelli, K.; Ardourel, D.; et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur. J. Immunol. 2011, 41, 902–915. [Google Scholar] [CrossRef]
- Bottino, C.; Castriconi, R.; Pende, D.; Rivera, P.; Nanni, M.; Carnemolla, B.; Cantoni, C.; Grassi, J.; Marcenaro, S.; Reymond, N.; et al. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J. Exp. Med. 2003, 198, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Seth, S.; Maier, M.K.; Qiu, Q.; Ravens, I.; Kremmer, E.; Förster, R.; Bernhardt, G. The murine pan T cell marker CD96 is an adhesion receptor for CD155 and nectin-1. Biochem. Biophys. Res. Commun. 2007, 364, 959–965. [Google Scholar] [CrossRef]
- Zhu, Y.; Paniccia, A.; Schulick, A.C.; Chen, W.; Koenig, M.R.; Byers, J.T.; Yao, S.; Bevers, S.; Edil, B.H. Identification of CD112R as a novel checkpoint for human T cells. J. Exp. Med. 2016, 213, 167–176. [Google Scholar] [CrossRef]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Zhang, H.; Han, F.; Chen, X.; Lin, R.; Wang, W.; Qiu, H.; Zhuang, Z.; Liao, Q.; Zhang, W.; et al. CD155T/TIGIT Signaling Regulates CD8. Cancer Res. 2017, 77, 6375–6388. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Y.; Das, I.; Lepletier, A.; Addala, V.; Bald, T.; Stannard, K.; Barkauskas, D.; Liu, J.; Aguilera, A.R.; Takeda, K.; et al. CD155 loss enhances tumor suppression via combined host and tumor-intrinsic mechanisms. J. Clin. Investig. 2018, 128, 2613–2625. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef]
- Muller, S.; Victoria Lai, W.; Adusumilli, P.S.; Desmeules, P.; Frosina, D.; Jungbluth, A.; Ni, A.; Eguchi, T.; Travis, W.D.; Ladanyi, M.; et al. V-domain Ig-containing suppressor of T-cell activation (VISTA), a potentially targetable immune checkpoint molecule, is highly expressed in epithelioid malignant pleural mesothelioma. Mod. Pathol. 2020, 33, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, S.R.; Knecht, M.; Mollaee, M.; Zhong, Z.; Erkes, D.A.; McCue, P.A.; Chervoneva, I.; Berger, A.C.; Lo, J.A.; Fisher, D.E.; et al. FOXD3 Regulates VISTA Expression in Melanoma. Cell Rep. 2020, 30, 510–524.e516. [Google Scholar] [CrossRef]
- Villarroel-Espindola, F.; Yu, X.; Datar, I.; Mani, N.; Sanmamed, M.; Velcheti, V.; Syrigos, K.; Toki, M.; Zhao, H.; Chen, L.; et al. Spatially Resolved and Quantitative Analysis of VISTA/PD-1H as a Novel Immunotherapy Target in Human Non-Small Cell Lung Cancer. Clin. Cancer Res. 2018, 24, 1562–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Zhang, J.; Shi, Z.; Liu, W.; Hu, X.; Qie, C.; Chen, W.; Wang, Y.; Wang, L.; Jiang, J.; et al. The Expression Pattern and Clinical Significance of the Immune Checkpoint Regulator VISTA in Human Breast Cancer. Front. Immunol. 2020, 11, 563044. [Google Scholar] [CrossRef]
- Hong, S.; Yuan, Q.; Xia, H.; Zhu, G.; Feng, Y.; Wang, Q.; Zhang, Z.; He, W.; Lu, J.; Dong, C.; et al. Analysis of VISTA expression and function in renal cell carcinoma highlights VISTA as a potential target for immunotherapy. Protein Cell 2019, 10, 840–845. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Huang, J.; Qiao, Q.; Zang, W.; Hong, S.; Tan, H.; Dong, C.; Yang, Z.; Ni, L. Expression of the inhibitory B7 family molecule VISTA in human colorectal carcinoma tumors. Cancer Immunol. Immunother. 2018, 67, 1685–1694. [Google Scholar] [CrossRef]
- Mulati, K.; Hamanishi, J.; Matsumura, N.; Chamoto, K.; Mise, N.; Abiko, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Murakami, R.; et al. VISTA expressed in tumour cells regulates T cell function. Br. J. Cancer 2019, 120, 115–127. [Google Scholar] [CrossRef] [Green Version]
- Mehta, N.; Maddineni, S.; Mathews, I.I.; Andres Parra Sperberg, R.; Huang, P.S.; Cochran, J.R. Structure and Functional Binding Epitope of V-domain Ig Suppressor of T Cell Activation. Cell Rep. 2019, 28, 2509–2516.e2505. [Google Scholar] [CrossRef] [PubMed]
- Tagliamento, M.; Agostinetto, E.; Borea, R.; Brandão, M.; Poggio, F.; Addeo, A.; Lambertini, M. VISTA: A Promising Target for Cancer Immunotherapy? Immunotargets 2021, 10, 185–200. [Google Scholar] [CrossRef] [PubMed]
- Corda, D.; Di Girolamo, M. Functional aspects of protein mono-ADP-ribosylation. EMBO J. 2003, 22, 1953–1958. [Google Scholar] [CrossRef]
- Wennerberg, E.; Mukherjee, S.; Sainz, R.M.; Stiles, B.M. The ART of tumor immune escape. OncoImmunology 2022, 11, 2076310. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, E.; Mukherjee, S.; Spada, S.; Hung, C.; Agrusa, C.J.; Chen, C.; Valeta-Magara, A.; Rudqvist, N.-P.; Van Nest, S.J.; Kamel, M.K. Expression of the mono-ADP-ribosyltransferase ART1 by tumor cells mediates immune resistance in non–small cell lung cancer. Sci. Transl. Med. 2022, 14, eabe8195. [Google Scholar] [CrossRef]
- Mukherjee, S.; Wennerberg, E.; Hung, C.; Saadallah, N.; Kariyawasam, S.; Agrusa, C.; Valeta, A.; Altorki, N.; McGraw, T.; Demaria, S. Art1, an extracellular mono-ADP-ribosyltransferase, is upregulated in response to cellular stress and promotes lung cancer growth. Cancer Res. 2020, 80, 1820. [Google Scholar] [CrossRef]
- Mukherjee, S.; Wennerberg, E.; Hung, C.; Saadallah, N.; Kariyawasam, S.; Hussein, M.; Narula, N.; Adusumilli, P.; Borczuk, A.; Altorki, N. A05 ART1, a Mono-ADP-Ribosyltransferase, Regulates Tumor-Infiltrating CD8+ T Cells and Is Highly Expressed in EGFR Mutated Lung Cancers. J. Thorac. Oncol. 2020, 15, S13. [Google Scholar] [CrossRef]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Solinas, C.; Gu-Trantien, C.; Willard-Gallo, K. The rationale behind targeting the ICOS-ICOS ligand costimulatory pathway in cancer immunotherapy. ESMO Open 2020, 5, e000544. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Ren, X.; Galbo, P.M.; Moerdler, S.; Wang, H.; Sica, R.A.; Etemad-Gilbertson, B.; Shi, L.; Zhu, L.; Tang, X.; et al. KIR3DL3-HHLA2 is a human immunosuppressive pathway and a therapeutic target. Sci. Immunol. 2021, 6, eabf9792. [Google Scholar] [CrossRef]
- Ren, X.; Peng, M.; Xing, P.; Wei, Y.; Galbo, P.M.; Corrigan, D.; Wang, H.; Su, Y.; Dong, X.; Sun, Q.; et al. Blockade of the immunosuppressive KIR2DL5/PVR pathway elicits potent human NK cell-mediated antitumor immunity. J. Clin. Investig. 2022, 132, e163620. [Google Scholar] [CrossRef]
- Bilemjian, V.; Vlaming, M.R.; Álvarez Freile, J.; Huls, G.; De Bruyn, M.; Bremer, E. The Novel Immune Checkpoint GPR56 Is Expressed on Tumor-Infiltrating Lymphocytes and Selectively Upregulated upon TCR Signaling. Cancers 2022, 14, 3164. [Google Scholar] [CrossRef]
- Aubert, N.; Brunel, S.; Olive, D.; Marodon, G. Blockade of HVEM for Prostate Cancer Immunotherapy in Humanized Mice. Cancers 2021, 13, 3009. [Google Scholar] [CrossRef] [PubMed]
- Leichter, A.L.; Sullivan, M.J.; Eccles, M.R.; Chatterjee, A. MicroRNA expression patterns and signalling pathways in the development and progression of childhood solid tumours. Mol. Cancer 2017, 16, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin-Acevedo, J.A.; Soyano, A.E.; Dholaria, B.; Knutson, K.L.; Lou, Y. Cancer immunotherapy beyond immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, A.R.; Sandhu, V.; Sprauten, M.; Flote, V.G.; Kure, E.H.; Brustugun, O.T.; Helland, Å. Circulating microRNAs associated with prolonged overall survival in lung cancer patients treated with nivolumab. Acta Oncol. 2018, 57, 1225–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huber, V.; Vallacchi, V.; Fleming, V.; Hu, X.; Cova, A.; Dugo, M.; Shahaj, E.; Sulsenti, R.; Vergani, E.; Filipazzi, P. Tumor-derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J. Clin. Investig. 2018, 128, 5505–5516. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.-F.; Pei, X.; Li, K.-S.; Jin, L.-N.; Wang, F.; Wu, J.; Zhang, X.-M. Circular RNA circFGFR1 promotes progression and anti-PD-1 resistance by sponging miR-381-3p in non-small cell lung cancer cells. Mol. Cancer 2019, 18, 179. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, S.; Fukushima, S.; Okada, E.; Morinaga, J.; Kubo, Y.; Tokuzumi, A.; Matsumoto, S.; Tsuruta-Kadohisa, M.; Kimura, T.; Kuriyama, H. MicroRNAs that predict the effectiveness of anti-PD-1 therapies in patients with advanced melanoma. J. Dermatol. Sci. 2020, 97, 77–79. [Google Scholar] [CrossRef]
- Mollaei, H.; Safaralizadeh, R.; Rostami, Z. MicroRNA replacement therapy in cancer. J. Cell. Physiol. 2019, 234, 12369–12384. [Google Scholar] [CrossRef]
- Huemer, F.; Leisch, M.; Geisberger, R.; Zaborsky, N.; Greil, R. miRNA-based therapeutics in the era of immune-checkpoint inhibitors. Pharmaceuticals 2021, 14, 89. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.-K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef] [Green Version]
- van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- Seto, A.G.; Beatty, X.; Lynch, J.M.; Hermreck, M.; Tetzlaff, M.; Duvic, M.; Jackson, A.L. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br. J. Haematol. 2018, 183, 428–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasiadou, E.; Seto, A.G.; Beatty, X.; Hermreck, M.; Gilles, M.-E.; Stroopinsky, D.; Pinter-Brown, L.C.; Pestano, L.; Marchese, C.; Avigan, D. Cobomarsen, an Oligonucleotide Inhibitor of miR-155, Slows DLBCL Tumor Cell Growth In Vitro and In VivoCobomarsen, a miRNA-based Compound for DLBCL Treatment. Clin. Cancer Res. 2021, 27, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Türeci, Ö.; Vormehr, M.; Kranz, L.M. mRNA therapeutics in cancer immunotherapy. Mol. Cancer 2021, 20, 69. [Google Scholar] [CrossRef] [PubMed]
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weng, Y.; Li, C.; Yang, T.; Hu, B.; Zhang, M.; Guo, S.; Xiao, H.; Liang, X.-J.; Huang, Y. The challenge and prospect of mRNA therapeutics landscape. Biotechnol. Adv. 2020, 40, 107534. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef]
- Deng, Z.; Tian, Y.; Song, J.; An, G.; Yang, P. mRNA vaccines: The dawn of a new era of cancer immunotherapy. Front. Immunol. 2022, 13, 887125. [Google Scholar] [CrossRef] [PubMed]
- Howard, T.P.; Vazquez, F.; Tsherniak, A.; Hong, A.L.; Rinne, M.; Aguirre, A.J.; Boehm, J.S.; Hahn, W.C. Functional genomic characterization of cancer genomes. Cold Spring Harb. Symp. Quant. Biol. 2016, 81, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Cowley, G.S.; Weir, B.A.; Vazquez, F.; Tamayo, P.; Scott, J.A.; Rusin, S.; East-Seletsky, A.; Ali, L.D.; Gerath, W.F.; Pantel, S.E. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci. Data 2014, 1, 140035. [Google Scholar] [CrossRef] [Green Version]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef] [PubMed]
- Paulson, K.G.; Park, S.Y.; Vandeven, N.A.; Lachance, K.; Thomas, H.; Chapuis, A.G.; Harms, K.L.; Thompson, J.A.; Bhatia, S.; Stang, A. Merkel cell carcinoma: Current US incidence and projected increases based on changing demographics. J. Am. Acad. Dermatol. 2018, 78, 457–463.e452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sabio, E.; Krishna, C.; Ma, X.; Wang, J.; Jiang, H.; Havel, J.J.; Chan, T.A. Qa-1(b) Modulates Resistance to Anti-PD-1 Immune Checkpoint Blockade in Tumors with Defects in Antigen Processing. Mol. Cancer Res. 2021, 19, 1076–1084. [Google Scholar] [CrossRef]
- Khalaf, K.; Janowicz, K.; Dyszkiewicz-Konwińska, M.; Hutchings, G.; Dompe, C.; Moncrieff, L.; Jankowski, M.; Machnik, M.; Oleksiewicz, U.; Kocherova, I. CRISPR/Cas9 in cancer immunotherapy: Animal models and human clinical trials. Genes 2020, 11, 921. [Google Scholar] [CrossRef]
- Xia, A.-L.; He, Q.-F.; Wang, J.-C.; Zhu, J.; Sha, Y.-Q.; Sun, B.; Lu, X.-J. Applications and advances of CRISPR-Cas9 in cancer immunotherapy. J. Med. Genet. 2019, 56, 4–9. [Google Scholar] [CrossRef]


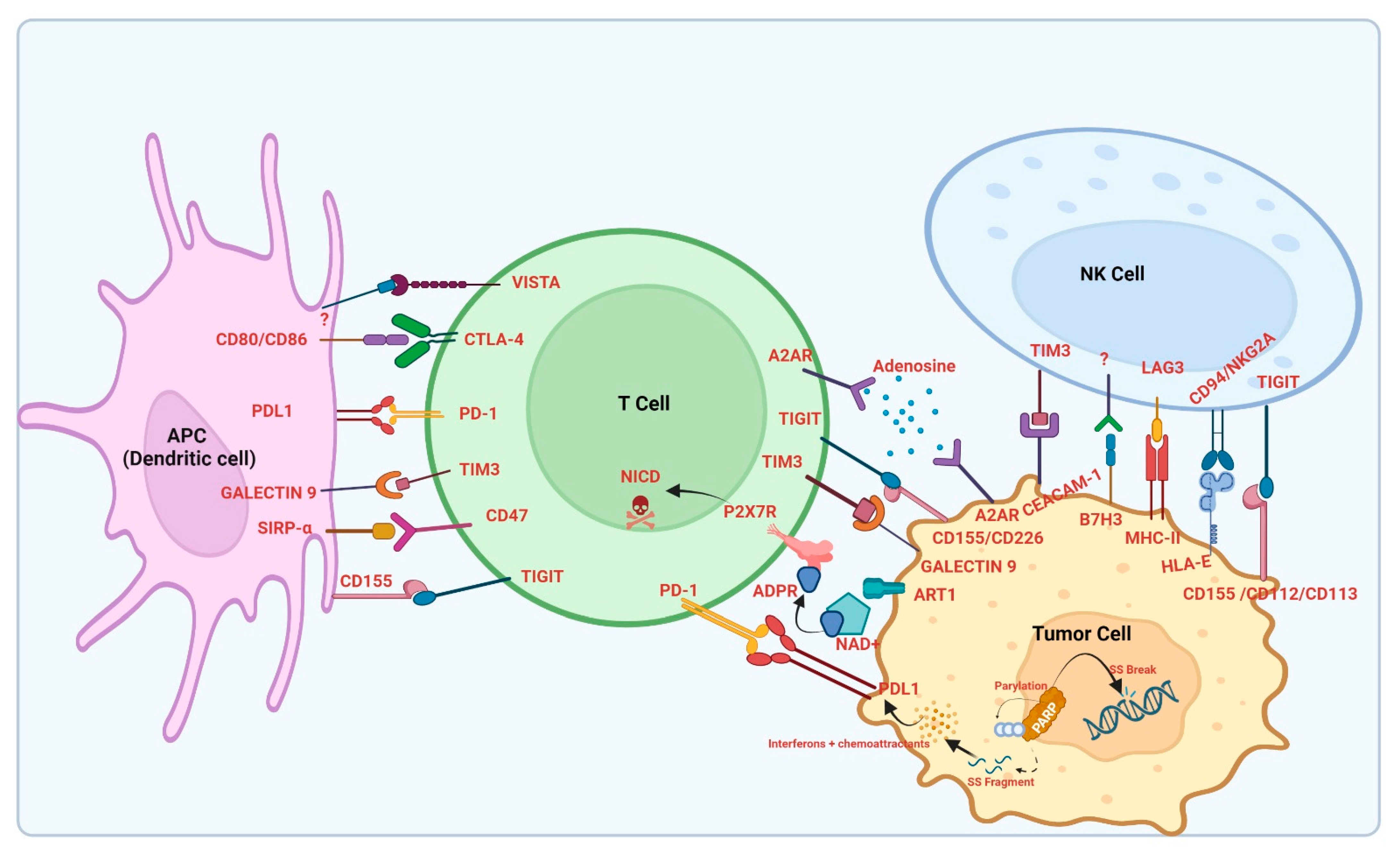{kind=link}
| Molecule/Target | Drug | Mechanism | Cancer Type | Trial Name/Phase | Estimated/Actual Completion Date |
|---|---|---|---|---|---|
| PD-1 | Retifanlimab + INCAGN02385 + INCAGN02390 | Anti-PD-1 + Anti-LAG-3 + Anti-TIM-3 | Head and Neck Cancer | (NCT05287113)/Phase 2 | 15 September 2024 |
| Pembrolizumab | Anti-PD-1 | Refractory melanomas, Non small cell lung carcinoma (NSCLC), Urothelial Carcinoma, Metastatic Head and Neck squamous cell carcinoma (HNSCC) etc. | KEYNOTE Trials: (NCT01295827) Phase 3 | 11 December 2018 | |
| Nivolumab | Anti-PD-1 | Metastatic Melanoma NSCLC, Urothelial carcinoma, Colorectal carcinoma | Checkmate studies (NCT02388906) (NCT01668784) Phase2/Phase3 | 30 January 2026 19 July 2021 | |
| Cemiplimab | Anti-PD-1 | Metastatic pancreatic cancer | (NCT04177810) | 1 August 2023 | |
| Malignant glioma | (NCT03690869) | 20 November 2024 | |||
| Hepatocellular carcinoma | (NCT03916627) | 3 September 2029 | |||
| NSCLC | (NCT03580694) | 4 December 2019 | |||
| Renal cancer | (NCT02394083) | 5 November 2023 | |||
| Lymphoma | (NCT02651662) | 19 August 2026 | |||
| Multiple myeloma | (NCT03194867) | 31 March 2023 | |||
| Prostate cancer | (NCT03951831) | December 2022 | |||
| Ovarian cancer | (NCT03564340) | December 2022 | |||
| Cervical cancer | (NCT03257267) | 9 July 2023 | |||
| Phase 2/Phase3 | |||||
| PD-L1 | Atezolizumab | Anti-PD-L1 | NSCLC | NCT02008227/Phase3 | 9 January 2019 |
| Avelumab | Anti-PD-L1/PD-1 | Renal cell carcinoma | NCT02684006/ Phase 3 | 21 May 2024 | |
| Durvalumab | Anti-PD-L1 | Urothelial carcinoma NSCLC | NCT01693562 NCT02125461 | 28 February 2020 30 December 2022 | |
| CTLA-4 | Anti-CTLA-4 Monoclonal Antibody BMS-986218 + Nivolumab | Anti-CTLA-4 + Anti-PD-1 | Advanced Lung Carcinoma, Advanced Malignant Solid Neoplasm, Malignant Adrenal Gland Neoplasm, Metastatic Liver Carcinoma, Metastatic Lung Carcinoma, Metastatic Malignant Solid Neoplasm | NCT04785287/Phase 1, Phase 2 | 27 May 2024 |
| HLA-E | TTX-080 | Anti-HLA-E | Refractory solid malignancies such as HNSCC, NSCLC, Colorectal cancer and triple negative breast cancer | NCT04485013/Phase 1 | 1 June 2024 |
| NKG2A | Monalizumab + Cetuximab + Anti-PD(L)-1 | Anti-NKG2A + Anti-EGFR | Head and Neck Neoplasms | NCT02643550/Phase 1, Phase 2 | September 2022 |
| Durvalumab + Monalizumab | Anti-PD-L1+ Anti-NKG2A | Stage III Non-small Cell Lung Cancer Unresectable | NCT03822351/Phase 2 | 21 June 2023 | |
| NKG2D | CM-CS1/CYAD-01 | CAR T cell | Acute Myeloid Leukemia, Multiple Myeloma, Myelodysplastic Syndromes | NCT02203825/ Phase 1 | March 2018 |
| A2AR & A2BR | AZD4635 + Oleclumab + Durvalumab | Anti-A2aR & A2bR + Anti-CD73+ Anti-PD-L1 | Prostate Cancer Metastatic Castration-Resistant Prostate Cancer (mCRPC) | NCT04089553/Phase 2 | 31 December 2023 |
| Etrumadenant + zimberelimab+ mFOLFOX-6 + bevacizumab + regorafenib + AB680 | Anti-A2aR and A2bR+ Anti-PD-1 + Anti-CD73 | Metastatic Colorectal Cancer | NCT04660812/Phase 1, Phase 2 | 18 December 2023 | |
| SIRPα/CD47 | CC-90002 | Anti-CD47 | Acute myeloid leukemia | NCT02641002/Phase1 | 18 July 2018 |
| HX009 | Anti-CD47/PD-1 bifunctional antibody | Unresectable locally advanced/ metastatic solid tumors | NCT04886271/Phase 2 | 10 February 2023 | |
| TTI-621 | Anti-SIRPa | R/R Hematologic malignancies and selected solid tumors (PTCL, CTCL) | NCT02663518/Phase 1 | 31 December 2022 | |
| BI-765063/OSE172 | Anti-SIRPa | Advanced solid tumors (NSCLC, TNBC, pancreatic cancer, melanoma, HNSCC, RCC, UC, SCL, gastric cancer, CRC and OC) | NCT03990233/Phase 1 | 31 December 2022 | |
| TIM-3 | TSR022 | Anti-TIM-3 | High Grade serous ovarian cancer | NCT04139902/Phase 1 | October 2027 |
| LAG-3 | IERAMILImAB (LAG525) | Anti-LAG-3 | Advanced/Metastatic solid tumors | NCT02460224/Phase 1 | 31 December 2020 |
| Relatlimab | Anti-LAG-3 | Previously untreated metastatic/uresectable melanoma | NCT03470922/Phase 1/2 | 16 December 2025 | |
| REGN3767/Fianlimab + Cemiplimab | Anti-LAG-3 + Anti-PD-1/PD-L1 | Previously Untreated Unresectable Locally Advanced or Metastatic Melanoma | NCT05352672/Phase 3 | 20 April 2031 | |
| B7-H3 | Enoblituzumabn (MGA271) | Anti-B7-H3 | Prostate Cancer | NCT02923180/Phase 2 | 30 July 2023 |
| MGD009/Orlotamab | Anti-B7-H3 | Mesothelioma, Bladder Cancer, Melanoma,Squamous Cell Carcinoma of the Head and Neck,NSCLCr,Clear Cell Renal Cell Carcinoma Ovarian Cancer,Thyroid Cancer, Breast Cancer,Pancreatic Cancer,Prostate Cancer, Colon Cancer, Soft Tissue Sarcoma | NCT04145622/Phase 1 | 1 December 2023 | |
| MGC018 +/− MGA012 | Anti-B7-H3 +/− Anti-PD-1 | Squamous Cell Carcinoma of Head &Neck, Triple Negative Breast Cancer, Melanoma, Advanced Solid Tumor, Adult Metastatic Castrate Resistant Prostate Cancer, NSCLC | NCT03729596/Phase 1, 2 | May 2023 | |
| PARPs | Olaparib (AZD2281) | PARP inhibitor | Patients with ovarian cancer that recurred within 12 months of prior platinum therapy and with confirmed germline BRCA1/BRCA2 mutation | NCT00753545/Phase 2 | 29 December 2023 |
| Rucaparib | PARP inhibitor | Advanced gynecologic cancer and triple negative breast cancer | NCT03101280/Phase 1 | 11 August 2020 | |
| TIGIT | COM701 in combination with BMS-986207 and nivolumab. | Anti-TIGIT Antibody | Endometrial Neoplasms,Ovarian Cancer,Solid Tumor,Head and Neck Cancer | NCT04570839/Phase1 Phase 2 | December 2023 |
| Ociperlimab (BGB-A1217) + Tislelizumab | Anti-TIGIT Antibody | Locally Advanced and Metastatic Solid Tumors | NCT04047862/Phase 1 | October 2024 | |
| Tiragolumab + Atezolizumab | Anti-TIGIT Antibody | Non-small Cell Lung Cancer | NCT03563716/Phase 2 | 30 September 2023 | |
| VISTA | JNJ-61610588 | Anti-VISTA | Advanced Cancer | NCT02671955/Phase 1 | July 2017 |
| CA-170 | Oral PD-L1, PD-L2 and VISTA Checkpoint Antagonist | Advanced Solid Tumors or Lymphomas | NCT02812875/Phase 1 | 7 May 2020 | |
| CI-8993 | Anti-VISTA | Solid Tumor | NCT04475523/Phase 1 | 1 July 2023 | |
| microRNA | Cobomarsen | Immunotherapeutic MicroRNA | Cutaneous T-cell Lymphoma Mycosis Fungoides Chronic Lymphocytic Leukemia Diffuse Large B-Cell Lymphoma, ABC Subtype Adult T-Cell Leukemia/Lymphoma | NCT02580552/Phase 1 | 6 October 2020 |
| TargomiRs | miR-16 Mimic | Malignant Pleural Mesothelioma Non-Small Cell Lung Cancer | NCT02369198/Phase 1 | 4 January 2017 | |
| mRNA | Lipo-MERIT | RNA-lipoplex Cancer Vaccine | Melanoma | NCT02410733/Phase 1 | May 2023 |
| mRNA-4157 + Pembrolizumab | Personalized cancer vaccine | Solid Tumors | NCT03313778/Phase 1 | 30 June 2025 | |
| CRISPR/CAS9 | Tumor-Infiltrating Lymphocytes (TIL) | Tumor Infiltrating Lymphocytes in Which the Gene Encoding the Intracellular Immune Checkpoint CISH Is Inhibited | Gastrointestinal Epithelial Cancer, Gastrointestinal Neoplasms, Gastrointestinal Cancer, Colo-rectal Cancer, Pancreatic Cancer, Gall Bladder Cancer, Colon Cancer, Esophageal Cancer, Stomach Cancer | NCT04426669/Phase 1 | January 2023 |
| CTX110 | CD19-directed chimeric antigen receptor (CAR) T cell immunotherapy | B-cell Malignancy, Non-Hodgkin Lymphoma, B-cell Lymphoma, Adult B Cell ALL | NCT04035434/Phase 1 | August 2026 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dutta, S.; Ganguly, A.; Chatterjee, K.; Spada, S.; Mukherjee, S. Targets of Immune Escape Mechanisms in Cancer: Basis for Development and Evolution of Cancer Immune Checkpoint Inhibitors. Biology 2023, 12, 218. https://doi.org/10.3390/biology12020218
Dutta S, Ganguly A, Chatterjee K, Spada S, Mukherjee S. Targets of Immune Escape Mechanisms in Cancer: Basis for Development and Evolution of Cancer Immune Checkpoint Inhibitors. Biology. 2023; 12(2):218. https://doi.org/10.3390/biology12020218
Chicago/Turabian StyleDutta, Shovan, Anirban Ganguly, Kaushiki Chatterjee, Sheila Spada, and Sumit Mukherjee. 2023. "Targets of Immune Escape Mechanisms in Cancer: Basis for Development and Evolution of Cancer Immune Checkpoint Inhibitors" Biology 12, no. 2: 218. https://doi.org/10.3390/biology12020218