
Hemochromatosis Mimicked Gaucher Disease: Role of Hyperferritinemia in Evaluation of a Clinical Case

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Glucocerebrosidase Activity Assay
2.3. Chitotriosidase Assay
2.4. DNA Extraction
2.5. PCR and Sequencing
2.6. Multiplex Ligation Probe Amplification
2.7. Glucosylsphingosine Determination
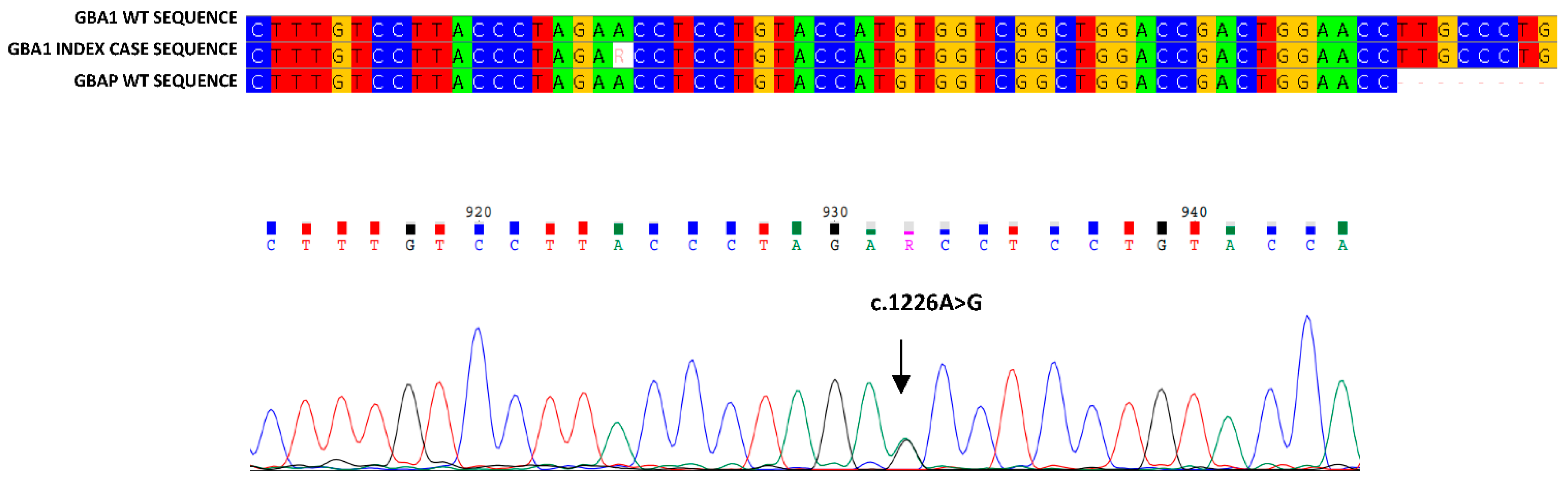
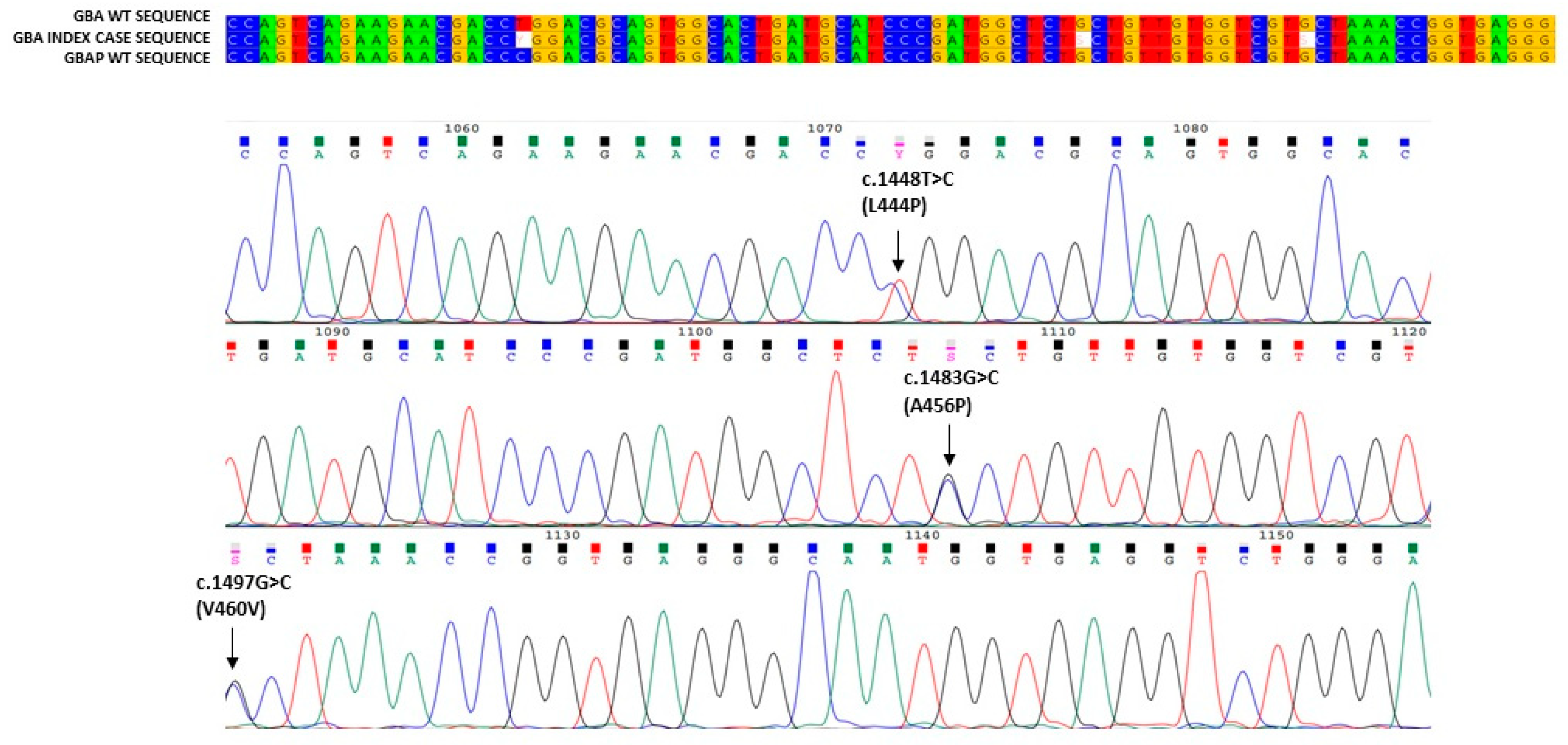
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Rosenbloom, B.E.; Weinreb, N.J. Gaucher disease: A comprehensive review. Crit. Rev. Oncog. 2013, 18, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Nalysnyk, L.; Rotella, P.; Simeone, J.C.; Hamed, A.; Weinreb, N. Gaucher disease epidemiology and natural history: A comprehensive review of the literature. Hematology 2017, 22, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabowski, G.A. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef]
- Stirnemann, J.; Vigan, M.; Hamroun, D.; Heraoui, D.; Rossi-Semerano, L.; Berger, M.G.; Rose, C.; Camou, F.; de Roux-Serratrice, C.; Grosbois, B.; et al. The French Gaucher’s disease registry: Clinical characteristics, complications and treatment of 562 patients. Orphanet. J. Rare Dis. 2012, 7, 77. [Google Scholar] [CrossRef] [Green Version]
- Sidransky, E. Gaucher disease: Insights from a rare Mendelian disorder. Discov. Med. 2012, 14, 273–281. [Google Scholar]
- Marchi, G.; Nascimbeni, F.; Motta, I.; Busti, F.; Carubbi, F.; Cappellini, M.D.; Pietrangelo, A.; Corradini, E.; Piperno, A.; Girelli, D. Hyperferritinemia and diagnosis of type 1 Gaucher disease. Am. J. Hematol. 2020, 95, 570–576. [Google Scholar] [CrossRef]
- Chamoles, N.A.; Blanco, M.; Gaggioli, D.; Casentini, C. Gaucher and Niemann–Pick diseases—Enzymatic diagnosis in dried blood spots on filter paper: Retrospective diagnoses in newborn-screening cards. Clin. Chim. Acta 2002, 317, 191–197. [Google Scholar] [CrossRef]
- Rodrigues, M.D.; de Oliveira, A.C.; Müller, K.B.; Martins, A.M.; D’Almeida, V. Chitotriosidase determination in plasma and in dried blood spots: A comparison using two different substrates in a microplate assay. Clin. Chim. Acta 2009, 406, 86–88. [Google Scholar] [CrossRef]
- Tang, C.; Jia, X.; Tang, F.; Liu, S.; Jiang, X.; Zhao, X.; Sheng, H.; Peng, M.; Liu, L.; Huang, Y. Detection of glucosylsphingosine in dried blood spots for diagnosis of Gaucher disease by LC-MS/MS. Clin. Biochem. 2020, 87, 79–84. [Google Scholar] [CrossRef]
- Burlina, A.B.; Polo, G.; Rubert, L.; Gueraldi, D.; Cazzorla, C.; Duro, G.; Salviati, L.; Burlina, A.P. Implementation of Second-Tier Tests in Newborn Screening for Lysosomal Disorders in North Eastern Italy. Int. J. Neonatal Screen. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polo, G.; Burlina, A.P.; Ranieri, E.; Colucci, F.; Rubert, L.; Pascarella, A.; Duro, G.; Tummolo, A.; Padoan, A.; Plebani, M.; et al. Plasma and dried blood spot lysosphingolipids for the diagnosis of different sphingolipidoses: A comparative study. Clin. Chem. Lab. Med. 2019, 57, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in North East Italy. J. Inherit. Metab. Dis. 2017, 41, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Cherif, W.; Ben Turkia, H.; Ben Rhouma, F.; Riahi, I.; Chemli, J.; Amaral, O.; Sá Miranda, M.C.; Caillaud, C.; Kaabachi, N.; Tebib, N.; et al. Molecular diagnosis of Gaucher disease in Tunisia. Pathol. Biol. 2013, 61, 59–63. [Google Scholar] [CrossRef]
- Erdos, M.; Hodanova, K.; Taskó, S.; Palicz, A.; Stolnaja, L.; Dvorakova, L.; Hrebicek, M.; Maródi, L. Genetic and clinical features of patients with Gaucher disease in Hungary. Blood Cells Mol. Dis. 2007, 39, 119–123. [Google Scholar] [CrossRef]
- Cherif, W.; Ben Turkia, H.; Tebib, N.; Amaral, O.; Ben Rhouma, F.; Abdelmoula, M.S.; Azzouz, H.; Caillaud, C.; Sà Miranda, M.C.; Abdelhak, S.; et al. Mutation spectrum of Gaucher disease in Tunisia: High frequency of N370S/Rec NciI compound heterozygous. Arch. Inst. Pasteur. Tunis. 2007, 84, 65–70. [Google Scholar]
- Hodanová, K.; Hrebícek, M.; Cervenková, M.; Mrázová, L.; Vepreková, L.; Zemen, J. Analysis of the beta-glucocerebrosidase gene in Czech and Slovak Gaucher patients: Mutation profile and description of six novel mutant alleles. Blood Cells Mol. Dis. 1999, 25, 287–298. [Google Scholar] [CrossRef]
- Tsuji, S.; Martin, B.M.; Barranger, J.A.; Stubblefield, B.K.; La Marca, M.E.; Ginns, E.I. Genetic heterogeneity in type 1 Gaucher disease: Multiple genotypes in Ashkenazic and non-Ashkenazic individuals. Proc. Natl. Acad. Sci. USA 1988, 85, 2349–2352. [Google Scholar] [CrossRef] [Green Version]
- Latham, T.; Grabowski, G.A.; Theophilus, B.D.; Smith, F.I. Complex alleles of the acid beta-glucosidase gene in Gaucher disease. Am. J. Hum. Genet. 1990, 47, 79–86. [Google Scholar]
- Hollak, C.E.; Evers, L.; Aerts, J.M.; van Oers, M.H. Elevated levels of M-CSF, sCD14 and IL8 in type 1 Gaucher disease. Blood Cells Mol. Dis. 1997, 23, 201–212. [Google Scholar] [CrossRef] [Green Version]
- van Breemen, M.J.; de Fost, M.; Voerman, J.S.; Laman, J.D.; Boot, R.G.; Maas, M.; Hollak, C.E.; Aerts, J.M.; Rezaee, F. Increased plasma macrophage inflammatory protein (MIP)-1alpha and MIP-1beta levels in type 1 Gaucher disease. Biochim. Biophys. Acta. 2007, 1772, 788–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barak, V.; Acker, M.; Nisman, B.; Kalickman, I.; Abrahamov, A.; Zimran, A.; Yatziv, S. Cytokines in Gaucher’s disease. Eur. Cytokine Netw. 1999, 10, 205–210. [Google Scholar] [PubMed]
- Mekinian, A.; Stirnemann, J.; Belmatoug, N.; Heraoui, D.; Fantin, B.; Fain, O.; Charpentier, A.; Rose, C. Ferritinemia during type 1 Gaucher disease: Mechanisms and progression under treat-ment. Blood Cells Mol. Dis. 2012, 49, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Cohen, L.A.; Gutierrez, L.; Weiss, A.; Leichtmann-Bardoogo, Y.; Zhang, D.L.; Crooks, D.R.; Sougrat, R.; Morgenstern, A.; Galy, B.; Hentze, M.W.; et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood 2010, 116, 1574–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, F.; Pawłowicz, E.; Klimkowska, M.; Beshara, S.; Bulanda Brustad, A.; Skotnicki, A.B.; Wahlin, A.; Machaczka, M. Ferritinemia and serum inflammatory cytokines in Swedish adults with Gaucher disease type 1. Blood Cells Mol. Dis. 2018, 68, 35–42. [Google Scholar] [CrossRef]
- Medrano-Engay, B.; Irun, P.; Gervas-Arruga, J.; Andrade-Campos, M.; Andreu, V.; Alfonso, P.; Pocovi, M.; Giraldo, P. Iron homeostasis and infIammatory biomarker analysis in patients with type 1 Gaucher disease. Blood Cells Mol. Dis. 2014, 53, 171–175. [Google Scholar] [CrossRef]
- Camaschella, C.; De Gobbi, M.; Roetto, A. Hereditary hemochromatosis: Progress and perspectives. Rev. Clin. Exp. Hematol. 2000, 4, 302–321. [Google Scholar] [CrossRef]
- Stål, P.; Hultcrantz, R. Hereditary hemochromatosis—a common genetic disease. Lakartidningen 2012, 109, 2097–2099. [Google Scholar]
- Mistry, P.K.; Cappellini, M.D.; Lukina, E.; Ozsan, H.; Mach Pascual, S.; Rosenbaum, H.; Solano, M.H.; Spigelman, Z.; Villarrubia, J.; Watman, N.P.; et al. A reappraisal of Gaucher disease-diagnosis and disease management algorithms. Am. J. Hematol. 2011, 86, 110–115. [Google Scholar] [CrossRef]
- Revel-Vilk, S.; Szer, J.; Mehta, A.; Zimran, A. How we manage Gaucher Disease in the era of choices. Br. J. Haematol. 2018, 182, 467–480. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Kinship | Age | Gender | GCase Activity | GBA1 Mutations | Status | |
|---|---|---|---|---|---|---|
| Index case | 38 | M | 0.6 | N370S | RecNci I | Hyperferritinemia, thrombocytopenia, splenomegaly |
| Mother | 66 | F | 5.6 | N370S | wt | Asymptomatic |
| Father | 67 | M | 4.8 | wt | RecNci I | Asymptomatic |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zizzo, C.; Ruggeri, I.; Colomba, P.; Argano, C.; Francofonte, D.; Zora, M.; Marsana, E.M.; Duro, G.; Corrao, S. Hemochromatosis Mimicked Gaucher Disease: Role of Hyperferritinemia in Evaluation of a Clinical Case. Biology 2022, 11, 914. https://doi.org/10.3390/biology11060914
Zizzo C, Ruggeri I, Colomba P, Argano C, Francofonte D, Zora M, Marsana EM, Duro G, Corrao S. Hemochromatosis Mimicked Gaucher Disease: Role of Hyperferritinemia in Evaluation of a Clinical Case. Biology. 2022; 11(6):914. https://doi.org/10.3390/biology11060914
Chicago/Turabian StyleZizzo, Carmela, Irene Ruggeri, Paolo Colomba, Christiano Argano, Daniele Francofonte, Marcomaria Zora, Emanuela Maria Marsana, Giovanni Duro, and Salvatore Corrao. 2022. "Hemochromatosis Mimicked Gaucher Disease: Role of Hyperferritinemia in Evaluation of a Clinical Case" Biology 11, no. 6: 914. https://doi.org/10.3390/biology11060914