
Making the Most of Its Short Reads: A Bioinformatics Workflow for Analysing the Short-Read-Only Data of Leishmania orientalis (Formerly Named Leishmania siamensis) Isolate PCM2 in Thailand

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Culture of Leishmania orientalis Isolate PCM2
2.2. Genomic DNA Preparation
2.3. Quality Check and Processing of the Raw Sequence Reads
2.4. Bioinformatic Workflow for Assembling the Genomic Reads of Leishmania orientalis Isolate PCM2 by Hybrid Methods and Salvaging the Unmapped Reads
2.5. Quality Examination of L. orientalis PCM2 Genomic Contigs by Genomic Comparison with the Reference Genomes
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M.; Team, W.L.C. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Lainson, R.; Ward, R.; Shaw, J. Leishmania in phlebotomid sandflies: VI. Importance of hindgut development in distinguishing between parasites of the Leishmania mexicana and L. braziliensis complexes. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1977, 199, 309–320. [Google Scholar]
- Murray, H.W.; Berman, J.D.; Davies, C.R.; Saravia, N.G. Advances in leishmaniasis. Lancet 2005, 366, 1561–1577. [Google Scholar] [CrossRef]
- Steverding, D. The history of leishmaniasis. Parasites Vectors 2017, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Belo, V.S.; Struchiner, C.J.; Barbosa, D.S.; Nascimento, B.W.L.; Horta, M.A.P.; da Silva, E.S.; Werneck, G.L. Risk factors for adverse prognosis and death in American visceral leishmaniasis: A meta-analysis. PLoS Negl. Trop. Dis. 2014, 8, e2982. [Google Scholar] [CrossRef] [PubMed]
- Banuls, A.-L.; Bastien, P.; Pomares, C.; Arevalo, J.; Fisa, R.; Hide, M. Clinical pleiomorphism in human leishmaniases, with special mention of asymptomatic infection. Clin. Microbiol. Infect. 2011, 17, 1451–1461. [Google Scholar] [CrossRef]
- Ameen, M. Cutaneous leishmaniasis: Advances in disease pathogenesis, diagnostics and therapeutics. Clin. Exp. Dermatol. Clin. Dermatol. 2010, 35, 699–705. [Google Scholar] [CrossRef]
- de Oliveira Guerra, J.A.; Prestes, S.R.; Silveira, H.; Câmara, L.I.d.A.R.; Gama, P.; Moura, A.; Amato, V.; Barbosa, M.d.G.V.; de Lima Ferreira, L.C. Mucosal leishmaniasis caused by Leishmania (Viannia) braziliensis and Leishmania (Viannia) guyanensis in the Brazilian Amazon. PLoS Negl. Trop. Dis. 2011, 5, e980. [Google Scholar]
- Viriyavejakul, P.; Viravan, C.; Riganti, M.; Punpoowong, B. Imported cutaneous leishmaniasis in Thailand. Southeast Asian J. Trop. Med. Public Health 1997, 28, 558–562. [Google Scholar]
- Suttinont, P.; Thammanichanont, C.; Chantarakul, N. Visceral leishmaniasis: A case report. Southeast Asian J. Trop. Med. Public Health 1987, 18, 103–106. [Google Scholar]
- Sukmee, T.; Siripattanapipong, S.; Mungthin, M.; Worapong, J.; Rangsin, R.; Samung, Y.; Kongkaew, W.; Bumrungsana, K.; Chanachai, K.; Apiwathanasorn, C. A suspected new species of Leishmania, the causative agent of visceral leishmaniasis in a Thai patient. Int. J. Parasitol. 2008, 38, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Jariyapan, N.; Daroontum, T.; Jaiwong, K.; Chanmol, W.; Intakhan, N.; Sor-Suwan, S.; Siriyasatien, P.; Somboon, P.; Bates, M.D.; Bates, P.A. Leishmania (Mundinia) orientalis n. sp.(Trypanosomatidae), a parasite from Thailand responsible for localised cutaneous leishmaniasis. Parasites Vectors 2018, 11, 351. [Google Scholar] [CrossRef] [PubMed]
- Leelayoova, S.; Siripattanapipong, S.; Hitakarun, A.; Kato, H.; Tan-ariya, P.; Siriyasatien, P.; Osatakul, S.; Mungthin, M. Multilocus characterization and phylogenetic analysis of Leishmania siamensis isolated from autochthonous visceral leishmaniasis cases, southern Thailand. BMC Microbiol. 2013, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Müller, N.; Welle, M.; Lobsiger, L.; Stoffel, M.H.; Boghenbor, K.K.; Hilbe, M.; Gottstein, B.; Frey, C.F.; Geyer, C.; von Bomhard, W. Occurrence of Leishmania sp. in cutaneous lesions of horses in Central Europe. Vet. Parasitol. 2009, 166, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Lobsiger, L.; Müller, N.; Schweizer, T.; Frey, C.; Wiederkehr, D.; Zumkehr, B.; Gottstein, B. An autochthonous case of cutaneous bovine leishmaniasis in Switzerland. Vet. Parasitol. 2010, 169, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Reuss, S.M.; Dunbar, M.D.; Mays, M.B.C.; Owen, J.L.; Mallicote, M.F.; Archer, L.L.; Wellehan, J.F., Jr. Autochthonous Leishmania siamensis in horse, Florida, USA. Emerg. Infect. Dis. 2012, 18, 1545. [Google Scholar] [CrossRef]
- Chusri, S.; Thammapalo, S.; Silpapojakul, K.; Siriyasatien, P. Animal reservoirs and potential vectors of Leishmania siamensis in southern Thailand. Southeast Asian J. Trop. Med. Public Health 2014, 45, 13. [Google Scholar]
- Mukhtar, M.M.; Sharief, A.; El Saffi, S.; Harith, A.; Higazzi, T.; Adam, A.; Abdalla, H.S. Detection of antibodies to Leishmania donovani in animals in a kala-azar endemic region in eastern Sudan: A preliminary report. Trans. R. Soc. Trop. Med. Hyg. 2000, 94, 33–36. [Google Scholar] [CrossRef]
- Rohousova, I.; Talmi-Frank, D.; Kostalova, T.; Polanska, N.; Lestinova, T.; Kassahun, A.; Yasur-Landau, D.; Maia, C.; King, R.; Votypka, J. Exposure to Leishmania spp. and sand flies in domestic animals in northwestern Ethiopia. Parasites Vectors 2015, 8, 360. [Google Scholar] [CrossRef]
- Polseela, R.; Vitta, A.; Nateeworanart, S.; Apiwathnasorn, C. Distribution of cave-dwelling phlebotomine sand flies and their nocturnal and diurnal activity in Phitsanulok Province, Thailand. Southeast Asian J. Trop. Med. Public Health 2011, 42, 1395–1404. [Google Scholar]
- Manomat, J.; Leelayoova, S.; Bualert, L.; Tan-Ariya, P.; Siripattanapipong, S.; Mungthin, M.; Naaglor, T.; Piyaraj, P. Prevalence and risk factors associated with Leishmania infection in Trang Province, southern Thailand. PLoS Negl. Trop. Dis. 2017, 11, e0006095. [Google Scholar] [CrossRef] [PubMed]
- Lindoso, J.A.L.; Cunha, M.A.; Queiroz, I.T.; Moreira, C.H.V. Leishmaniasis–HIV coinfection: Current challenges. HIV/AIDS 2016, 8, 147. [Google Scholar]
- Faraut-Gambarelli, F.; Piarroux, R.; Deniau, M.; Giusiano, B.; Marty, P.; Michel, G.; Faugère, B.; Dumon, H. In vitro and in vivo resistance of Leishmania infantum to meglumine antimoniate: A study of 37 strains collected from patients with visceral leishmaniasis. Antimicrob. Agents Chemother. 1997, 41, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Sundar, S.; Fairlamb, A.H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [Google Scholar] [CrossRef]
- Sundar, S.; Sinha, P.K.; Rai, M.; Verma, D.K.; Nawin, K.; Alam, S.; Chakravarty, J.; Vaillant, M.; Verma, N.; Pandey, K. Comparison of short-course multidrug treatment with standard therapy for visceral leishmaniasis in India: An open-label, non-inferiority, randomised controlled trial. Lancet 2011, 377, 477–486. [Google Scholar] [CrossRef]
- Musa, A.; Khalil, E.; Hailu, A.; Olobo, J.; Balasegaram, M.; Omollo, R.; Edwards, T.; Rashid, J.; Mbui, J.; Musa, B. Sodium stibogluconate (SSG) & paromomycin combination compared to SSG for visceral leishmaniasis in East Africa: A randomised controlled trial. PLoS Negl. Trop. Dis. 2012, 6, e1674. [Google Scholar]
- van Griensven, J.; Gadisa, E.; Aseffa, A.; Hailu, A.; Beshah, A.M.; Diro, E. Treatment of cutaneous leishmaniasis caused by Leishmania aethiopica: A systematic review. PLoS Negl. Trop. Dis. 2016, 10, e0004495. [Google Scholar] [CrossRef]
- Lindoso, J.A.; Cota, G.F.; da Cruz, A.M.; Goto, H.; Maia-Elkhoury, A.N.S.; Romero, G.A.S.; de Sousa-Gomes, M.L.; Santos-Oliveira, J.R.; Rabello, A. Visceral leishmaniasis and HIV coinfection in Latin America. PLoS Negl. Trop. Dis. 2014, 8, e3136. [Google Scholar] [CrossRef] [Green Version]
- Sundar, S. Drug resistance in Indian visceral leishmaniasis. Trop. Med. Int. Health 2001, 6, 849–854. [Google Scholar] [CrossRef]
- Vanaerschot, M.; Dumetz, F.; Roy, S.; Ponte-Sucre, A.; Arevalo, J.; Dujardin, J.-C. Treatment failure in leishmaniasis: Drug-resistance or another (epi-) phenotype? Expert Rev. Anti-Infect. Ther. 2014, 12, 937–946. [Google Scholar] [CrossRef]
- Myler, P.J.; Audleman, L.; DeVos, T.; Hixson, G.; Kiser, P.; Lemley, C.; Magness, C.; Rickel, E.; Sisk, E.; Sunkin, S. Leishmania major Friedlin chromosome 1 has an unusual distribution of protein-coding genes. Proc. Natl. Acad. Sci. USA 1999, 96, 2902–2906. [Google Scholar] [CrossRef] [PubMed]
- Berriman, M.; Ghedin, E.; Hertz-Fowler, C.; Blandin, G.; Renauld, H.; Bartholomeu, D.C.; Lennard, N.J.; Caler, E.; Hamlin, N.E.; Haas, B. The genome of the African trypanosome Trypanosoma brucei. Science 2005, 309, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Peacock, C.S.; Seeger, K.; Harris, D.; Murphy, L.; Ruiz, J.C.; Quail, M.A.; Peters, N.; Adlem, E.; Tivey, A.; Aslett, M. Comparative genomic analysis of three Leishmania species that cause diverse human disease. Nat. Genet. 2007, 39, 839–847. [Google Scholar] [CrossRef]
- Cuypers, B.; Dumetz, F.; Meysman, P.; Laukens, K.; De Muylder, G.; Dujardin, J.-C.; Domagalska, M.A. The absence of C-5 DNA methylation in Leishmania donovani allows DNA enrichment from complex samples. Microorganisms 2020, 8, 1252. [Google Scholar] [CrossRef] [PubMed]
- Wincker, P.; Ravel, C.; Blaineau, C.; Pages, M.; Jauffret, Y.; Dedet, J.-P.; Bastien, P. The Leishmania genome comprises 36 chromosomes conserved across widely divergent human pathogenic species. Nucleic Acids Res. 1996, 24, 1688–1694. [Google Scholar] [CrossRef]
- Britto, C.; Ravel, C.; Bastien, P.; Blaineau, C.; Pagès, M.; Dedet, J.-P.; Wincker, P. Conserved linkage groups associated with large-scale chromosomal rearrangements between Old World and New World Leishmania genomes. Gene 1998, 222, 107–117. [Google Scholar] [CrossRef]
- Proudfoot, L.; O’Donnell, C.A.; Liew, F.Y. Glycoinositolphospholipids of Leishmania major inhibit nitric oxide synthesis and reduce leishmanicidal activity in murine macrophages. Eur. J. Immunol. 1995, 25, 745–750. [Google Scholar] [CrossRef]
- Argueta-Donohué, J.; Carrillo, N.; Valdés-Reyes, L.; Zentella, A.; Aguirre-García, M.; Becker, I.; Gutiérrez-Kobeh, L. Leishmania mexicana: Participation of NF-κB in the differential production of IL-12 in dendritic cells and monocytes induced by lipophosphoglycan (LPG). Exp. Parasitol. 2008, 120, 1–9. [Google Scholar] [CrossRef]
- Peters, C.; Stierhof, Y.-D.; Ilg, n.T. Proteophosphoglycan secreted by Leishmania mexicana amastigotes causes vacuole formation in macrophages. Infect. Immun. 1997, 65, 783–786. [Google Scholar] [CrossRef]
- Carvalho, L.P.; Passos, S.; Dutra, W.O.; Soto, M.; Alonso, C.; Gollob, K.; Carvalho, E.; Ribeiro de Jesus, A. Effect of LACK and KMP11 on IFN-γ Production by Peripheral Blood Mononuclear Cells from Cutaneous and Mucosal Leishmaniasis Patients. Scand. J. Immunol. 2005, 61, 337–342. [Google Scholar] [CrossRef]
- Almeida, M.S.; Pereira, B.A.S.; Guimarães, M.L.R.; Alves, C.R. Proteinases as virulence factors in Leishmania spp. infection in mammals. Parasites Vectors 2012, 5, 160. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.B.; Hilley, J.D.; Dickens, N.J.; Wilkes, J.; Bates, P.A.; Depledge, D.P.; Harris, D.; Her, Y.; Herzyk, P.; Imamura, H. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011, 21, 2129–2142. [Google Scholar] [CrossRef] [PubMed]
- Ivens, A.C.; Peacock, C.S.; Worthey, E.A.; Murphy, L.; Aggarwal, G.; Berriman, M.; Sisk, E.; Rajandream, M.-A.; Adlem, E.; Aert, R. The genome of the kinetoplastid parasite, Leishmania major. Science 2005, 309, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Worthey, E.; Martinez-Calvillo, S.; Schnaufer, A.; Aggarwal, G.; Cawthra, J.; Fazelinia, G.; Fong, C.; Fu, G.; Hassebrock, M.; Hixson, G. Leishmania major chromosome 3 contains two long convergent polycistronic gene clusters separated by a tRNA gene. Nucleic Acids Res. 2003, 31, 4201–4210. [Google Scholar] [CrossRef]
- Almutairi, H.; Urbaniak, M.D.; Bates, M.D.; Jariyapan, N.; Al-Salem, W.S.; Dillon, R.J.; Bates, P.A.; Gatherer, D. Chromosome-Scale Assembly of the Complete Genome Sequence of Leishmania (Mundinia) martiniquensis, Isolate LSCM1, Strain LV760. Microbiol. Resour. Announc. 2021, 10, e00058-21. [Google Scholar] [CrossRef]
- Almutairi, H.; Urbaniak, M.D.; Bates, M.D.; Jariyapan, N.; Kwakye-Nuako, G.; Thomaz Soccol, V.; Al-Salem, W.S.; Dillon, R.J.; Bates, P.A.; Gatherer, D. Chromosome-scale genome sequencing, assembly and annotation of six genomes from subfamily Leishmaniinae. Sci. Data 2021, 8, 1–9. [Google Scholar] [CrossRef]
- Almutairi, H.; Urbaniak, M.D.; Bates, M.D.; Jariyapan, N.; Kwakye-Nuako, G.; Thomaz-Soccol, V.; Al-Salem, W.S.; Dillon, R.J.; Bates, P.A.; Gatherer, D. LGAAP: Leishmaniinae genome assembly and annotation pipeline. Microbiol. Resour. Announc. 2021, 10, e00439-21. [Google Scholar] [CrossRef]
- Almutairi, H.; Urbaniak, M.D.; Bates, M.D.; Jariyapan, N.; Al-Salem, W.S.; Dillon, R.J.; Bates, P.A.; Gatherer, D. Chromosome-Scale Assembly of the Complete Genome Sequence of Leishmania (Mundinia) orientalis, Isolate LSCM4, Strain LV768. Microbiol. Resour. Announc. 2021, 10, e00574-21. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics; Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes de novo assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Zimin, A.V.; Marçais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA genome assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Keller, O.; Kollmar, M.; Stanke, M.; Waack, S. A novel hybrid gene prediction method employing protein multiple sequence alignments. Bioinformatics 2011, 27, 757–763. [Google Scholar] [CrossRef]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19, ii215–ii225. [Google Scholar] [CrossRef]
- Stanke, M.; Diekhans, M.; Baertsch, R.; Haussler, D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 2008, 24, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.F.; Waterman, M.S. Identification of common molecular subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Gel, B.; Serra, E. karyoploteR: An R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 2017, 33, 3088–3090. [Google Scholar] [CrossRef]
- Stanke, M.; Morgenstern, B. AUGUSTUS: A web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Res. 2005, 33, W465–W467. [Google Scholar] [CrossRef]
- Koskinen, J.P.; Holm, L. SANS: High-throughput retrieval of protein sequences allowing 50% mismatches. Bioinformatics 2012, 28, i438–i443. [Google Scholar] [CrossRef]
- Radivojac, P.; Clark, W.T.; Oron, T.R.; Schnoes, A.M.; Wittkop, T.; Sokolov, A.; Graim, K.; Funk, C.; Verspoor, K.; Ben-Hur, A. A large-scale evaluation of computational protein function prediction. Nat. Methods 2013, 10, 221–227. [Google Scholar] [CrossRef]
- Koskinen, P.; Törönen, P.; Nokso-Koivisto, J.; Holm, L. PANNZER: High-throughput functional annotation of uncharacterized proteins in an error-prone environment. Bioinformatics 2015, 31, 1544–1552. [Google Scholar] [CrossRef]
- Somervuo, P.; Holm, L. SANSparallel: Interactive homology search against Uniprot. Nucleic Acids Res. 2015, 43, W24–W29. [Google Scholar] [CrossRef]
- Törönen, P.; Medlar, A.; Holm, L. PANNZER2: A rapid functional annotation web server. Nucleic Acids Res. 2018, 46, W84–W88. [Google Scholar] [CrossRef]
- Törönen, P.; Holm, L. PANNZER—A practical tool for protein function prediction. Protein Sci. 2021, 31, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Sayols, S. rrvgo: A Bioconductor Package to Reduce and Visualize Gene Ontology Terms. 2020. Available online: https://bioconductor.org/packages/release/bioc/html/rrvgo.html (accessed on 20 July 2022).
- Leelayoova, S.; Siripattanapipong, S.; Manomat, J.; Piyaraj, P.; Tan-Ariya, P.; Bualert, L.; Mungthin, M. Leishmaniasis in Thailand: A review of causative agents and situations. Am. J. Trop. Med. Hyg. 2017, 96, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Patiño, L.H.; Muñoz, M.; Pavia, P.; Muskus, C.; Shaban, M.; Paniz-Mondolfi, A.; Ramírez, J.D. Filling the gaps in Leishmania naiffi and Leishmania guyanensis genome plasticity. G3 2022, 12, jkab377. [Google Scholar] [CrossRef]
- Coughlan, S.; Taylor, A.S.; Feane, E.; Sanders, M.; Schonian, G.; Cotton, J.A.; Downing, T. Leishmania naiffi and Leishmania guyanensis reference genomes highlight genome structure and gene evolution in the Viannia subgenus. R. Soc. Open Sci. 2018, 5, 172212. [Google Scholar] [CrossRef]
- Ravel, C.; Dubessay, P.; Bastien, P.; Blackwell, J.M.; Ivens, A.C. The complete chromosomal organization of the reference strain of the Leishmania genome project, L. major Friedlin’. Parasitol. Today 1998, 14, 301–303. [Google Scholar] [CrossRef]
- Dubessay, P.; Ravel, C.; Bastien, P.; Stuart, K.; Dedet, J.-P.; Blaineau, C.; Pagès, M. Mitotic stability of a coding DNA sequence-free version of Leishmania major chromosome 1 generated by targeted chromosome fragmentation. Gene 2002, 289, 151–159. [Google Scholar] [CrossRef]
- Cruz, A.K.; Titus, R.; Beverley, S.M. Plasticity in chromosome number and testing of essential genes in Leishmania by targeting. Proc. Natl. Acad. Sci. USA 1993, 90, 1599–1603. [Google Scholar] [CrossRef]
- Martínez-Calvillo, S.; Stuart, K.; Myler, P.J. Ploidy changes associated with disruption of two adjacent genes on Leishmania major chromosome 1. Int. J. Parasitol. 2005, 35, 419–429. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
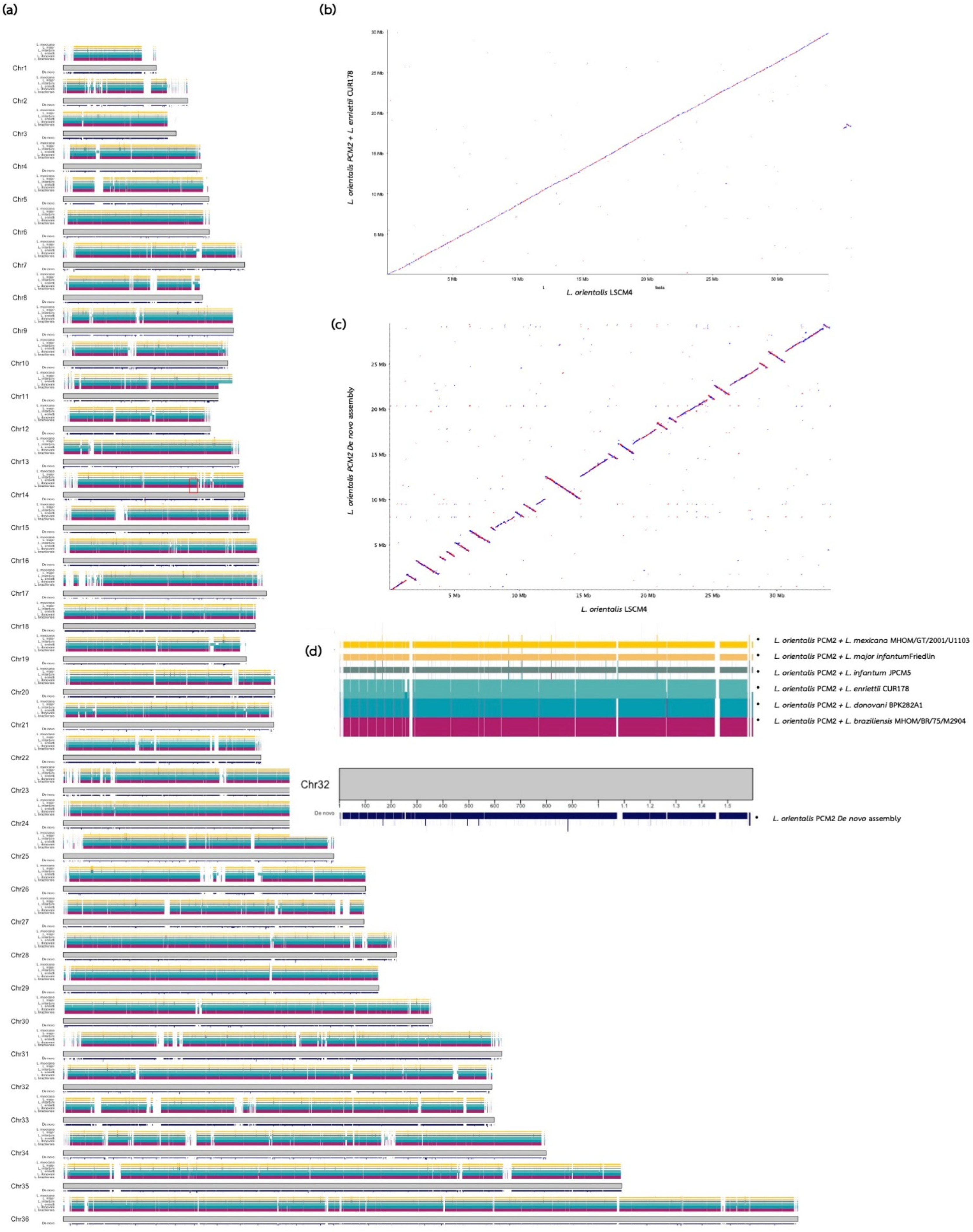{kind=link}
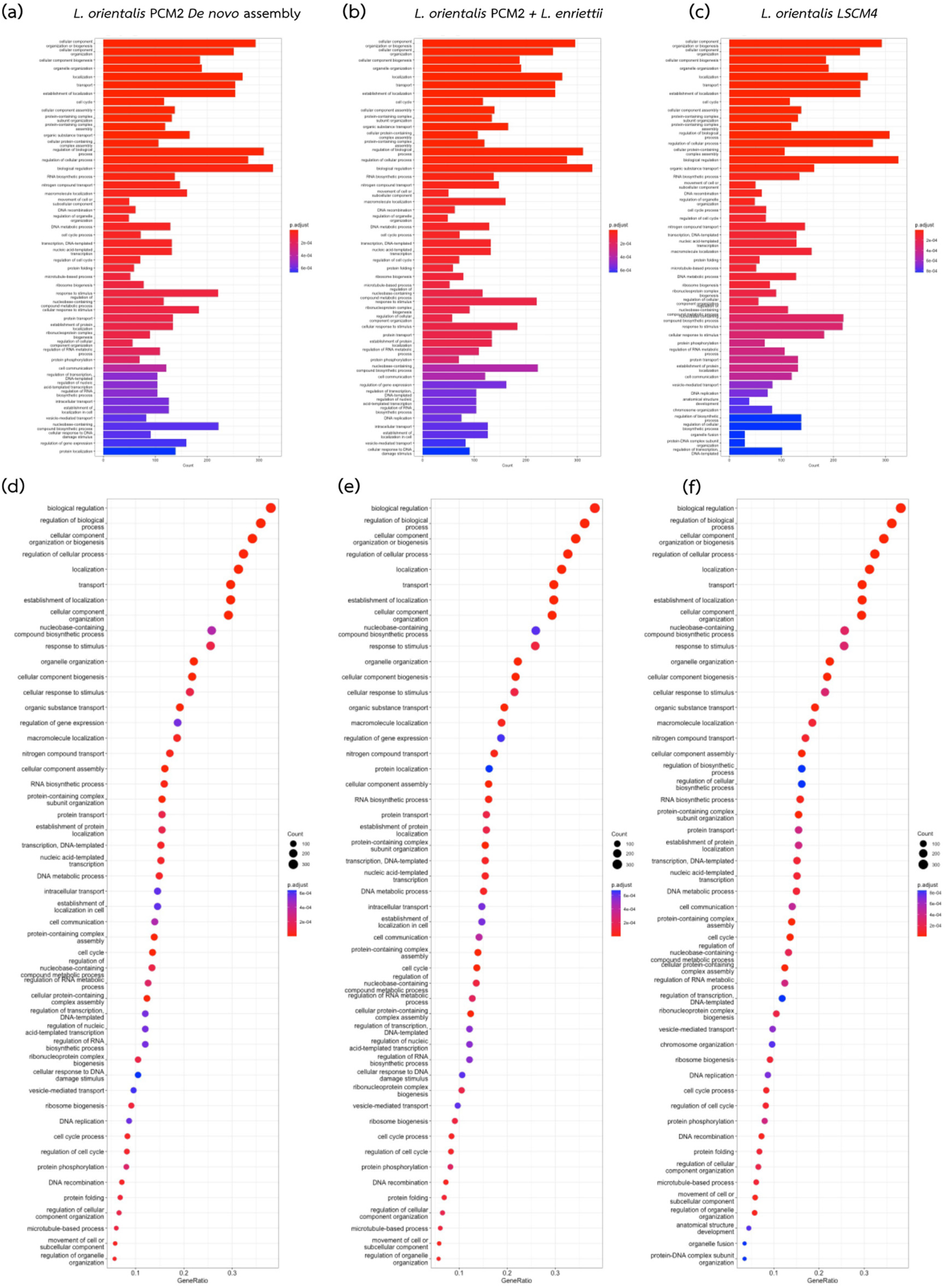{kind=link}
| Features | Sequence Assemblers | |||
|---|---|---|---|---|
| SPAdes | MEGAHIT | MaSuRCA | Velvet | |
| Total base in the assembly (Mb) | 30.15 | 29.94 | 28.50 | 29.05 |
| No. of contigs | 5565 | 6470 | 11,241 | 18,409 |
| Largest contigs (kb) | 60.19 | 85.32 | 33.26 | 27.81 |
| N50 | 12,259 | 13,737 | 4883 | 2919 |
| N75 | 6229 | 6714 | 2547 | 1450 |
| %GC content | 59.02 | 59.07 | 59.15 | 59.03 |
| Assembly Methods | Genes | Contigs | Largest Contigs | Total Length (Mb) | N50 | N75 | L50 | L75 | GC (%) | |
|---|---|---|---|---|---|---|---|---|---|---|
| MEGAHIT (De novo assembly) | Scaffold (A) | 7653 | 4989 | 90,086 | 29.94 | 11,804 | 5994 | 767 | 1663 | 59.03 |
| L. braziliensis MHOM/BR/75/M2904 | Scaffold (B) | 2265 | 138 | 2,686,643 | 32.07 | 992,961 | 641,930 | 11 | 22 | 58.68 |
| Unmapped (C) | 3670 | 12,897 | 30,803 | 13.17 | 967 | 683 | 3112 | 7219 | 60.02 | |
| Merged (D) | 8187 | 5001 | 99,540 | 30.09 | 17,861 | 8663 | 500 | 1100 | 59 | |
| L. donovani BPK282A1 | Scaffold (B) | 2977 | 36 | 2,713,248 | 32.44 | 1,024,085 | 671,483 | 11 | 21 | 59.22 |
| Unmapped (C) | 3672 | 12,929 | 30,803 | 13.17 | 968 | 683 | 3144 | 7250 | 60.02 | |
| Merged (D) | 8174 | 4973 | 99,540 | 30.08 | 18,120 | 8651 | 492 | 1087 | 59 | |
| L. enriettii CUR178 | Scaffold (B) | 7863 | 54 | 2,730,217 | 33.32 | 1,075,649 | 709,397 | 11 | 21 | 59.77 |
| Unmapped (C) | 944 | 12,331 | 10,814 | 9.84 | 798 | 627 | 4427 | 7922 | 58.51 | |
| Merged (D) | 8887 | 3367 | 206,463 | 30.27 | 24,513 | 13,215 | 359 | 779 | 59.05 | |
| L. infantum JPCM5 | Scaffold (B) | 3101 | 76 | 2,673,956 | 32.12 | 1,043,848 | 659,512 | 11 | 21 | 59.29 |
| Unmapped (C) | 3668 | 12,910 | 33,294 | 13.17 | 968 | 683 | 3133 | 7235 | 60.02 | |
| Merged (D) | 8181 | 4982 | 99,540 | 30.08 | 18,053 | 8696 | 498 | 1095 | 59 | |
| L. major Friedlin | Scaffold (B) | 3030 | 36 | 2,682,151 | 32.86 | 1,091,540 | 684,829 | 11 | 21 | 59.31 |
| Unmapped (C) | 3670 | 12,928 | 33,294 | 13.17 | 967 | 682 | 3135 | 7247 | 60.02 | |
| Merged (D) | 8178 | 4995 | 99,540 | 30.09 | 17,924 | 8632 | 495 | 1095 | 59 | |
| L. mexicana MHOM/GT/2001/U1103 | Scaffold (B) | 2930 | 575 | 3,343,498 | 32.10 | 1,044,075 | 655,046 | 10 | 20 | 59.41 |
| Unmapped (C) | 3674 | 12,924 | 22,679 | 13.17 | 968 | 683 | 3140 | 7246 | 60 | |
| Merged (D) | 8184 | 4984 | 99,540 | 30.09 | 18,053 | 8615 | 496 | 1093 | 59 | |
| L. orientalis LSCM4 | Scaffold (E) | 8158 | 98 | 2,735,713 | 34.19 | 1,120,138 | 682,718 | 11 | 22 | 59.72 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anuntasomboon, P.; Siripattanapipong, S.; Unajak, S.; Choowongkomon, K.; Burchmore, R.; Leelayoova, S.; Mungthin, M.; E-kobon, T. Making the Most of Its Short Reads: A Bioinformatics Workflow for Analysing the Short-Read-Only Data of Leishmania orientalis (Formerly Named Leishmania siamensis) Isolate PCM2 in Thailand. Biology 2022, 11, 1272. https://doi.org/10.3390/biology11091272
Anuntasomboon P, Siripattanapipong S, Unajak S, Choowongkomon K, Burchmore R, Leelayoova S, Mungthin M, E-kobon T. Making the Most of Its Short Reads: A Bioinformatics Workflow for Analysing the Short-Read-Only Data of Leishmania orientalis (Formerly Named Leishmania siamensis) Isolate PCM2 in Thailand. Biology. 2022; 11(9):1272. https://doi.org/10.3390/biology11091272
Chicago/Turabian StyleAnuntasomboon, Pornchai, Suradej Siripattanapipong, Sasimanas Unajak, Kiattawee Choowongkomon, Richard Burchmore, Saovanee Leelayoova, Mathirut Mungthin, and Teerasak E-kobon. 2022. "Making the Most of Its Short Reads: A Bioinformatics Workflow for Analysing the Short-Read-Only Data of Leishmania orientalis (Formerly Named Leishmania siamensis) Isolate PCM2 in Thailand" Biology 11, no. 9: 1272. https://doi.org/10.3390/biology11091272