
Phylogeny and Metabolic Potential of the Candidate Phylum SAR324

 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
2.1. Determining the Relative 16S rRNA Gene Sequence Abundance of SAR324 in the Oceanic Water Column
2.2. De Novo Assembly and Binning of SAR324 Genomes from under the Ross Ice Shelf (RIS)
2.3. Obtaining Existing SAR324 Genomes and Re-Assembly and Binning of SAR324 Genomes from Metagenomic Collections
2.4. De-Replication and Genome Characteristics
2.5. Phylogeny Clustering
2.6. Annotation and Database Generation
2.7. Mapping to TARA Ocean Metatranscriptome
3. Results and Discussion
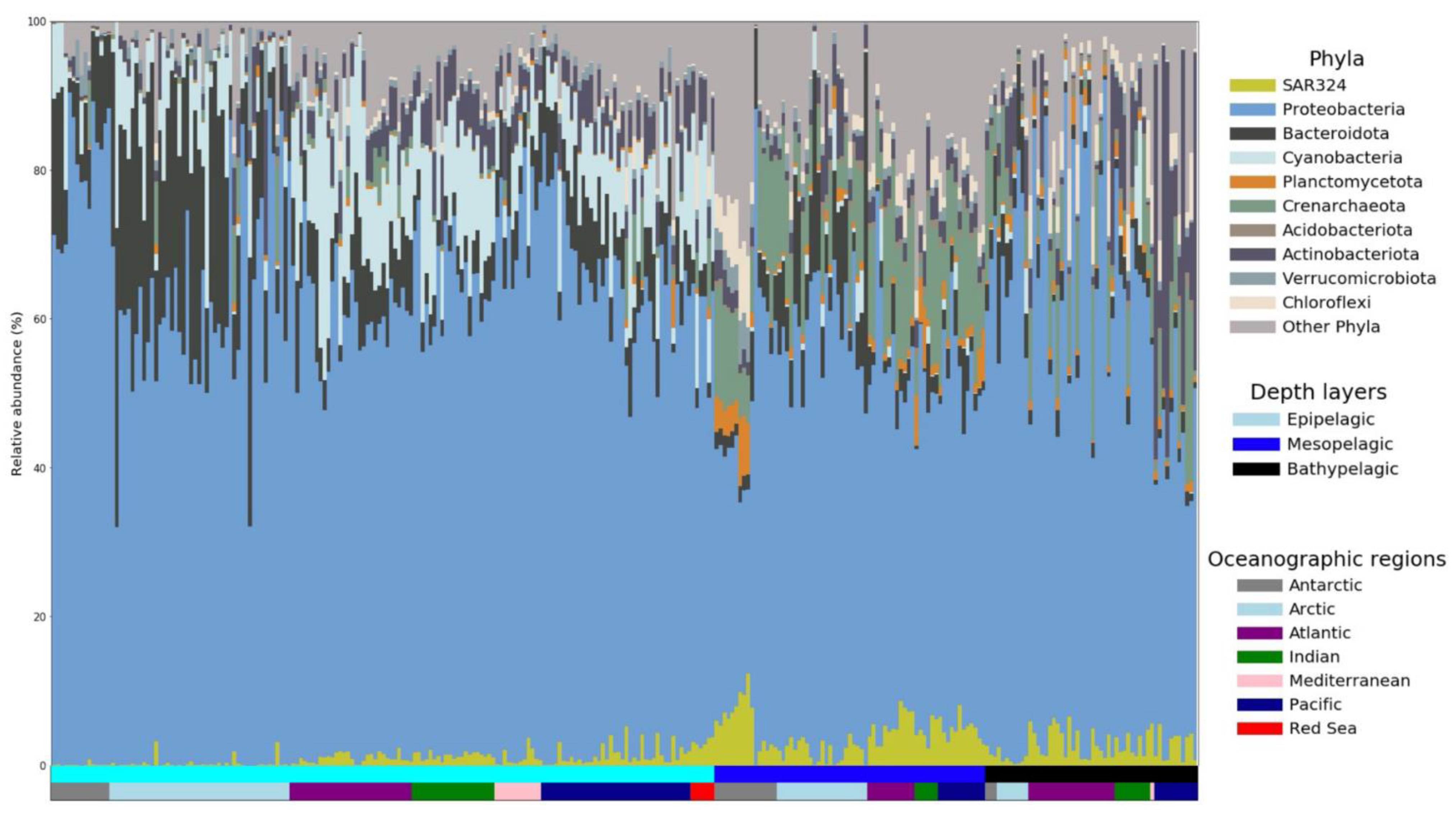
3.1. Distribution and Relative Sequence Abundance of SAR324 in the Ocean
3.2. SAR324 Genomic Characteristics
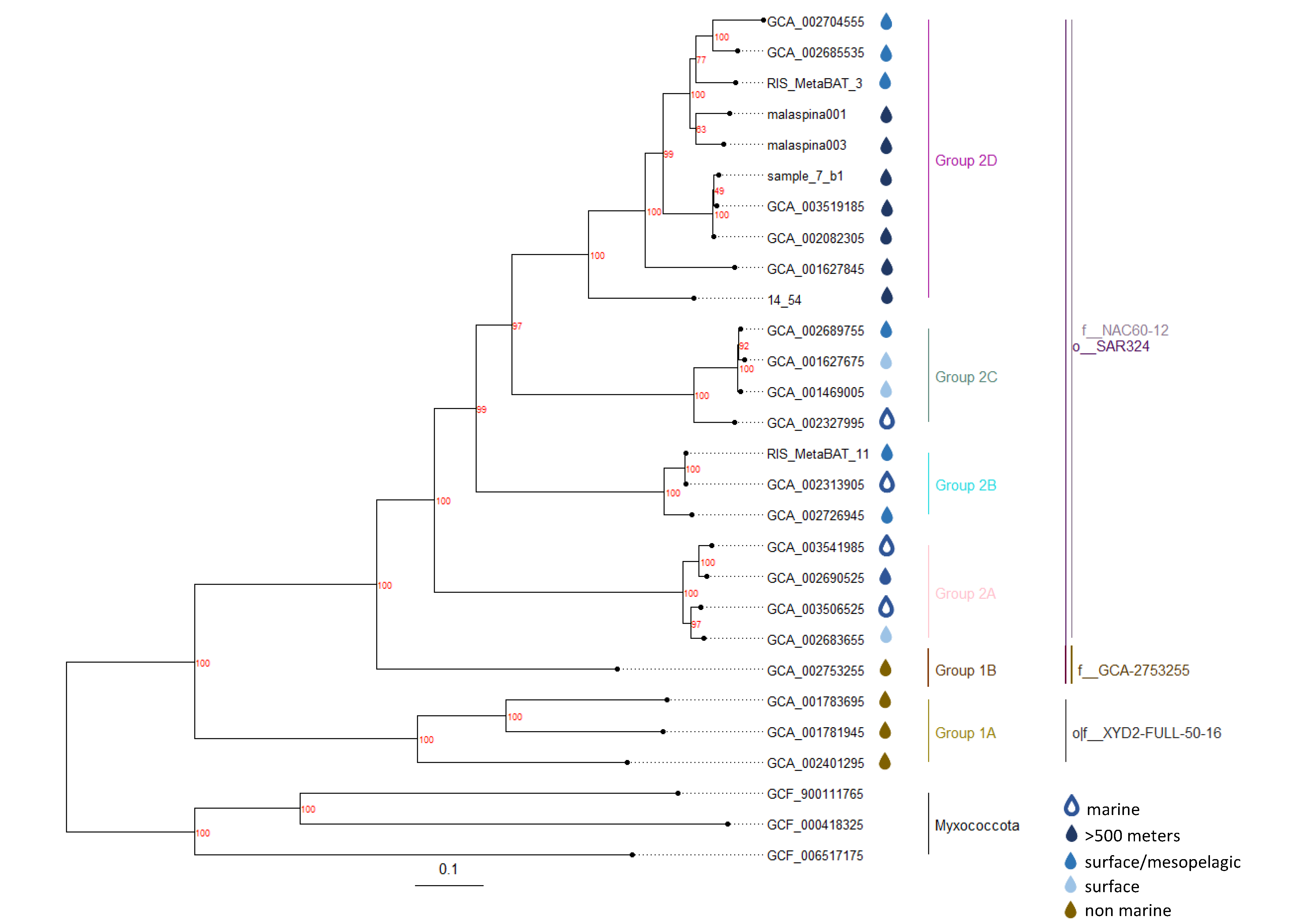
3.3. Global Phylogeny of SAR324
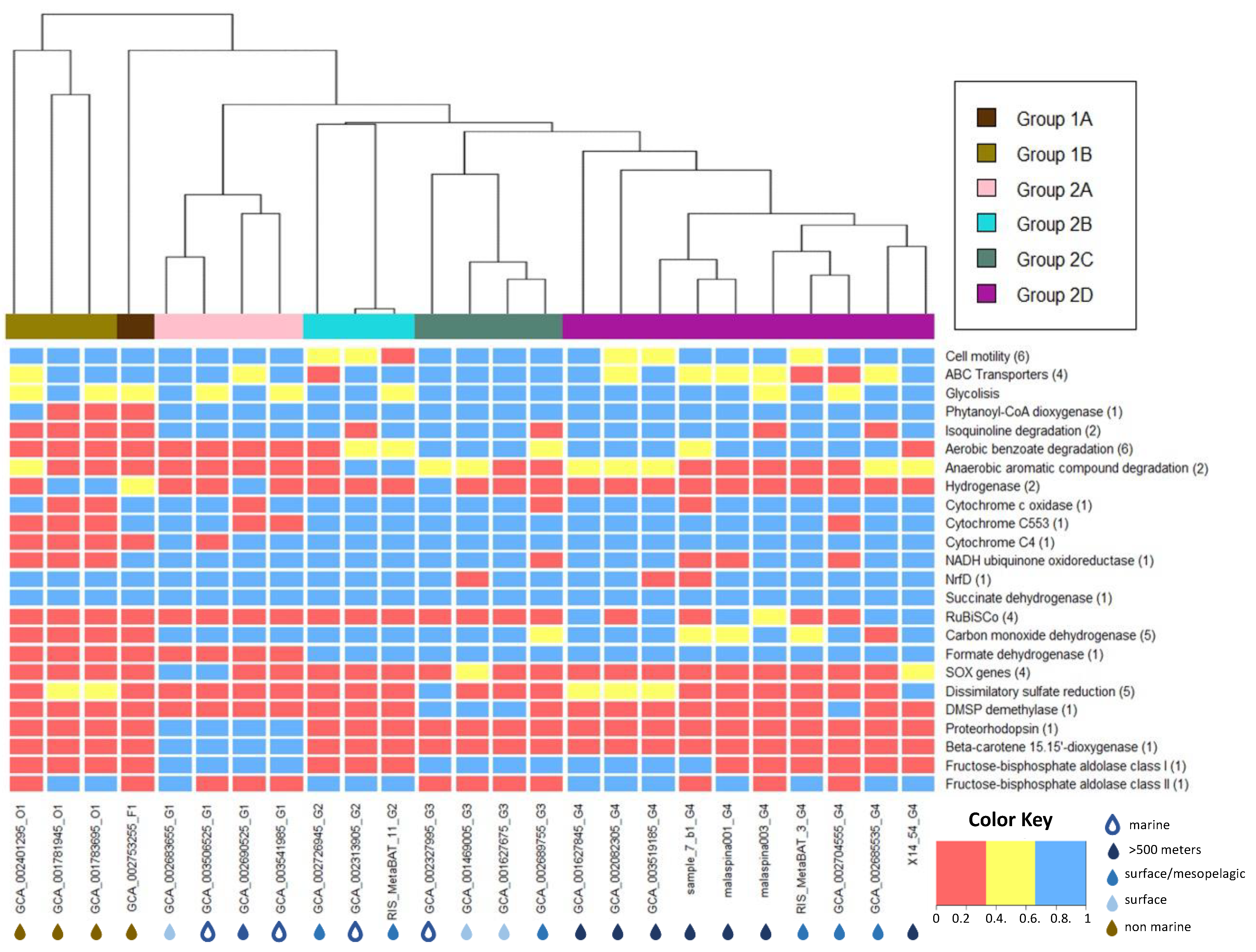
3.4. Metabolic Potential of SAR324
3.4.1. Energy Transport Chain
3.4.2. Central Carbon Metabolism
3.4.3. Role of Nitrogen and Sulfur Compounds in SAR324
3.4.4. Additional Traits
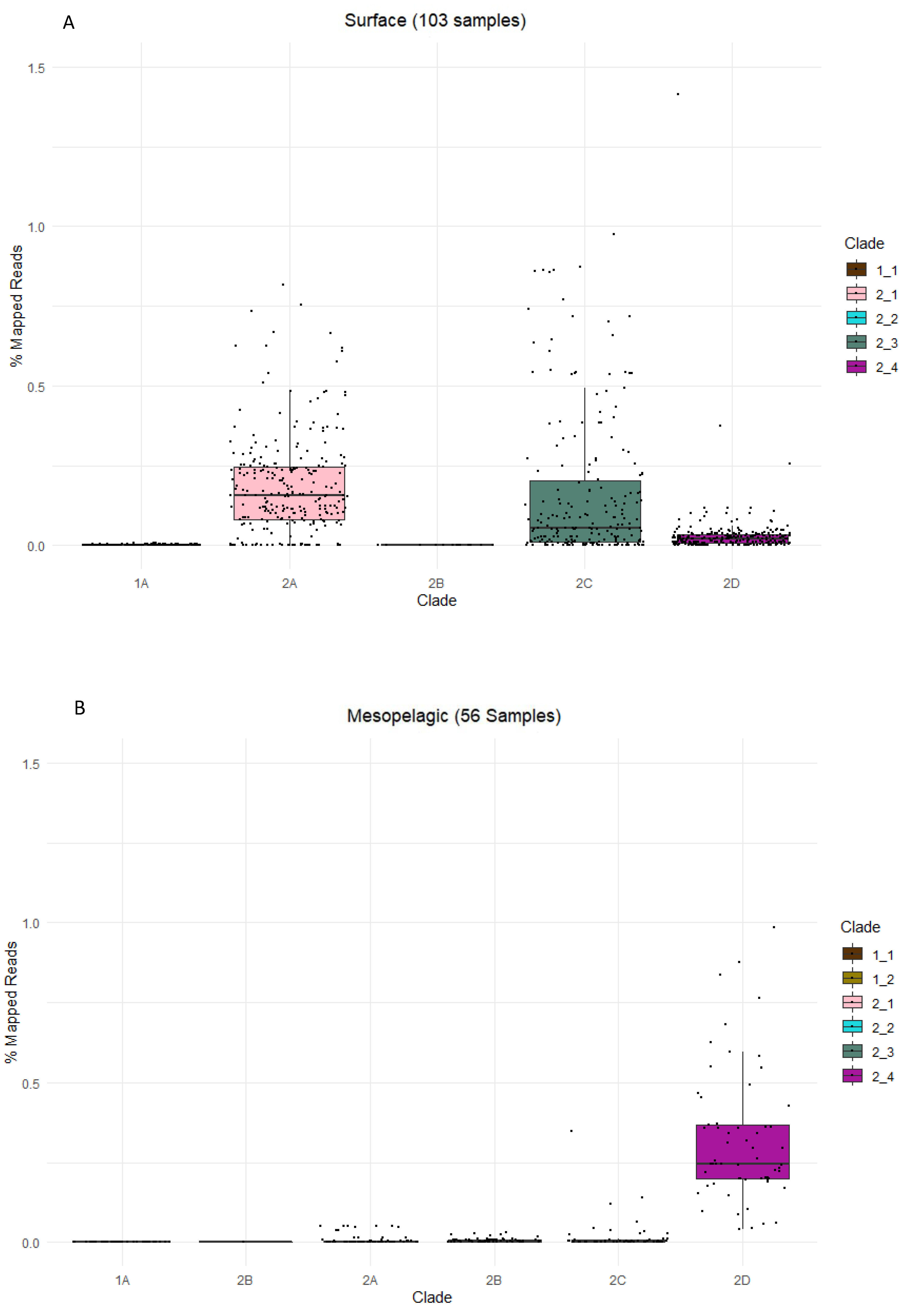
3.5. Global Ocean Metatranscriptomic Analysis of SAR324
3.6. Recent Insights into SAR324 at the ALOHA Station off Hawaii
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Azam, F.; Fenchel, T.; Field, J.; Gray, J.; Meyer-Reil, L.; Thingstad, F. The Ecological Role of Water-Column Microbes in the Sea. Mar. Ecol. Prog. Ser. 1983, 10, 257–263. [Google Scholar] [CrossRef]
- Herndl, G.J.; Reinthaler, T. Microbial control of the dark end of the biological pump. Nat. Geosci. 2013, 6, 718–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partensky, F.; Hess, W.R.; Vaulot, D. Prochlorococcus, a Marine Photosynthetic Prokaryote of Global Significance. Microbiol. Mol. Biol. Rev. 1999, 63, 106–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, Z.I.; Zinser, E.R.; Coe, A.; McNulty, N.P.; Woodward, E.M.S.; Chisholm, S.W. Niche Partitioning Among Prochlorococcus Ecotypes Along Ocean-Scale Environmental Gradients. Science 2006, 311, 1737–1740. [Google Scholar] [CrossRef] [Green Version]
- Newton, R.J.; Griffin, L.E.; Bowles, K.M.; Meile, C.; Gifford, S.; Givens, C.E.; Howard, E.C.; King, E.; Oakley, C.A.; Reisch, C.R.; et al. Genome characteristics of a generalist marine bacterial lineage. ISME J. 2010, 4, 784–798. [Google Scholar] [CrossRef] [Green Version]
- Shiba, T. Roseobacter litoralis gen. nov., sp. nov., and Roseobacter denitrificans sp. nov., Aerobic Pink-Pigmented Bacteria which Contain Bacteriochlorophyll a. Syst. Appl. Microbiol. 1991, 14, 140–145. [Google Scholar] [CrossRef]
- Morris, R.M.; Rappé, M.S.; Connon, S.A.; Vergin, K.; Siebold, W.A.; Carlson, C.A.; Giovannoni, S.J. SAR11 clade dominates ocean surface bacterioplankton communities. Nature 2002, 420, 806–810. [Google Scholar] [CrossRef]
- Herndl, G.; Agogué, H.; Baltar, F.; Reinthaler, T.; Sintes, E.; Varela, M. Regulation of aquatic microbial processes: The ‘microbial loop’ of the sunlit surface waters and the dark ocean dissected. Aquat. Microb. Ecol. 2008, 53, 59–68. [Google Scholar] [CrossRef]
- Sharma, R.; Ranjan, R.; Kapardar, R.K. “Unculturable” bacterial diversity: An untapped resource. Microb. Divers. 2022, 7, 72–77. [Google Scholar]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [Green Version]
- Danovaro, R.; Snelgrove, P.V.R.; Tyler, P. Challenging the paradigms of deep-sea ecology. Trends Ecol. Evol. 2014, 29, 465–475. [Google Scholar] [CrossRef]
- Streit, W.R.; Schmitz, R.A. Metagenomics—The key to the uncultured microbes. Curr. Opin. Microbiol. 2004, 7, 492–498. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Baltar, F.; Bayer, B.; Bednarsek, N.; Deppeler, S.; Escribano, R.; Gonzalez, C.E.; Hansman, R.L.; Mishra, R.K.; Moran, M.A.; Repeta, D.J.; et al. Towards Integrating Evolution, Metabolism, and Climate Change Studies of Marine Ecosystems. Trends Ecol. Evol. 2019, 34, 1022–1033. [Google Scholar] [CrossRef] [Green Version]
- Boetius, A. Global change microbiology—Big questions about small life for our future. Nat. Rev. Microbiol. 2019, 17, 331–332. [Google Scholar] [CrossRef]
- Hutchins, D.A.; Jansson, J.K.; Remais, J.V.; Rich, V.I.; Singh, B.K.; Trivedi, P. Climate change microbiology—Problems and perspectives. Nat. Rev. Microbiol. 2019, 17, 391–396. [Google Scholar] [CrossRef]
- Pommier, T.; Pinhassi, J.; Hagström, Å. Biogeographic analysis of ribosomal RNA clusters from marine bacterioplankton. Aquat. Microb. Ecol. 2005, 41, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Wright, T.D.; Vergin, K.L.; Boyd, P.W.; Giovannoni, S.J. A Novel Delta-Subdivision Proteobacterial Lineage from the Lower Ocean Surface Layer. Appl. Environ. Microbiol. 1997, 63, 8. [Google Scholar] [CrossRef] [Green Version]
- Salazar, G.; Cornejo-Castillo, F.M.; Benítez-Barrios, V.; Fraile-Nuez, E.; Álvarez-Salgado, X.A.; Duarte, C.M.; Gasol, J.M.; Acinas, S.G. Global diversity and biogeography of deep-sea pelagic prokaryotes. ISME J. 2016, 10, 596–608. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Chuvochina, M.; Waite, D.W.; Rinke, C.; Skarshewski, A.; Chaumeil, P.-A.; Hugenholtz, P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 2018, 36, 996–1004. [Google Scholar] [CrossRef]
- Swan, B.K.; Martinez-Garcia, M.; Preston, C.M.; Sczyrba, A.; Woyke, T.; Lamy, D.; Reinthaler, T.; Poulton, N.J.; Masland, E.D.P.; Gomez, M.L.; et al. Potential for Chemolithoautotrophy Among Ubiquitous Bacteria Lineages in the Dark Ocean. Science 2011, 333, 1296–1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheik, C.S.; Jain, S.; Dick, G.J. Metabolic flexibility of enigmatic SAR324 revealed through metagenomics and metatranscriptomics: Disentangling the ecophysiological role of SAR324. Environ. Microbiol. 2014, 16, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Dong, C.; Bougouffa, S.; Li, J.; Zhang, W.; Shao, Z.; Bajic, V.B.; Qian, P.-Y. Delta-proteobacterial SAR324 group in hydrothermal plumes on the South Mid-Atlantic Ridge. Sci. Rep. 2016, 6, 22842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quero, G.M.; Celussi, M. Inorganic and Organic Carbon Uptake Processes and Their Connection to Microbial Diversity in Meso- and Bathypelagic Arctic Waters (Eastern Fram Strait). Microb. Ecol. 2019, 79, 823–839. [Google Scholar] [CrossRef]
- Boeuf, D.; Eppley, J.M.; Mende, D.R.; Malmstrom, R.R.; Woyke, T.; DeLong, E.F. Metapangenomics reveals depth-dependent shifts in metabolic potential for the ubiquitous marine bacterial SAR324 lineage. Microbiome 2021, 9, 172. [Google Scholar] [CrossRef]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; et al. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef] [Green Version]
- Duarte, C.M. Seafaring in the 21St Century: The Malaspina 2010 Circumnavigation Expedition. Limnol. Oceanogr. Bull. 2015, 24, 11–14. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Zhang, W.; Ding, W.; Wang, M.; Fan, S.; Yang, B.; Mcminn, A.; Wang, M.; Xie, B.; Qin, Q.-L.; et al. Structure and function of the Arctic and Antarctic marine microbiota as revealed by metagenomics. Microbiome 2020, 8, 47. [Google Scholar] [CrossRef] [Green Version]
- Salazar, G.; Paoli, L.; Alberti, A.; Huerta-Cepas, J.; Ruscheweyh, H.-J.; Cuenca, M.; Field, C.M.; Coelho, L.P.; Cruaud, C.; Engelen, S.; et al. Gene Expression Changes and Community Turnover Differentially Shape the Global Ocean Metatranscriptome. Cell 2019, 179, 1068–1083.e21. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Pérez, C.; Greening, C.; Bay, S.K.; Lappan, R.J.; Zhao, Z.; De Corte, D.; Hulbe, C.; Ohneiser, C.; Stevens, C.; Thomson, B.; et al. Phylogenetically and functionally diverse microorganisms reside under the Ross Ice Shelf. Nat. Commun. 2022, 13, 117. [Google Scholar] [CrossRef]
- Logares, R.; Sunagawa, S.; Salazar, G.; Cornejo-Castillo, F.M.; Ferrera, I.; Sarmento, H.; Hingamp, P.; Ogata, H.; de Vargas, C.; Lima-Mendez, G.; et al. Metagenomic 16S rDNA Illumina tags are a powerful alternative to amplicon sequencing to explore diversity and structure of microbial communities: Using mi tag s to explore microbial communities. Environ. Microbiol. 2014, 16, 2659–2671. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Gruber-Vodicka, H.R.; Seah, B.K.B.; Pruesse, E. PhyloFlash: Rapid Small-Subunit RRNA Profiling and Targeted Assembly from Metagenomes. mSystems 2020, 5, e00920–e01020. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2. WIREs Comp. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Andrews, S. FastQC. Babraham Bioinforma. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 April 2022).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Strous, M.; Kraft, B.; Bisdorf, R.; Tegetmeyer, H.E. The Binning of Metagenomic Contigs for Microbial Physiology of Mixed Cultures. Front. Microbio. 2012, 3, 410. [Google Scholar] [CrossRef] [Green Version]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.-W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ‘omics data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Baltar, F.; Herndl, G.J. Linking extracellular enzymes to phylogeny indicates a predominantly particle-associated lifestyle of deep-sea prokaryotes. Sci. Adv. 2020, 6, eaaz4354. [Google Scholar] [CrossRef] [Green Version]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A Tool for Fast and Accurate Genome De-Replication That Enables Tracking of Microbial Genotypes and Improved Genome Recovery from Metagenomes. Bioinformatics 2017, 108142. [Google Scholar]
- Huang, Y.; Gilna, P.; Li, W. Identification of ribosomal RNA genes in metagenomic fragments. Bioinformatics 2009, 25, 1338–1340. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T. ggtree: An R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [Green Version]
- Søndergaard, D.; Pedersen, C.N.S.; Greening, C. HydDB: A web tool for hydrogenase classification and analysis. Sci. Rep. 2016, 6, 34212. [Google Scholar] [CrossRef]
- Delmont, T.O.; Eren, E.M. Linking pangenomes and metagenomes: The Prochlorococcus metapangenome. PeerJ 2018, 6, e4320. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab.(LBNL): Berkeley, CA, USA, 2014. [Google Scholar]
- Wright, J.J.; Konwar, K.M.; Hallam, S.J. Microbial ecology of expanding oxygen minimum zones. Nat. Rev. Microbiol. 2012, 10, 381–394. [Google Scholar] [CrossRef]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Tully, B.J. Metabolic diversity within the globally abundant Marine Group II Euryarchaea offers insight into ecological patterns. Nat. Commun. 2019, 10, 271. [Google Scholar] [CrossRef] [Green Version]
- Cameron, T.J.; Temperton, B.; Swan, B.K.; Landry, Z.C.; Woyke, T.; Delong, E.F.; Stepanauskas, R.; Giovannoni, S.J. Single-cell enabled comparative genomics of a deep ocean SAR11 bathytype. ISME J. 2014, 8, 1440–1451. [Google Scholar] [CrossRef]
- Mehrshad, M.; Rodriguez-Valera, F.; Amoozegar, M.A.; López-García, P.; Ghai, R. The enigmatic SAR202 cluster up close: Shedding light on a globally distributed dark ocean lineage involved in sulfur cycling. ISME J. 2018, 12, 655–668. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, F.; Meyer, A.; Eyre-Walker, A. Evidence of selection upon genomic GC-content in bacteria. PLoS Genet. 2010, 6, e1001107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chitsaz, H.; Yee-Greenbaum, J.L.; Tesler, G.; Lombardo, M.-J.; Dupont, C.L.; Badger, J.H.; Novotny, M.; Rusch, D.B.; Fraser, L.J.; Gormley, N.A.; et al. Efficient de novo assembly of single-cell bacterial genomes from short-read data sets. Nat. Biotechnol. 2011, 29, 915–921. [Google Scholar] [CrossRef] [PubMed]
- Acinas, S.G.; Sánchez, P.; Salazar, G.; Cornejo-Castillo, F.M.; Sebastián, M.; Logares, R.; Sunagawa, S.; Hingamp, P.; Ogata, H.; Lima-Mendez, G.; et al. Metabolic Architecture of the Deep Ocean Microbiome. Microbiology 2019. [Google Scholar] [CrossRef]
- Cordero, P.R.F.; Bayly, K.; Man Leung, P.; Huang, C.; Islam, Z.F.; Schittenhelm, R.B.; King, G.M.; Greening, C. Atmospheric carbon monoxide oxidation is a widespread mechanism supporting microbial survival. ISME J. 2019, 13, 2868–2881. [Google Scholar] [CrossRef] [Green Version]
- Thorup, C.; Schramm, A.; Findlay, A.J.; Finster, K.W.; Schreiber, L. Disguised as a Sulfate Reducer: Growth of the Deltaproteobacterium Desulfurivibrio alkaliphilus by Sulfide Oxidation with Nitrate. mBio 2017, 8, e00671–e00717. [Google Scholar] [CrossRef] [Green Version]
- Berben, T.; Overmars, L.; Sorokin, D.Y.; Muyzer, G. Diversity and distribution of sulfur oxidation-related genes in thioalkalivibrio, a genus of chemolithoautotrophic and haloalkaliphilic sulfur-oxidizing bacteria. Front. Microbiol. 2019, 10, 160. [Google Scholar] [CrossRef]
- Olson, D.K.; Yoshizawa, S.; Boeuf, D.; Iwasaki, W.; DeLong, E.F. Proteorhodopsin variability and distribution in the North Pacific Subtropical Gyre. ISME J. 2018, 12, 1047–1060. [Google Scholar] [CrossRef] [Green Version]
- Sabehi, G.; Loy, A.; Jung, K.H.; Partha, R.; Spudich, J.L.; Isaacson, T.; Hirschberg, J.; Wagner, M.; Béjà, O. New insights into metabolic properties of marine bacteria encoding proteorhodopsins. PLoS Biol. 2005, 3, e273. [Google Scholar] [CrossRef]
- Gómez-Consarnau, L.; Akram, N.; Lindell, K.; Pedersen, A.; Neutze, R.; Milton, D.L.; González, J.M.; Pinhassi, J. Proteorhodopsin Phototrophy Promotes Survival of Marine Bacteria during Starvation. PLoS Biol. 2010, 8, e1000358. [Google Scholar] [CrossRef] [Green Version]
- Palovaara, J.; Akram, N.; Baltar, F.; Bunse, C.; Forsberg, J.; Pedrós-Alió, C.; González, J.M.; Pinhassi, J. Stimulation of growth by proteorhodopsin phototrophy involves regulation of central metabolic pathways in marine planktonic bacteria. Proc. Natl. Acad. Sci. USA 2014, 111, E3650–E3658. [Google Scholar] [CrossRef] [Green Version]
- Tara Oceans Coordinators; Sunagawa, S.; Acinas, S.G.; Bork, P.; Bowler, C.; Eveillard, D.; Gorsky, G.; Guidi, L.; Iudicone, D.; Karsenti, E.; et al. Tara Oceans: Towards global ocean ecosystems biology. Nat. Rev. Microbiol. 2020, 18, 428–445. [Google Scholar] [CrossRef]
- Schofield, C.J.; McDonough, M.A. Structural and mechanistic studies on the peroxisomal oxygenase phytanoyl-CoA 2-hydroxylase (PhyH). Biochem. Soc. Trans. 2007, 35, 870–875. [Google Scholar] [CrossRef]
- Fuchsman, C.A.; Kirkpatrick, J.B.; Brazelton, W.J.; Murray, J.W.; Staley, J.T. Metabolic strategies of free-living and aggregate-associated bacterial communities inferred from biologic and chemical profiles in the Black Sea suboxic zone. FEMS Microbiol. Ecol. 2011, 78, 586–603. [Google Scholar] [CrossRef] [Green Version]
- Carolan, M.T. Quantifying Distributions of and Modeling Interactions among Sulfur- and Nitrogen- Cycling Chemolithoautotrophs in the Largest Oxygen Minimum Zone of the Global Ocean. Ph.D. Thesis, University of California, Merced, CA, USA, 2014. 95343. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Environment | Completeness | Contamination | Genome Size (Mbp) | GC % | # Contigs | N50 Contigs (KBP) | Coding Density % |
|---|---|---|---|---|---|---|---|---|
| GCA_001469005 | Red Sea water column Station 34–depth 10 m | 89.09 | 0.0 | 3.49 | 47.1 | 312 | 16.27 | 90 |
| GCA_001627675 | Red Sea water column Station 34–depth 25 m | 72.17 | 2.94 | 2.54 | 47.46 | 440 | 6.54 | 91 |
| GCA_001627845 | Red Sea water column Station 192–depth 500 m | 89.54 | 0.0 | 3.17 | 42.89 | 90 | 58.32 | 89 |
| GCA_001781945 | Rifle well CD01 at 16ft depth; 0.1 μm filter at time point D, USA: Rifle, CO | 91.96 | 1.68 | 3.12 | 56.51 | 84 | 65.77 | 91 |
| GCA_001783695 | Rifle well CD01 at 16ft depth; 0.1 μm filter at time point D, USA: Rifle, CO | 91.83 | 1.68 | 3.14 | 49.59 | 149 | 37.4 | 91 |
| GCA_002082305 | hydrothermal plumes on the South Mid-Atlantic Ridge | 90.35 | 0.0 | 2.8 | 42.3 | 148 | 26.08 | 89 |
| GCA_002313905 | marine | 81.27 | 0.84 | 3.15 | 55.43 | 595 | 8.63 | 88 |
| GCA_002327995 | marine | 87.04 | 0.84 | 4.04 | 45.73 | 470 | 14.94 | 86 |
| GCA_002401295 | Atlantic Ocean: North Pond, oxic subseafloor aquifer | 69.22 | 2.95 | 3.08 | 41.71 | 182 | 23.04 | 87 |
| GCA_002683655 | Chile-Peru Current Coastal Province, 5–1000 m (TARA) | 80.16 | 2.33 | 3.15 | 46.19 | 109 | 42.34 | 89 |
| GCA_002685535 * | Chile-Peru Current Coastal Province, 5–1000 m (TARA) | 87.85 | 2.75 | 7.29 | 46.09 | 245 | 51.04 | 77 |
| GCA_002689755 | Mediterranean Sea, 5–1000 m (TARA) | 65.12 | 0.85 | 2.05 | 46.5 | 161 | 12.88 | 92 |
| GCA_002690525 | Mediterranean Sea, 5–1000 m (TARA) | 80.24 | 1.68 | 3.12 | 44.81 | 187 | 18.62 | 88 |
| GCA_002704555 | Mediterranean Sea, 5–1000 m (TARA) | 69.42 | 1.05 | 1.97 | 38.89 | 114 | 19.51 | 89 |
| GCA_002726945 | North Pacific Ocean, 5–1000 m (TARA) | 89.88 | 2.2 | 2.6 | 57.34 | 156 | 18.45 | 89 |
| GCA_002753255 * | Australia: Punkally Creek Sediment | 78.03 | 0.96 | 5.06 | 41.83 | 530 | 14.91 | 90 |
| GCA_003506525 | marine | 72.8 | 0.95 | 3.48 | 44.67 | 720 | 7.44 | 83 |
| GCA_003519185 | Neamphius huxleyi metagenome | 68.15 | 0.0 | 2.13 | 40.87 | 1465 | 2.25 | 73 |
| GCA_003541985 | marine | 67.36 | 0.84 | 2.48 | 44.62 | 592 | 5.66 | 83 |
| 14_54 | Red Sea brine pool | 87.34 | 0.17 | 3.45 | 43.43 | 374 | 12.52 | 87 |
| RIS_MetaBAT_11 | Ross Ice Shelf Antarctica | 79.08 | 0.89 | 2.47 | 57.09 | 297 | 10.42 | 92 |
| RIS_MetaBAT_3 | Ross Ice Shelf Antarctica | 81.27 | 1.01 | 1.93 | 40.12 | 285 | 8.09 | 90 |
| malaspina001 | Malaspina Deep Sea Samples | 91.15 | 2.69 | 2.61 | 42.46 | 284 | 20.73 | 89 |
| malaspina003 | Malaspina Deep Sea Samples | 91.26 | 0.63 | 2.77 | 42.98 | 222 | 14.14 | 90 |
| sample_7_b1 | 45° N, 178° E, Deep Sea | 65.19 | 1.58 | 1.8 | 40.91 | 1138 | 1.57 | 88 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malfertheiner, L.; Martínez-Pérez, C.; Zhao, Z.; Herndl, G.J.; Baltar, F. Phylogeny and Metabolic Potential of the Candidate Phylum SAR324. Biology 2022, 11, 599. https://doi.org/10.3390/biology11040599
Malfertheiner L, Martínez-Pérez C, Zhao Z, Herndl GJ, Baltar F. Phylogeny and Metabolic Potential of the Candidate Phylum SAR324. Biology. 2022; 11(4):599. https://doi.org/10.3390/biology11040599
Chicago/Turabian StyleMalfertheiner, Lukas, Clara Martínez-Pérez, Zihao Zhao, Gerhard J. Herndl, and Federico Baltar. 2022. "Phylogeny and Metabolic Potential of the Candidate Phylum SAR324" Biology 11, no. 4: 599. https://doi.org/10.3390/biology11040599