
The Basolateral Amygdala: The Core of a Network for Threat Conditioning, Extinction, and Second-Order Threat Conditioning

{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
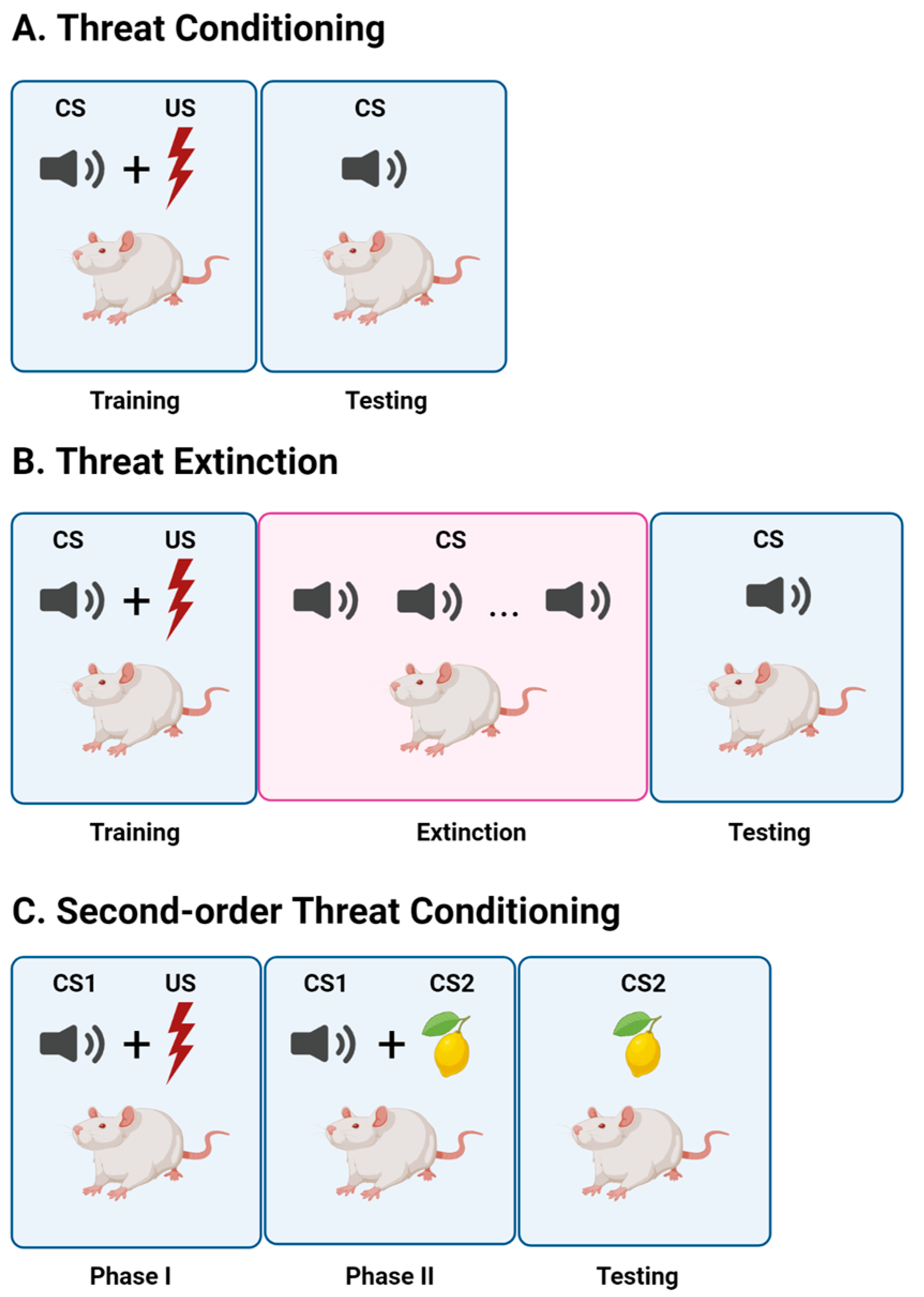
2. Threat Conditioning
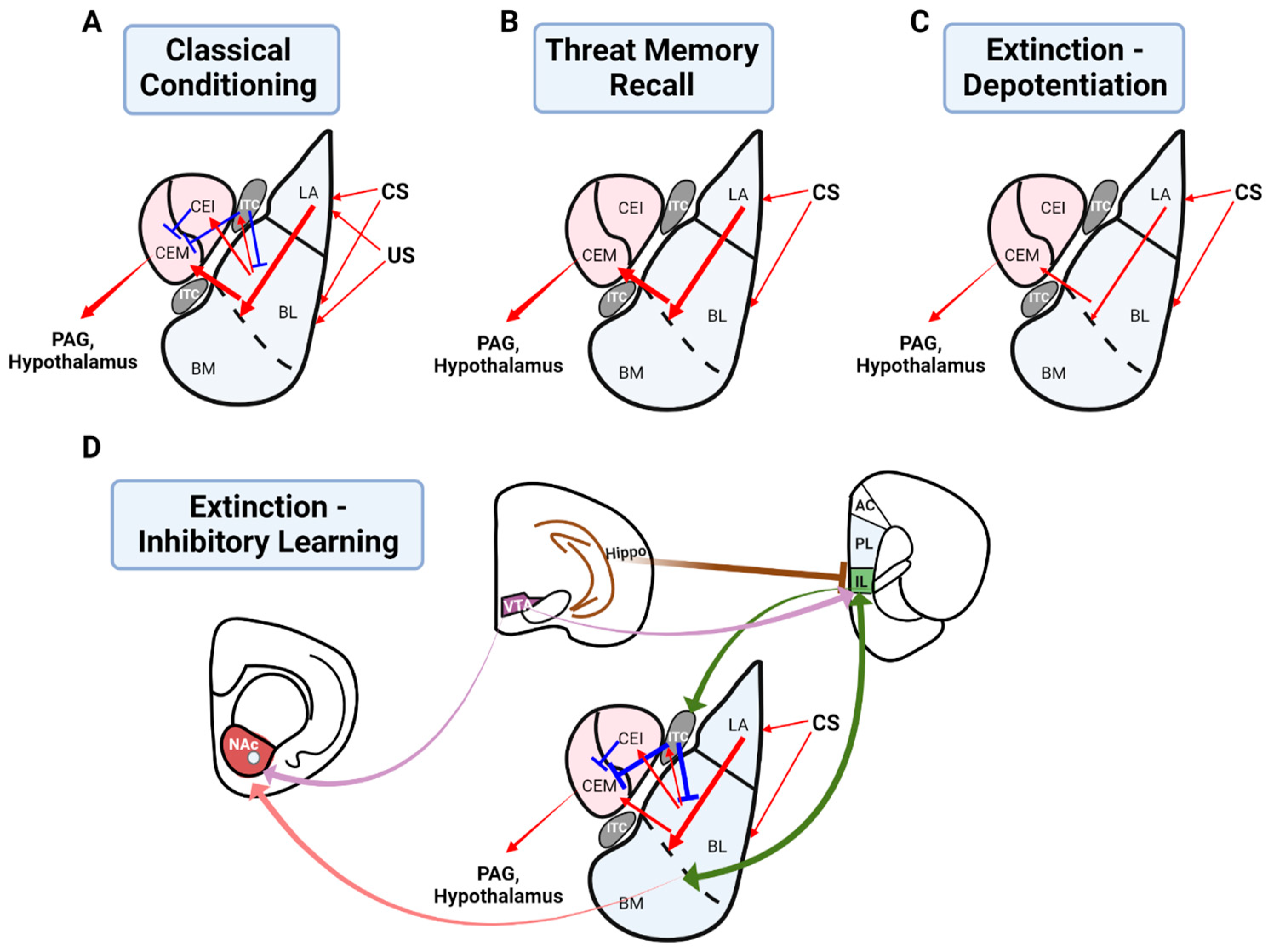
2.1. The Role of the Basolateral Amygdala in Threat Conditioning
2.2. Neuromodulation of the Basolateral Amygdala in Threat Conditioning
2.3. Greater Circuitry Involved in Threat Conditioning
3. Threat Extinction
3.1. The Role of the Basolateral Amygdala in Threat Extinction
3.1.1. Depotentiation of the Basolateral Amygdala as a Mechanism of Extinction Learning
3.1.2. Inhibitory Plasticity in the Basolateral Amygdala Underlying Extinction Learning
3.2. Greater Circuitry Involved in Threat Extinction: An Inhibitory Learning Model
3.2.1. The Infralimbic Cortex in Threat Extinction
3.2.2. An Interaction between the Infralimbic Cortex and Basolateral Amygdala for Threat Extinction
3.2.3. The Role of Intercalated Cells in Threat Extinction
3.2.4. The Role of the Hippocampus in Threat Extinction
3.3. Neuromodulation in Threat Extinction
3.4. Experimental Modifications Determine the Strength and Mechanism of Extinction Learning
4. Second-Order Threat Conditioning
Circuit and Molecular Mechanisms of Second-Order Threat Conditioning and Extinction
5. Discussion
5.1. Prediction Error-Driven Learning
5.2. Clinical Relevance
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pape, H.C.; Pare, D. Plastic synaptic networks of the amygdala for the acquisition, expression, and extinction of conditioned fear. Physiol. Rev. 2010, 90, 419–463. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, P.I. Conditioned reflexes: An investigation of the physiological activity of the cerebral cortex. Ann. Neurosci. 2010, 17, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, I.P. Conditioned Reflexes: An Investigation of the Physiological Activity of the Cerebral Cortex; Anrep, G.V., Ed.; Oxford University Press: London, UK, 1927. [Google Scholar]
- Graham, B.M.; Milad, M.R. The study of fear extinction: Implications for anxiety disorders. Am. J. Psychiatry 2011, 168, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Barrett, L.F. The theory of constructed emotion: An active inference account of interoception and categorization. Soc. Cogn. Affect. Neurosci. 2017, 12, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.T.; Haigh, E.A. Advances in cognitive theory and therapy: The generic cognitive model. Annu. Rev. Clin. Psychol. 2014, 10, 1–24. [Google Scholar] [CrossRef] [PubMed]
- LeDoux, J.E.; Brown, R. A higher-order theory of emotional consciousness. Proc. Natl. Acad. Sci. USA 2017, 114, E2016–E2025. [Google Scholar] [CrossRef]
- Romanski, L.M.; LeDoux, J.E. Information cascade from primary auditory cortex to the amygdala: Corticocortical and corticoamygdaloid projections of temporal cortex in the rat. Cereb. Cortex 1993, 3, 515–532. [Google Scholar] [CrossRef]
- Romanski, L.M.; Clugnet, M.C.; Bordi, F.; LeDoux, J.E. Somatosensory and auditory convergence in the lateral nucleus of the amygdala. Behav. Neurosci. 1993, 107, 444–450. [Google Scholar] [CrossRef]
- Johansen, J.P.; Cain, C.K.; Ostroff, L.E.; LeDoux, J.E. Molecular mechanisms of fear learning and memory. Cell 2011, 147, 509–524. [Google Scholar] [CrossRef]
- Lee, S.; Kim, S.J.; Kwon, O.B.; Lee, J.H.; Kim, J.H. Inhibitory networks of the amygdala for emotional memory. Front. Neural Circuits 2013, 7, 129. [Google Scholar] [CrossRef]
- Pare, D.; Duvarci, S. Amygdala microcircuits mediating fear expression and extinction. Curr. Opin. Neurobiol. 2012, 22, 717–723. [Google Scholar] [CrossRef]
- Pare, D.; Smith, Y. The intercalated cell masses project to the central and medial nuclei of the amygdala in cats. Neuroscience 1993, 57, 1077–1090. [Google Scholar] [CrossRef] [PubMed]
- Pare, D.; Royer, S.; Smith, Y.; Lang, E.J. Contextual inhibitory gating of impulse traffic in the intra-amygdaloid network. Ann. N. Y. Acad. Sci. 2003, 985, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, D.A.; Holstege, G. Amygdaloid projections to the mesencephalon, pons and medulla oblongata in the cat. Exp. Brain Res. 1978, 32, 529–547. [Google Scholar] [CrossRef]
- Sigurdsson, T.; Doyere, V.; Cain, C.K.; LeDoux, J.E. Long-term potentiation in the amygdala: A cellular mechanism of fear learning and memory. Neuropharmacology 2007, 52, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Pare, D.; Quirk, G.J.; Ledoux, J.E. New vistas on amygdala networks in conditioned fear. J. Neurophysiol. 2004, 92, 1–9. [Google Scholar] [CrossRef]
- Maren, S. Neurobiology of Pavlovian fear conditioning. Annu. Rev. Neurosci. 2001, 24, 897–931. [Google Scholar] [CrossRef]
- Rodrigues, S.M.; Schafe, G.E.; LeDoux, J.E. Molecular mechanisms underlying emotional learning and memory in the lateral amygdala. Neuron 2004, 44, 75–91. [Google Scholar] [CrossRef]
- Hagihara, K.M.; Bukalo, O.; Zeller, M.; Aksoy-Aksel, A.; Karalis, N.; Limoges, A.; Rigg, T.; Campbell, T.; Mendez, A.; Weinholtz, C.; et al. Intercalated amygdala clusters orchestrate a switch in fear state. Nature 2021, 594, 403–407. [Google Scholar] [CrossRef]
- Bissiere, S.; Humeau, Y.; Luthi, A. Dopamine gates LTP induction in lateral amygdala by suppressing feedforward inhibition. Nat. Neurosci. 2003, 6, 587–592. [Google Scholar] [CrossRef]
- Tully, K.; Li, Y.; Tsvetkov, E.; Bolshakov, V.Y. Norepinephrine enables the induction of associative long-term potentiation at thalamo-amygdala synapses. Proc. Natl. Acad. Sci. USA 2007, 104, 14146–14150. [Google Scholar] [CrossRef]
- Maren, S.; Aharonov, G.; Stote, D.L.; Fanselow, M.S. N-methyl-D-aspartate receptors in the basolateral amygdala are required for both acquisition and expression of conditional fear in rats. Behav. Neurosci. 1996, 110, 1365–1374. [Google Scholar] [CrossRef]
- Miserendino, M.J.; Sananes, C.B.; Melia, K.R.; Davis, M. Blocking of acquisition but not expression of conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature 1990, 345, 716–718. [Google Scholar] [CrossRef]
- Rodrigues, S.M.; Schafe, G.E.; LeDoux, J.E. Intra-amygdala blockade of the NR2B subunit of the NMDA receptor disrupts the acquisition but not the expression of fear conditioning. J. Neurosci. 2001, 21, 6889–6896. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.J. Molecular and cellular cognitive studies of the role of synaptic plasticity in memory. J. Neurobiol. 2003, 54, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, S.M.; Farb, C.R.; Bauer, E.P.; LeDoux, J.E.; Schafe, G.E. Pavlovian fear conditioning regulates Thr286 autophosphorylation of Ca2+/calmodulin-dependent protein kinase II at lateral amygdala synapses. J. Neurosci. 2004, 24, 3281–3288. [Google Scholar] [CrossRef]
- Nader, K.; Hardt, O. A single standard for memory: The case for reconsolidation. Nat. Rev. Neurosci. 2009, 10, 224–234. [Google Scholar] [CrossRef] [PubMed]
- Monfils, M.H.; Cowansage, K.K.; Klann, E.; LeDoux, J.E. Extinction-reconsolidation boundaries: Key to persistent attenuation of fear memories. Science 2009, 324, 951–955. [Google Scholar] [CrossRef]
- Schafe, G.E.; Nadel, N.V.; Sullivan, G.M.; Harris, A.; LeDoux, J.E. Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn. Mem. 1999, 6, 97–110. [Google Scholar] [CrossRef]
- Monsey, M.S.; Ota, K.T.; Akingbade, I.F.; Hong, E.S.; Schafe, G.E. Epigenetic alterations are critical for fear memory consolidation and synaptic plasticity in the lateral amygdala. PLoS ONE 2011, 6, e19958. [Google Scholar] [CrossRef]
- Berridge, C.W.; Waterhouse, B.D. The locus coeruleus-noradrenergic system: Modulation of behavioral state and state-dependent cognitive processes. Brain Res. Rev. 2003, 42, 33–84. [Google Scholar] [CrossRef] [PubMed]
- Fallon, J.H.; Koziell, D.A.; Moore, R.Y. Catecholamine innervation of the basal forebrain. II. Amygdala, suprarhinal cortex and entorhinal cortex. J. Comp. Neurol. 1978, 180, 509–532. [Google Scholar] [CrossRef] [PubMed]
- Bush, D.E.; Caparosa, E.M.; Gekker, A.; Ledoux, J. Beta-adrenergic receptors in the lateral nucleus of the amygdala contribute to the acquisition but not the consolidation of auditory fear conditioning. Front. Behav. Neurosci. 2010, 4, 154. [Google Scholar] [CrossRef] [PubMed]
- Johansen, J.P.; Diaz-Mataix, L.; Hamanaka, H.; Ozawa, T.; Ycu, E.; Koivumaa, J.; Kumar, A.; Hou, M.; Deisseroth, K.; Boyden, E.S.; et al. Hebbian and neuromodulatory mechanisms interact to trigger associative memory formation. Proc. Natl. Acad. Sci. USA 2014, 111, E5584–E5592. [Google Scholar] [CrossRef]
- Muller, J.; Corodimas, K.P.; Fridel, Z.; LeDoux, J.E. Functional inactivation of the lateral and basal nuclei of the amygdala by muscimol infusion prevents fear conditioning to an explicit conditioned stimulus and to contextual stimuli. Behav. Neurosci. 1997, 111, 683–691. [Google Scholar] [CrossRef]
- Wilensky, A.E.; Schafe, G.E.; LeDoux, J.E. Functional inactivation of the amygdala before but not after auditory fear conditioning prevents memory formation. J. Neurosci. 1999, 19, RC48. [Google Scholar] [CrossRef]
- Bouret, S.; Duvel, A.; Onat, S.; Sara, S.J. Phasic activation of locus ceruleus neurons by the central nucleus of the amygdala. J. Neurosci. 2003, 23, 3491–3497. [Google Scholar] [CrossRef]
- Giustino, T.F.; Ramanathan, K.R.; Totty, M.S.; Miles, O.W.; Maren, S. Locus Coeruleus Norepinephrine Drives Stress-Induced Increases in Basolateral Amygdala Firing and Impairs Extinction Learning. J. Neurosci. 2020, 40, 907–916. [Google Scholar] [CrossRef]
- Soya, S.; Takahashi, T.M.; McHugh, T.J.; Maejima, T.; Herlitze, S.; Abe, M.; Sakimura, K.; Sakurai, T. Orexin modulates behavioral fear expression through the locus coeruleus. Nat. Commun. 2017, 8, 1606. [Google Scholar] [CrossRef]
- Uematsu, A.; Tan, B.Z.; Ycu, E.A.; Cuevas, J.S.; Koivumaa, J.; Junyent, F.; Kremer, E.J.; Witten, I.B.; Deisseroth, K.; Johansen, J.P. Modular organization of the brainstem noradrenaline system coordinates opposing learning states. Nat. Neurosci. 2017, 20, 1602–1611. [Google Scholar] [CrossRef]
- Gonzales, C.; Chesselet, M.F. Amygdalonigral pathway: An anterograde study in the rat with Phaseolus vulgaris leucoagglutinin (PHA-L). J. Comp. Neurol. 1990, 297, 182–200. [Google Scholar] [CrossRef] [PubMed]
- Inglis, F.M.; Moghaddam, B. Dopaminergic innervation of the amygdala is highly responsive to stress. J. Neurochem. 1999, 72, 1088–1094. [Google Scholar] [CrossRef] [PubMed]
- Gore, B.B.; Soden, M.E.; Zweifel, L.S. Visualization of plasticity in fear-evoked calcium signals in midbrain dopamine neurons. Learn. Mem. 2014, 21, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Guarraci, F.A.; Frohardt, R.J.; Kapp, B.S. Amygdaloid D1 dopamine receptor involvement in Pavlovian fear conditioning. Brain Res. 1999, 827, 28–40. [Google Scholar] [CrossRef]
- Rosenkranz, J.A.; Grace, A.A. Dopamine-mediated modulation of odour-evoked amygdala potentials during pavlovian conditioning. Nature 2002, 417, 282–287. [Google Scholar] [CrossRef]
- Tang, W.; Kochubey, O.; Kintscher, M.; Schneggenburger, R. A VTA to Basal Amygdala Dopamine Projection Contributes to Signal Salient Somatosensory Events during Fear Learning. J. Neurosci. 2020, 40, 3969–3980. [Google Scholar] [CrossRef]
- Fanselow, M.S.; LeDoux, J.E. Why we think plasticity underlying Pavlovian fear conditioning occurs in the basolateral amygdala. Neuron 1999, 23, 229–232. [Google Scholar] [CrossRef]
- Li, W.; Wilson, D.A. Threat Memory in the Sensory Cortex: Insights from Olfaction. Neuroscientist 2023, 10738584221148994. [Google Scholar] [CrossRef]
- Josselyn, S.A.; Tonegawa, S. Memory engrams: Recalling the past and imagining the future. Science 2020, 367, eaaw4325. [Google Scholar] [CrossRef]
- Weinberger, N.M. Specific long-term memory traces in primary auditory cortex. Nat. Rev. Neurosci. 2004, 5, 279–290. [Google Scholar] [CrossRef]
- Bouton, M.E.; Maren, S.; McNally, G.P. Behavioral and Neurobiological Mechanisms of Pavlovian and Instrumental Extinction Learning. Physiol. Rev. 2021, 101, 611–681. [Google Scholar] [CrossRef] [PubMed]
- Senn, V.; Wolff, S.B.; Herry, C.; Grenier, F.; Ehrlich, I.; Grundemann, J.; Fadok, J.P.; Muller, C.; Letzkus, J.J.; Luthi, A. Long-range connectivity defines behavioral specificity of amygdala neurons. Neuron 2014, 81, 428–437. [Google Scholar] [CrossRef]
- Gilmartin, M.R.; McEchron, M.D. Single neurons in the medial prefrontal cortex of the rat exhibit tonic and phasic coding during trace fear conditioning. Behav. Neurosci. 2005, 119, 1496–1510. [Google Scholar] [CrossRef] [PubMed]
- Lacagnina, A.F.; Brockway, E.T.; Crovetti, C.R.; Shue, F.; McCarty, M.J.; Sattler, K.P.; Lim, S.C.; Santos, S.L.; Denny, C.A.; Drew, M.R. Distinct hippocampal engrams control extinction and relapse of fear memory. Nat. Neurosci. 2019, 22, 753–761. [Google Scholar] [CrossRef]
- Maren, S.; Fanselow, M.S. Synaptic plasticity in the basolateral amygdala induced by hippocampal formation stimulation in vivo. J. Neurosci. 1995, 15, 7548–7564. [Google Scholar] [CrossRef]
- Laurent, V.; Marchand, A.R.; Westbrook, R.F. The basolateral amygdala is necessary for learning but not relearning extinction of context conditioned fear. Learn. Mem. 2008, 15, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Laurent, V.; Westbrook, R.F. Distinct contributions of the basolateral amygdala and the medial prefrontal cortex to learning and relearning extinction of context conditioned fear. Learn. Mem. 2008, 15, 657–666. [Google Scholar] [CrossRef]
- Sacco, T.; Sacchetti, B. Role of secondary sensory cortices in emotional memory storage and retrieval in rats. Science 2010, 329, 649–656. [Google Scholar] [CrossRef]
- Cambiaghi, M.; Grosso, A.; Renna, A.; Sacchetti, B. Differential Recruitment of Auditory Cortices in the Consolidation of Recent Auditory Fearful Memories. J. Neurosci. 2016, 36, 8586–8597. [Google Scholar] [CrossRef]
- Mouly, A.M.; Bouillot, C.; Costes, N.; Zimmer, L.; Ravel, N.; Litaudon, P. PET Metabolic Imaging of Time-Dependent Reorganization of Olfactory Cued Fear Memory Networks in Rats. Cereb. Cortex 2022, 32, 2717–2728. [Google Scholar] [CrossRef]
- You, Y.; Novak, L.R.; Clancy, K.J.; Li, W. Pattern differentiation and tuning shift in human sensory cortex underlie long-term threat memory. Curr. Biol. 2022, 32, 2067–2075.e2064. [Google Scholar] [CrossRef] [PubMed]
- East, B.S.; Fleming, G.; Vervoordt, S.; Shah, P.; Sullivan, R.M.; Wilson, D.A. Basolateral amygdala to posterior piriform cortex connectivity ensures precision in learned odor threat. Sci. Rep. 2021, 11, 21746. [Google Scholar] [CrossRef] [PubMed]
- Hegoburu, C.; Parrot, S.; Ferreira, G.; Mouly, A.M. Differential involvement of amygdala and cortical NMDA receptors activation upon encoding in odor fear memory. Learn. Mem. 2014, 21, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Meissner-Bernard, C.; Dembitskaya, Y.; Venance, L.; Fleischmann, A. Encoding of Odor Fear Memories in the Mouse Olfactory Cortex. Curr. Biol. 2019, 29, 367–380.e364. [Google Scholar] [CrossRef] [PubMed]
- Concina, G.; Renna, A.; Grosso, A.; Sacchetti, B. The auditory cortex and the emotional valence of sounds. Neurosci. Biobehav. Rev. 2019, 98, 256–264. [Google Scholar] [CrossRef]
- Headley, D.B.; Kanta, V.; Kyriazi, P.; Pare, D. Embracing Complexity in Defensive Networks. Neuron 2019, 103, 189–201. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, D.Q.; Huang, W.; Deng, J.; Sun, Y.; Zuo, Y.; Poo, M.M. Selective synaptic remodeling of amygdalocortical connections associated with fear memory. Nat. Neurosci. 2016, 19, 1348–1355. [Google Scholar] [CrossRef]
- Tsukano, H.; Hou, X.; Horie, M.; Kitaura, H.; Nishio, N.; Hishida, R.; Takahashi, K.; Kakita, A.; Takebayashi, H.; Sugiyama, S.; et al. Reciprocal connectivity between secondary auditory cortical field and amygdala in mice. Sci. Rep. 2019, 9, 19610. [Google Scholar] [CrossRef]
- Majak, K.; Ronkko, S.; Kemppainen, S.; Pitkanen, A. Projections from the amygdaloid complex to the piriform cortex: A PHA-L study in the rat. J. Comp. Neurol. 2004, 476, 414–428. [Google Scholar] [CrossRef]
- Armony, J.L.; Quirk, G.J.; LeDoux, J.E. Differential effects of amygdala lesions on early and late plastic components of auditory cortex spike trains during fear conditioning. J. Neurosci. 1998, 18, 2592–2601. [Google Scholar] [CrossRef]
- Fourcaudot, E.; Gambino, F.; Casassus, G.; Poulain, B.; Humeau, Y.; Luthi, A. L-type voltage-dependent Ca(2+) channels mediate expression of presynaptic LTP in amygdala. Nat. Neurosci. 2009, 12, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Humeau, Y.; Shaban, H.; Bissiere, S.; Luthi, A. Presynaptic induction of heterosynaptic associative plasticity in the mammalian brain. Nature 2003, 426, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Chavez, C.M.; McGaugh, J.L.; Weinberger, N.M. The basolateral amygdala modulates specific sensory memory representations in the cerebral cortex. Neurobiol. Learn. Mem. 2009, 91, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.F.; Barnes, D.C.; Wilson, D.A. Generalized vs. stimulus-specific learned fear differentially modifies stimulus encoding in primary sensory cortex of awake rats. J. Neurophysiol. 2011, 106, 3136–3144. [Google Scholar] [CrossRef] [PubMed]
- Cambiaghi, M.; Grosso, A.; Likhtik, E.; Mazziotti, R.; Concina, G.; Renna, A.; Sacco, T.; Gordon, J.A.; Sacchetti, B. Higher-Order Sensory Cortex Drives Basolateral Amygdala Activity during the Recall of Remote, but Not Recently Learned Fearful Memories. J. Neurosci. 2016, 36, 1647–1659. [Google Scholar] [CrossRef]
- Letzkus, J.J.; Wolff, S.B.; Meyer, E.M.; Tovote, P.; Courtin, J.; Herry, C.; Luthi, A. A disinhibitory microcircuit for associative fear learning in the auditory cortex. Nature 2011, 480, 331–335. [Google Scholar] [CrossRef]
- Moczulska, K.E.; Tinter-Thiede, J.; Peter, M.; Ushakova, L.; Wernle, T.; Bathellier, B.; Rumpel, S. Dynamics of dendritic spines in the mouse auditory cortex during memory formation and memory recall. Proc. Natl. Acad. Sci. USA 2013, 110, 18315–18320. [Google Scholar] [CrossRef]
- Quirk, G.J.; Armony, J.L.; LeDoux, J.E. Fear conditioning enhances different temporal components of tone-evoked spike trains in auditory cortex and lateral amygdala. Neuron 1997, 19, 613–624. [Google Scholar] [CrossRef]
- Brosch, M.; Selezneva, E.; Scheich, H. Nonauditory events of a behavioral procedure activate auditory cortex of highly trained monkeys. J. Neurosci. 2005, 25, 6797–6806. [Google Scholar] [CrossRef]
- Brosch, M.; Selezneva, E.; Scheich, H. Formation of associations in auditory cortex by slow changes of tonic firing. Hear. Res. 2011, 271, 66–73. [Google Scholar] [CrossRef]
- Brosch, M.; Selezneva, E.; Scheich, H. Representation of reward feedback in primate auditory cortex. Front. Syst. Neurosci. 2011, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.B.; Stettler, D.D.; Kallman, B.R.; Bhaskar, S.T.; Fleischmann, A.; Axel, R. Driving opposing behaviors with ensembles of piriform neurons. Cell 2011, 146, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Frankland, P.W.; Bontempi, B.; Talton, L.E.; Kaczmarek, L.; Silva, A.J. The involvement of the anterior cingulate cortex in remote contextual fear memory. Science 2004, 304, 881–883. [Google Scholar] [CrossRef]
- Bontempi, B.; Laurent-Demir, C.; Destrade, C.; Jaffard, R. Time-dependent reorganization of brain circuitry underlying long-term memory storage. Nature 1999, 400, 671–675. [Google Scholar] [CrossRef]
- Li, W. Learning to smell danger: Acquired associative representation of threat in the olfactory cortex. Front. Behav. Neurosci. 2014, 8, 98. [Google Scholar] [CrossRef]
- Applegate, C.D.; Frysinger, R.C.; Kapp, B.S.; Gallagher, M. Multiple unit activity recorded from amygdala central nucleus during Pavlovian heart rate conditioning in rabbit. Brain Res. 1982, 238, 457–462. [Google Scholar] [CrossRef]
- McEchron, M.D.; McCabe, P.M.; Green, E.J.; Llabre, M.M.; Schneiderman, N. Simultaneous single unit recording in the medial nucleus of the medial geniculate nucleus and amygdaloid central nucleus throughout habituation, acquisition, and extinction of the rabbit’s classically conditioned heart rate. Brain Res. 1995, 682, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Falls, W.A.; Miserendino, M.J.; Davis, M. Extinction of fear-potentiated startle: Blockade by infusion of an NMDA antagonist into the amygdala. J. Neurosci. 1992, 12, 854–863. [Google Scholar] [CrossRef]
- Lee, H.; Kim, J.J. Amygdalar NMDA receptors are critical for new fear learning in previously fear-conditioned rats. J. Neurosci. 1998, 18, 8444–8454. [Google Scholar] [CrossRef]
- Lee, J.L.; Milton, A.L.; Everitt, B.J. Reconsolidation and extinction of conditioned fear: Inhibition and potentiation. J. Neurosci. 2006, 26, 10051–10056. [Google Scholar] [CrossRef]
- Sotres-Bayon, F.; Bush, D.E.; LeDoux, J.E. Acquisition of fear extinction requires activation of NR2B-containing NMDA receptors in the lateral amygdala. Neuropsychopharmacology 2007, 32, 1929–1940. [Google Scholar] [CrossRef]
- Zimmerman, J.M.; Maren, S. NMDA receptor antagonism in the basolateral but not central amygdala blocks the extinction of Pavlovian fear conditioning in rats. Eur. J. Neurosci. 2010, 31, 1664–1670. [Google Scholar] [CrossRef] [PubMed]
- Hong, I.; Song, B.; Lee, S.; Kim, J.; Kim, J.; Choi, S. Extinction of cued fear memory involves a distinct form of depotentiation at cortical input synapses onto the lateral amygdala. Eur. J. Neurosci. 2009, 30, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, S.; Park, K.; Hong, I.; Song, B.; Son, G.; Park, H.; Kim, W.R.; Park, E.; Choe, H.K.; et al. Amygdala depotentiation and fear extinction. Proc. Natl. Acad. Sci. USA 2007, 104, 20955–20960. [Google Scholar] [CrossRef]
- Lange, M.D.; Doengi, M.; Lesting, J.; Pape, H.C.; Jungling, K. Heterosynaptic long-term potentiation at interneuron-principal neuron synapses in the amygdala requires nitric oxide signalling. J. Physiol. 2012, 590, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Bloodgood, D.W.; Sugam, J.A.; Holmes, A.; Kash, T.L. Fear extinction requires infralimbic cortex projections to the basolateral amygdala. Transl. Psychiatry 2018, 8, 60. [Google Scholar] [CrossRef]
- Cho, J.H.; Deisseroth, K.; Bolshakov, V.Y. Synaptic encoding of fear extinction in mPFC-amygdala circuits. Neuron 2013, 80, 1491–1507. [Google Scholar] [CrossRef]
- Kim, W.B.; Cho, J.H. Encoding of Discriminative Fear Memory by Input-Specific LTP in the Amygdala. Neuron 2017, 95, 1129–1146.e1125. [Google Scholar] [CrossRef]
- Repa, J.C.; Muller, J.; Apergis, J.; Desrochers, T.M.; Zhou, Y.; LeDoux, J.E. Two different lateral amygdala cell populations contribute to the initiation and storage of memory. Nat. Neurosci. 2001, 4, 724–731. [Google Scholar] [CrossRef]
- Bouton, M.E. Context and behavioral processes in extinction. Learn. Mem. 2004, 11, 485–494. [Google Scholar] [CrossRef]
- Clem, R.L.; Schiller, D. New Learning and Unlearning: Strangers or Accomplices in Threat Memory Attenuation? Trends Neurosci. 2016, 39, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Luchkina, N.V.; Bolshakov, V.Y. Mechanisms of fear learning and extinction: Synaptic plasticity-fear memory connection. Psychopharmacology 2019, 236, 163–182. [Google Scholar] [CrossRef] [PubMed]
- Dunsmoor, J.E.; Niv, Y.; Daw, N.; Phelps, E.A. Rethinking Extinction. Neuron 2015, 88, 47–63. [Google Scholar] [CrossRef]
- Nabavi, S.; Fox, R.; Proulx, C.D.; Lin, J.Y.; Tsien, R.Y.; Malinow, R. Engineering a memory with LTD and LTP. Nature 2014, 511, 348–352. [Google Scholar] [CrossRef]
- Lin, C.H.; Yeh, S.H.; Lu, H.Y.; Gean, P.W. The similarities and diversities of signal pathways leading to consolidation of conditioning and consolidation of extinction of fear memory. J. Neurosci. 2003, 23, 8310–8317. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Lee, C.C.; Gean, P.W. Involvement of a calcineurin cascade in amygdala depotentiation and quenching of fear memory. Mol. Pharmacol. 2003, 63, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Yeh, S.H.; Leu, T.H.; Chang, W.C.; Wang, S.T.; Gean, P.W. Identification of calcineurin as a key signal in the extinction of fear memory. J. Neurosci. 2003, 23, 1574–1579. [Google Scholar] [CrossRef] [PubMed]
- Trouche, S.; Sasaki, J.M.; Tu, T.; Reijmers, L.G. Fear extinction causes target-specific remodeling of perisomatic inhibitory synapses. Neuron 2013, 80, 1054–1065. [Google Scholar] [CrossRef]
- Chhatwal, J.P.; Myers, K.M.; Ressler, K.J.; Davis, M. Regulation of gephyrin and GABAA receptor binding within the amygdala after fear acquisition and extinction. J. Neurosci. 2005, 25, 502–506. [Google Scholar] [CrossRef]
- Gabbott, P.L.; Warner, T.A.; Jays, P.R.; Salway, P.; Busby, S.J. Prefrontal cortex in the rat: Projections to subcortical autonomic, motor, and limbic centers. J. Comp. Neurol. 2005, 492, 145–177. [Google Scholar] [CrossRef]
- McGarry, L.M.; Carter, A.G. Inhibitory Gating of Basolateral Amygdala Inputs to the Prefrontal Cortex. J. Neurosci. 2016, 36, 9391–9406. [Google Scholar] [CrossRef] [PubMed]
- Milad, M.R.; Quirk, G.J. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 2002, 420, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Robles, A.; Vidal-Gonzalez, I.; Santini, E.; Quirk, G.J. Consolidation of fear extinction requires NMDA receptor-dependent bursting in the ventromedial prefrontal cortex. Neuron 2007, 53, 871–880. [Google Scholar] [CrossRef] [PubMed]
- Marek, R.; Jin, J.; Goode, T.D.; Giustino, T.F.; Wang, Q.; Acca, G.M.; Holehonnur, R.; Ploski, J.E.; Fitzgerald, P.J.; Lynagh, T.; et al. Hippocampus-driven feed-forward inhibition of the prefrontal cortex mediates relapse of extinguished fear. Nat. Neurosci. 2018, 21, 384–392. [Google Scholar] [CrossRef]
- Sotres-Bayon, F.; Diaz-Mataix, L.; Bush, D.E.; LeDoux, J.E. Dissociable roles for the ventromedial prefrontal cortex and amygdala in fear extinction: NR2B contribution. Cereb. Cortex 2009, 19, 474–482. [Google Scholar] [CrossRef]
- Sepulveda-Orengo, M.T.; Lopez, A.V.; Soler-Cedeno, O.; Porter, J.T. Fear extinction induces mGluR5-mediated synaptic and intrinsic plasticity in infralimbic neurons. J. Neurosci. 2013, 33, 7184–7193. [Google Scholar] [CrossRef]
- Mueller, D.; Porter, J.T.; Quirk, G.J. Noradrenergic signaling in infralimbic cortex increases cell excitability and strengthens memory for fear extinction. J. Neurosci. 2008, 28, 369–375. [Google Scholar] [CrossRef]
- Herry, C.; Mons, N. Resistance to extinction is associated with impaired immediate early gene induction in medial prefrontal cortex and amygdala. Eur. J. Neurosci. 2004, 20, 781–790. [Google Scholar] [CrossRef]
- Bauer, E.P.; LeDoux, J.E. Heterosynaptic long-term potentiation of inhibitory interneurons in the lateral amygdala. J. Neurosci. 2004, 24, 9507–9512. [Google Scholar] [CrossRef]
- Mahanty, N.K.; Sah, P. Calcium-permeable AMPA receptors mediate long-term potentiation in interneurons in the amygdala. Nature 1998, 394, 683–687. [Google Scholar] [CrossRef]
- Polepalli, J.S.; Sullivan, R.K.; Yanagawa, Y.; Sah, P. A specific class of interneuron mediates inhibitory plasticity in the lateral amygdala. J. Neurosci. 2010, 30, 14619–14629. [Google Scholar] [CrossRef] [PubMed]
- Hobin, J.A.; Goosens, K.A.; Maren, S. Context-dependent neuronal activity in the lateral amygdala represents fear memories after extinction. J. Neurosci. 2003, 23, 8410–8416. [Google Scholar] [CrossRef] [PubMed]
- Santini, E.; Ge, H.; Ren, K.; Pena de Ortiz, S.; Quirk, G.J. Consolidation of fear extinction requires protein synthesis in the medial prefrontal cortex. J. Neurosci. 2004, 24, 5704–5710. [Google Scholar] [CrossRef] [PubMed]
- Duvarci, S.; Mamou, C.B.; Nader, K. Extinction is not a sufficient condition to prevent fear memories from undergoing reconsolidation in the basolateral amygdala. Eur. J. Neurosci. 2006, 24, 249–260. [Google Scholar] [CrossRef]
- Quirk, G.J.; Likhtik, E.; Pelletier, J.G.; Pare, D. Stimulation of medial prefrontal cortex decreases the responsiveness of central amygdala output neurons. J. Neurosci. 2003, 23, 8800–8807. [Google Scholar] [CrossRef]
- Berretta, S.; Pantazopoulos, H.; Caldera, M.; Pantazopoulos, P.; Pare, D. Infralimbic cortex activation increases c-Fos expression in intercalated neurons of the amygdala. Neuroscience 2005, 132, 943–953. [Google Scholar] [CrossRef]
- Amano, T.; Unal, C.T.; Pare, D. Synaptic correlates of fear extinction in the amygdala. Nat. Neurosci. 2010, 13, 489–494. [Google Scholar] [CrossRef]
- Royer, S.; Pare, D. Bidirectional synaptic plasticity in intercalated amygdala neurons and the extinction of conditioned fear responses. Neuroscience 2002, 115, 455–462. [Google Scholar] [CrossRef]
- Whittle, N.; Fadok, J.; MacPherson, K.P.; Nguyen, R.; Botta, P.; Wolff, S.B.E.; Muller, C.; Herry, C.; Tovote, P.; Holmes, A.; et al. Central amygdala micro-circuits mediate fear extinction. Nat. Commun. 2021, 12, 4156. [Google Scholar] [CrossRef]
- Pitkanen, A.; Pikkarainen, M.; Nurminen, N.; Ylinen, A. Reciprocal connections between the amygdala and the hippocampal formation, perirhinal cortex, and postrhinal cortex in rat. A review. Ann. N. Y. Acad. Sci. 2000, 911, 369–391. [Google Scholar] [CrossRef]
- Corcoran, K.A.; Maren, S. Hippocampal inactivation disrupts contextual retrieval of fear memory after extinction. J. Neurosci. 2001, 21, 1720–1726. [Google Scholar] [CrossRef] [PubMed]
- Knapska, E.; Maren, S. Reciprocal patterns of c-Fos expression in the medial prefrontal cortex and amygdala after extinction and renewal of conditioned fear. Learn. Mem. 2009, 16, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Mamiya, N.; Fukushima, H.; Suzuki, A.; Matsuyama, Z.; Homma, S.; Frankland, P.W.; Kida, S. Brain region-specific gene expression activation required for reconsolidation and extinction of contextual fear memory. J. Neurosci. 2009, 29, 402–413. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente, V.; Federman, N.; Fustinana, M.S.; Zalcman, G.; Romano, A. Calcineurin phosphatase as a negative regulator of fear memory in hippocampus: Control on nuclear factor-kappaB signaling in consolidation and reconsolidation. Hippocampus 2014, 24, 1549–1561. [Google Scholar] [CrossRef]
- Fischer, A.; Radulovic, M.; Schrick, C.; Sananbenesi, F.; Godovac-Zimmermann, J.; Radulovic, J. Hippocampal Mek/Erk signaling mediates extinction of contextual freezing behavior. Neurobiol. Learn. Mem. 2007, 87, 149–158. [Google Scholar] [CrossRef]
- Giustino, T.F.; Maren, S. Noradrenergic Modulation of Fear Conditioning and Extinction. Front. Behav. Neurosci. 2018, 12, 43. [Google Scholar] [CrossRef]
- Fitzgerald, P.J.; Giustino, T.F.; Seemann, J.R.; Maren, S. Noradrenergic blockade stabilizes prefrontal activity and enables fear extinction under stress. Proc. Natl. Acad. Sci. USA 2015, 112, E3729–E3737. [Google Scholar] [CrossRef]
- Giustino, T.F.; Seemann, J.R.; Acca, G.M.; Goode, T.D.; Fitzgerald, P.J.; Maren, S. beta-Adrenoceptor Blockade in the Basolateral Amygdala, But Not the Medial Prefrontal Cortex, Rescues the Immediate Extinction Deficit. Neuropsychopharmacology 2017, 42, 2537–2544. [Google Scholar] [CrossRef]
- Salinas-Hernandez, X.I.; Vogel, P.; Betz, S.; Kalisch, R.; Sigurdsson, T.; Duvarci, S. Dopamine neurons drive fear extinction learning by signaling the omission of expected aversive outcomes. eLife 2018, 7, e3881. [Google Scholar] [CrossRef]
- Badrinarayan, A.; Wescott, S.A.; Vander Weele, C.M.; Saunders, B.T.; Couturier, B.E.; Maren, S.; Aragona, B.J. Aversive stimuli differentially modulate real-time dopamine transmission dynamics within the nucleus accumbens core and shell. J. Neurosci. 2012, 32, 15779–15790. [Google Scholar] [CrossRef]
- Luo, R.; Uematsu, A.; Weitemier, A.; Aquili, L.; Koivumaa, J.; McHugh, T.J.; Johansen, J.P. A dopaminergic switch for fear to safety transitions. Nat. Commun. 2018, 9, 2483. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.; Bravo-Rivera, C.; Quirk, G.J. Infralimbic D2 receptors are necessary for fear extinction and extinction-related tone responses. Biol. Psychiatry 2010, 68, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Holtzman-Assif, O.; Laurent, V.; Westbrook, R.F. Blockade of dopamine activity in the nucleus accumbens impairs learning extinction of conditioned fear. Learn. Mem. 2010, 17, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Correia, S.S.; McGrath, A.G.; Lee, A.; Graybiel, A.M.; Goosens, K.A. Amygdala-ventral striatum circuit activation decreases long-term fear. eLife 2016, 5, e12669. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhu, J.J.; Wang, L.; Kan, Y.P.; Liu, Y.M.; Wu, Y.J.; Gu, X.; Yi, X.; Lin, Z.J.; Wang, Q.; et al. Insular cortical circuits as an executive gateway to decipher threat or extinction memory via distinct subcortical pathways. Nat. Commun. 2022, 13, 5540. [Google Scholar] [CrossRef]
- Urcelay, G.P.; Wheeler, D.S.; Miller, R.R. Spacing extinction trials alleviates renewal and spontaneous recovery. Learn. Behav. 2009, 37, 60–73. [Google Scholar] [CrossRef]
- Clem, R.L.; Huganir, R.L. Calcium-permeable AMPA receptor dynamics mediate fear memory erasure. Science 2010, 330, 1108–1112. [Google Scholar] [CrossRef]
- An, B.; Kim, J.; Park, K.; Lee, S.; Song, S.; Choi, S. Amount of fear extinction changes its underlying mechanisms. eLife 2017, 6, e25224. [Google Scholar] [CrossRef]
- Gogolla, N.; Caroni, P.; Luthi, A.; Herry, C. Perineuronal nets protect fear memories from erasure. Science 2009, 325, 1258–1261. [Google Scholar] [CrossRef]
- Kim, J.H.; Richardson, R. New findings on extinction of conditioned fear early in development: Theoretical and clinical implications. Biol. Psychiatry 2010, 67, 297–303. [Google Scholar] [CrossRef]
- Rizley, R.C.; Rescorla, R.A. Associations in second-order conditioning and sensory preconditioning. J. Comp. Physiol. Psychol. 1972, 81, 1–11. [Google Scholar] [CrossRef]
- Debiec, J.; Doyere, V.; Nader, K.; Ledoux, J.E. Directly reactivated, but not indirectly reactivated, memories undergo reconsolidation in the amygdala. Proc. Natl. Acad. Sci. USA 2006, 103, 3428–3433. [Google Scholar] [CrossRef] [PubMed]
- Rescorla, R.A. Effect of US habituation following conditioning. J. Comp. Physiol. Psychol. 1973, 82, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Holmes, N.M.; Cai, S.Y.; Lay, B.P.; Watts, N.R.; Westbrook, R.F. Extinguished second-order conditioned fear responses are renewed but not reinstated. J. Exp. Psychol. Anim. Learn. Cogn. 2014, 40, 440–456. [Google Scholar] [CrossRef]
- Luettgau, L.; Porcu, E.; Tempelmann, C.; Jocham, G. Reinstatement of Cortical Outcome Representations during Higher-Order Learning. Cereb. Cortex 2021, 32, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, J.C.; Davis, M. Using pavlovian higher-order conditioning paradigms to investigate the neural substrates of emotional learning and memory. Learn. Mem. 2000, 7, 257–266. [Google Scholar] [CrossRef]
- Parkes, S.L.; Westbrook, R.F. Role of the basolateral amygdala and NMDA receptors in higher-order conditioned fear. Rev. Neurosci. 2011, 22, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Rescorla, R.A.; Furrow, D.R. Stimulus similarity as a determinant of Pavlovian conditioning. J. Exp. Psychol. Anim. Behav. Process. 1977, 3, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Rescorla, R.A. Simultaneous second-order conditioning produces S-S learning in conditioned suppression. J. Exp. Psychol. Anim. Behav. Process. 1982, 8, 23–32. [Google Scholar] [CrossRef]
- Pearce, J.M. Evaluation and development of a connectionist theory of configural learning. Anim. Learn. Behav. 2002, 30, 73–95. [Google Scholar] [CrossRef]
- Lingawi, N.W.; Laurent, V.; Westbrook, R.F.; Holmes, N.M. Acquisition and extinction of second-order context conditioned fear: Role of the amygdala. Neurobiol. Learn. Mem. 2021, 183, 107485. [Google Scholar] [CrossRef] [PubMed]
- Holmes, N.M.; Parkes, S.L.; Killcross, A.S.; Westbrook, R.F. The basolateral amygdala is critical for learning about neutral stimuli in the presence of danger, and the perirhinal cortex is critical in the absence of danger. J. Neurosci. 2013, 33, 13112–13125. [Google Scholar] [CrossRef] [PubMed]
- Parkes, S.L.; Westbrook, R.F. The basolateral amygdala is critical for the acquisition and extinction of associations between a neutral stimulus and a learned danger signal but not between two neutral stimuli. J. Neurosci. 2010, 30, 12608–12618. [Google Scholar] [CrossRef] [PubMed]
- Todd, T.P.; Huszar, R.; DeAngeli, N.E.; Bucci, D.J. Higher-order conditioning and the retrosplenial cortex. Neurobiol. Learn. Mem. 2016, 133, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Lay, B.P.P.; Westbrook, R.F.; Glanzman, D.L.; Holmes, N.M. Commonalities and Differences in the Substrates Underlying Consolidation of First- and Second-Order Conditioned Fear. J. Neurosci. 2018, 38, 1926–1941. [Google Scholar] [CrossRef]
- Gewirtz, J.C.; Davis, M. Second-order fear conditioning prevented by blocking NMDA receptors in amygdala. Nature 1997, 388, 471–474. [Google Scholar] [CrossRef]
- Williams-Spooner, M.J.; Delaney, A.J.; Westbrook, R.F.; Holmes, N.M. Prediction Error Determines Whether NMDA Receptors in the Basolateral Amygdala Complex Are Involved in Pavlovian Fear Conditioning. J. Neurosci. 2022, 42, 4360–4379. [Google Scholar] [CrossRef]
- Leidl, D.M.; Lay, B.P.P.; Chakouch, C.; Westbrook, R.F.; Holmes, N.M. Protein synthesis in the basolateral amygdala complex is required for consolidation of a first-order fear memory, but not for consolidation of a higher-order fear memory. Neurobiol. Learn. Mem. 2018, 153, 153–165. [Google Scholar] [CrossRef]
- Williams-Spooner, M.J.; Westbrook, R.F.; Holmes, N.M. The Conditions under Which Consolidation of Serial-Order Conditioned Fear Requires De Novo Protein Synthesis in the Basolateral Amygdala Complex. J. Neurosci. 2019, 39, 7357–7368. [Google Scholar] [CrossRef]
- Nader, K.; LeDoux, J.E. Inhibition of the mesoamygdala dopaminergic pathway impairs the retrieval of conditioned fear associations. Behav. Neurosci. 1999, 113, 891–901. [Google Scholar] [CrossRef]
- Holmes, N.M.; Raipuria, M.; Qureshi, O.A.; Killcross, S.; Westbrook, F. Danger Changes the Way the Mammalian Brain Stores Information about Innocuous Events: A Study of Sensory Preconditioning in Rats. eNeuro 2018, 5, ENEURO.0381-17.2017. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.S.; Westbrook, R.F.; Holmes, N.M. ‘Online’ integration of sensory and fear memories in the rat medial temporal lobe. eLife 2019, 8, e47085. [Google Scholar] [CrossRef] [PubMed]
- Schultz, W.; Dickinson, A. Neuronal coding of prediction errors. Annu. Rev. Neurosci. 2000, 23, 473–500. [Google Scholar] [CrossRef] [PubMed]
- Clark, A. Whatever next? Predictive brains, situated agents, and the future of cognitive science. Behav. Brain Sci. 2013, 36, 181–204. [Google Scholar] [CrossRef]
- Hutchinson, J.B.; Barrett, L.F. The power of predictions: An emerging paradigm for psychological research. Curr. Dir. Psychol. Sci. 2019, 28, 280–291. [Google Scholar] [CrossRef]
- Keller, G.B.; Mrsic-Flogel, T.D. Predictive Processing: A Canonical Cortical Computation. Neuron 2018, 100, 424–435. [Google Scholar] [CrossRef]
- Harley, C.W. Norepinephrine and dopamine as learning signals. Neural Plast. 2004, 11, 191–204. [Google Scholar] [CrossRef]
- Dayan, P.; Yu, A.J. Phasic norepinephrine: A neural interrupt signal for unexpected events. Network 2006, 17, 335–350. [Google Scholar] [CrossRef]
- Jordan, R.; Keller, G.B. The locus coeruleus broadcasts prediction errors across the cortex to promote sensorimotor plasticity. eLife 2023, 12, e85111. [Google Scholar] [CrossRef]
- Mather, M.; Clewett, D.; Sakaki, M.; Harley, C.W. Norepinephrine ignites local hotspots of neuronal excitation: How arousal amplifies selectivity in perception and memory. Behav. Brain Sci. 2016, 39, e200. [Google Scholar] [CrossRef]
- Phillips, W.A.; Larkum, M.E.; Harley, C.W.; Silverstein, S.M. The effects of arousal on apical amplification and conscious state. Neurosci. Conscious. 2016, 2016, niw015. [Google Scholar] [CrossRef]
- Omoluabi, T.; Power, K.D.; Sepahvand, T.; Yuan, Q. Phasic and Tonic Locus Coeruleus Stimulation Associated Valence Learning Engages Distinct Adrenoceptors in the Rat Basolateral Amygdala. Front. Cell Neurosci. 2022, 16, 886803. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Massaeli, F.; Power, K.D.; Omoluabi, T.; Torraville, S.E.; Pritchett, J.B.; Sepahvand, T.; Strong, V.D.; Reinhardt, C.; Chen, X.; et al. Locus Coeruleus Activation Patterns Differentially Modulate Odor Discrimination Learning and Odor Valence in Rats. Cereb. Cortex Commun. 2021, 2, tgab026. [Google Scholar] [CrossRef] [PubMed]
- Quirk, G.J.; Mueller, D. Neural mechanisms of extinction learning and retrieval. Neuropsychopharmacology 2008, 33, 56–72. [Google Scholar] [CrossRef] [PubMed]
- Orsini, C.A.; Maren, S. Neural and cellular mechanisms of fear and extinction memory formation. Neurosci. Biobehav. Rev. 2012, 36, 1773–1802. [Google Scholar] [CrossRef]
- Hermann, A.; Stark, R.; Blecker, C.R.; Milad, M.R.; Merz, C.J. Brain structural connectivity and context-dependent extinction memory. Hippocampus 2017, 27, 883–889. [Google Scholar] [CrossRef]
- Walker, D.L.; Ressler, K.J.; Lu, K.T.; Davis, M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of D-cycloserine as assessed with fear-potentiated startle in rats. J. Neurosci. 2002, 22, 2343–2351. [Google Scholar] [CrossRef] [PubMed]
- Bouton, M.E.; Vurbic, D.; Woods, A.M. D-cycloserine facilitates context-specific fear extinction learning. Neurobiol. Learn. Mem. 2008, 90, 504–510. [Google Scholar] [CrossRef]
- Davis, M.; Ressler, K.; Rothbaum, B.O.; Richardson, R. Effects of D-cycloserine on extinction: Translation from preclinical to clinical work. Biol. Psychiatry 2006, 60, 369–375. [Google Scholar] [CrossRef]
- Ressler, K.J.; Rothbaum, B.O.; Tannenbaum, L.; Anderson, P.; Graap, K.; Zimand, E.; Hodges, L.; Davis, M. Cognitive enhancers as adjuncts to psychotherapy: Use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch. Gen. Psychiatry 2004, 61, 1136–1144. [Google Scholar] [CrossRef]
- Haaker, J.; Lonsdorf, T.B.; Kalisch, R. Effects of post-extinction l-DOPA administration on the spontaneous recovery and reinstatement of fear in a human fMRI study. Eur. Neuropsychopharmacol. 2015, 25, 1544–1555. [Google Scholar] [CrossRef]
- Kaczorowski, C.C.; Davis, S.J.; Moyer, J.R., Jr. Aging redistributes medial prefrontal neuronal excitability and impedes extinction of trace fear conditioning. Neurobiol. Aging 2012, 33, 1744–1757. [Google Scholar] [CrossRef] [PubMed]
- Oler, J.A.; Markus, E.J. Age-related deficits on the radial maze and in fear conditioning: Hippocampal processing and consolidation. Hippocampus 1998, 8, 402–415. [Google Scholar] [CrossRef]
- Hernandez, C.M.; Jackson, N.L.; Hernandez, A.R.; McMahon, L.L. Impairments in Fear Extinction Memory and Basolateral Amygdala Plasticity in the TgF344-AD Rat Model of Alzheimer’s Disease Are Distinct from Nonpathological Aging. eNeuro 2022, 9, ENEURO.0181-22.2022. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Garofalo, S.; di Pellegrino, G. Context-dependent extinction of threat memories: Influences of healthy aging. Sci. Rep. 2018, 8, 12592. [Google Scholar] [CrossRef]
- Nasrouei, S.; Rattel, J.A.; Liedlgruber, M.; Marksteiner, J.; Wilhelm, F.H. Fear acquisition and extinction deficits in amnestic mild cognitive impairment and early Alzheimer’s disease. Neurobiol. Aging 2020, 87, 26–34. [Google Scholar] [CrossRef]
- Rainer, C.; Nasrouei, S.; Tschofen, S.; Bliem, H.R.; Wilhelm, F.H.; Marksteiner, J. Fear acquisition and extinction in elderly patients with depression. J. Affect. Disord. 2020, 276, 197–204. [Google Scholar] [CrossRef]
- Yehuda, R.; Golier, J.A.; Tischler, L.; Stavitsky, K.; Harvey, P.D. Learning and memory in aging combat veterans with PTSD. J. Clin. Exp. Neuropsychol. 2005, 27, 504–515. [Google Scholar] [CrossRef]
- Bremner, J.D.; Vermetten, E.; Schmahl, C.; Vaccarino, V.; Vythilingam, M.; Afzal, N.; Grillon, C.; Charney, D.S. Positron emission tomographic imaging of neural correlates of a fear acquisition and extinction paradigm in women with childhood sexual-abuse-related post-traumatic stress disorder. Psychol. Med. 2005, 35, 791–806. [Google Scholar] [CrossRef]
- Lanius, R.A.; Williamson, P.C.; Densmore, M.; Boksman, K.; Gupta, M.A.; Neufeld, R.W.; Gati, J.S.; Menon, R.S. Neural correlates of traumatic memories in posttraumatic stress disorder: A functional MRI investigation. Am. J. Psychiatry 2001, 158, 1920–1922. [Google Scholar] [CrossRef]
- Orr, S.P.; Metzger, L.J.; Lasko, N.B.; Macklin, M.L.; Peri, T.; Pitman, R.K. De novo conditioning in trauma-exposed individuals with and without posttraumatic stress disorder. J. Abnorm. Psychol. 2000, 109, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Wessa, M.; Flor, H. Failure of extinction of fear responses in posttraumatic stress disorder: Evidence from second-order conditioning. Am. J. Psychiatry 2007, 164, 1684–1692. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.X.; Yin, B. A new understanding of the cognitive reappraisal technique: An extension based on the schema theory. Front. Behav. Neurosci. 2023, 17, 1174585. [Google Scholar] [CrossRef] [PubMed]
- Cutuli, D. Cognitive reappraisal and expressive suppression strategies role in the emotion regulation: An overview on their modulatory effects and neural correlates. Front. Syst. Neurosci. 2014, 8, 175. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sepahvand, T.; Power, K.D.; Qin, T.; Yuan, Q. The Basolateral Amygdala: The Core of a Network for Threat Conditioning, Extinction, and Second-Order Threat Conditioning. Biology 2023, 12, 1274. https://doi.org/10.3390/biology12101274
Sepahvand T, Power KD, Qin T, Yuan Q. The Basolateral Amygdala: The Core of a Network for Threat Conditioning, Extinction, and Second-Order Threat Conditioning. Biology. 2023; 12(10):1274. https://doi.org/10.3390/biology12101274
Chicago/Turabian StyleSepahvand, Tayebeh, Kyron D. Power, Tian Qin, and Qi Yuan. 2023. "The Basolateral Amygdala: The Core of a Network for Threat Conditioning, Extinction, and Second-Order Threat Conditioning" Biology 12, no. 10: 1274. https://doi.org/10.3390/biology12101274