

The Nature of Functional Features of Different Classes of G-Protein-Coupled Receptors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
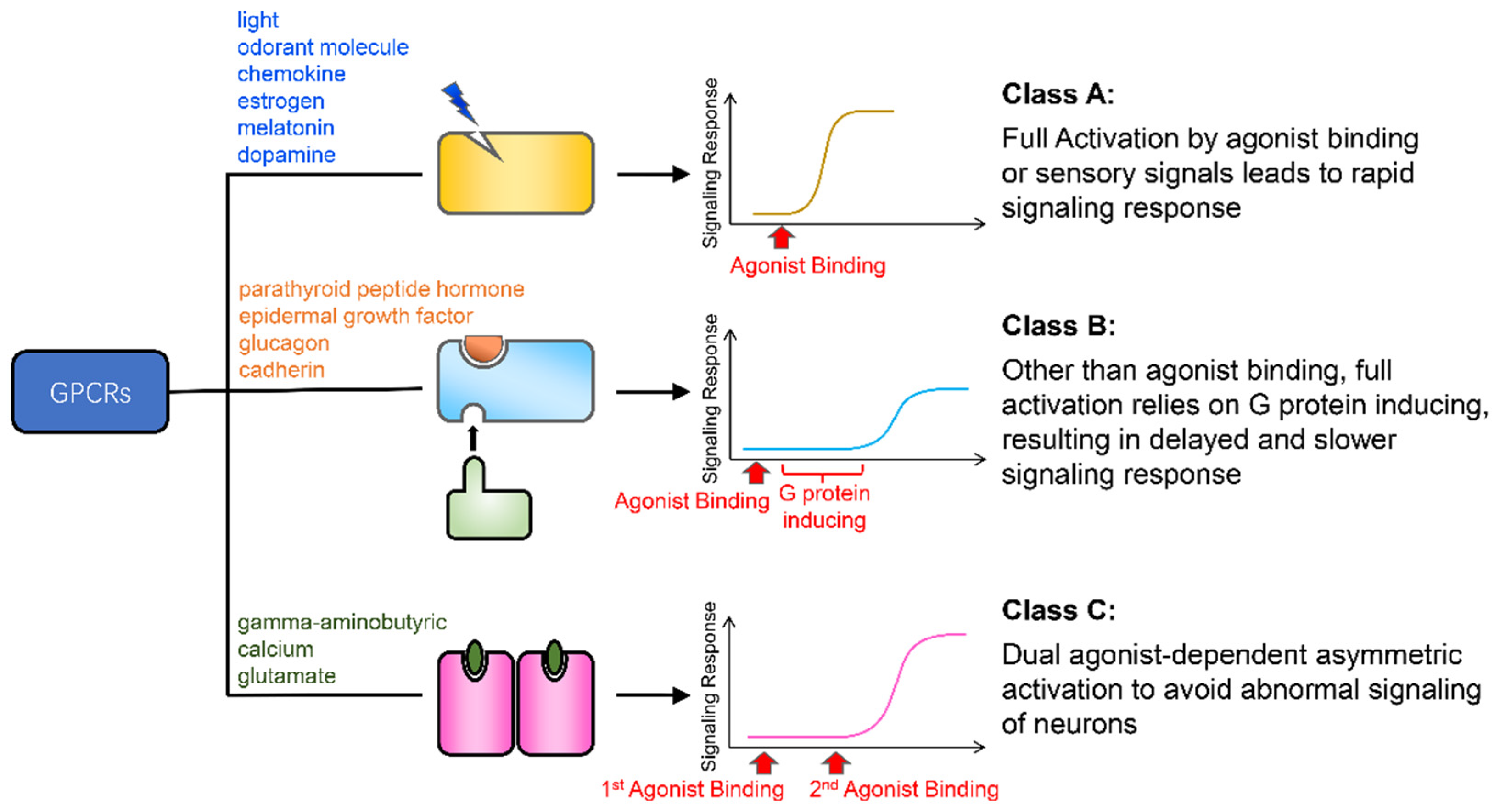
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Modeling the Structures
2.2. Calculating the Folding Free Energy of Protein
2.3. Ligand Docking and Calculating the Binding Free Energy
3. Results
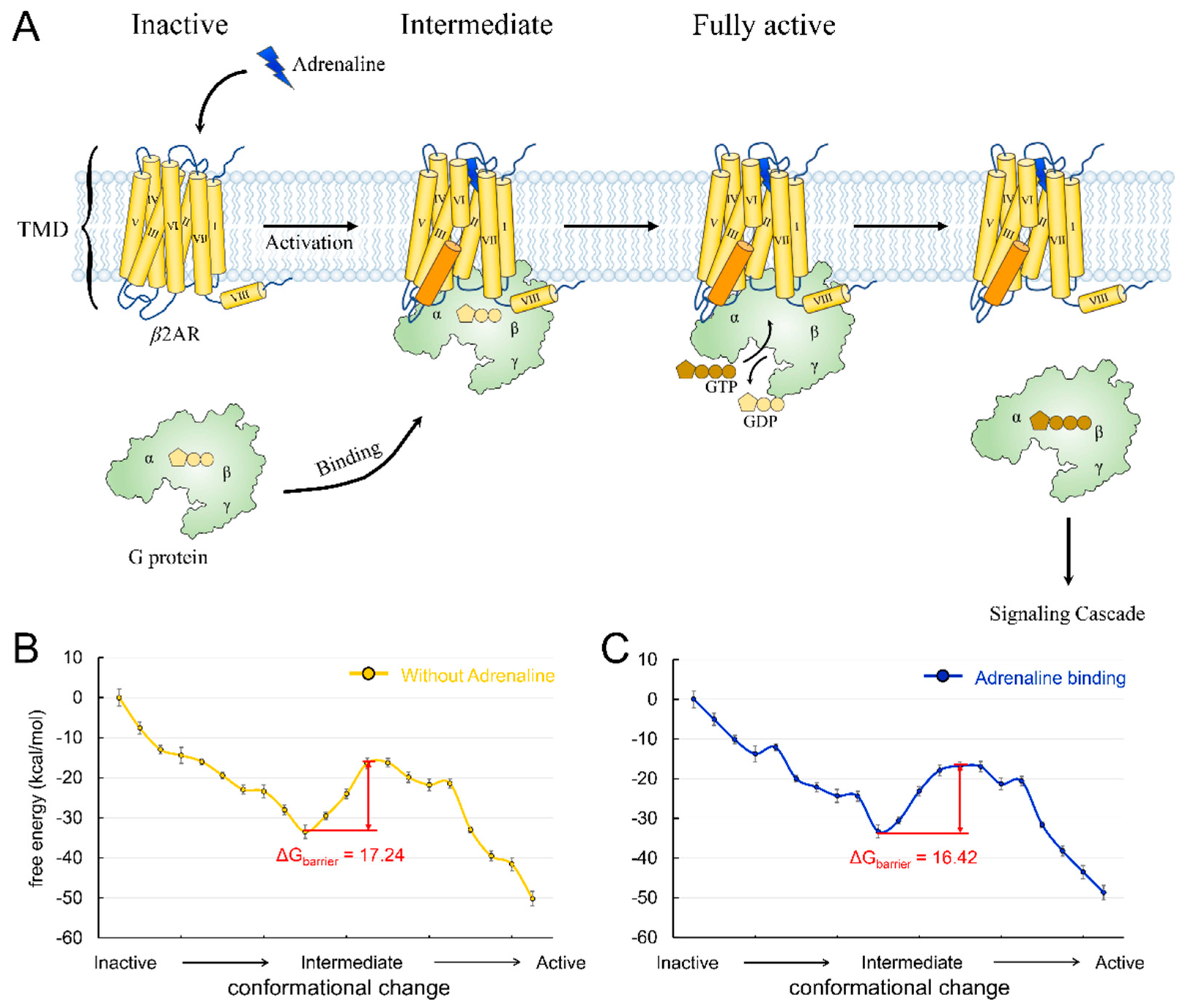
3.1. Agonist Binding Reduces the Activation Energy Barrier of β2AR Adrenergic Receptor
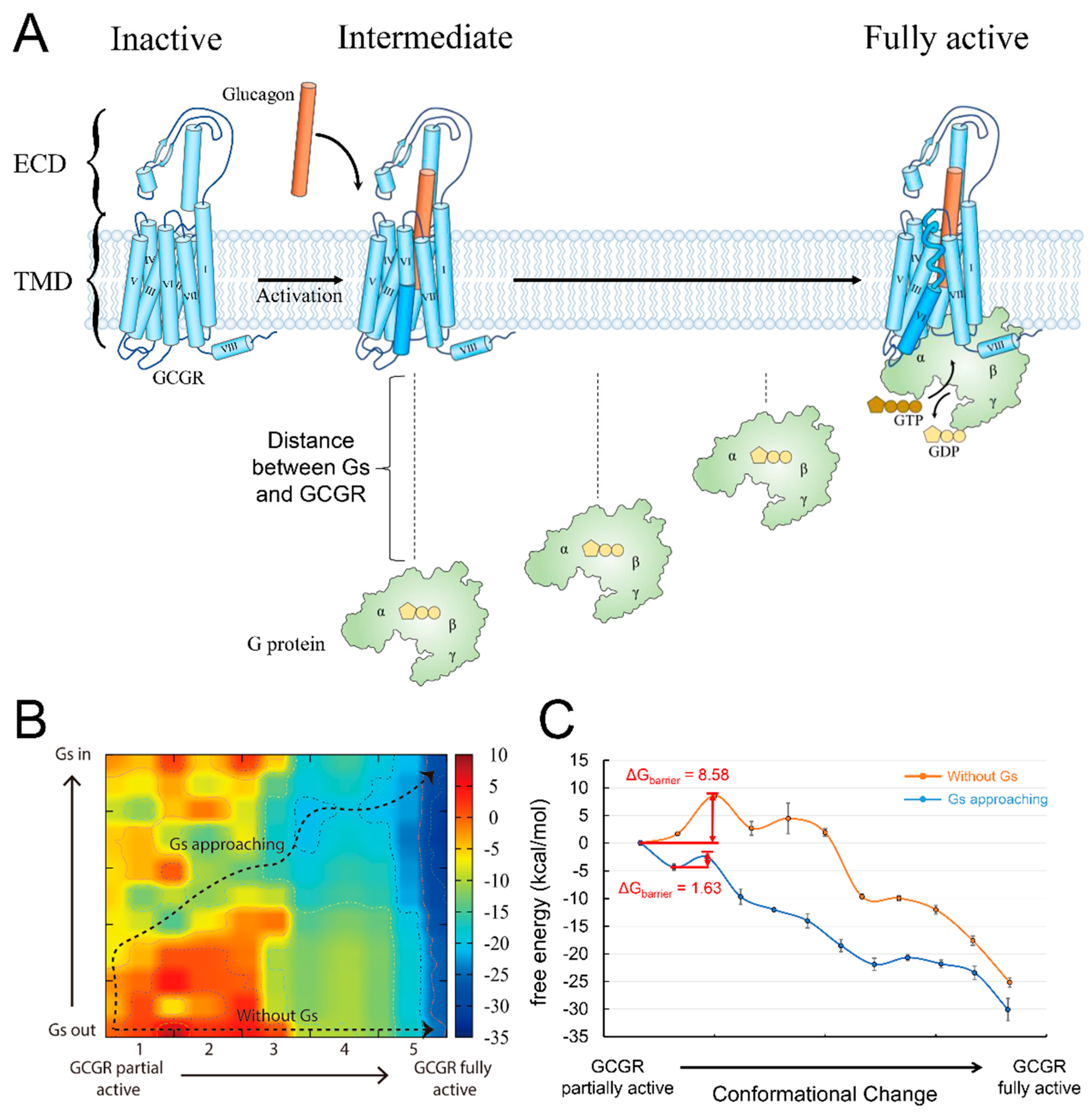
3.2. G protein Approaching Plays an Essential Role in Glucagon Receptor Activation
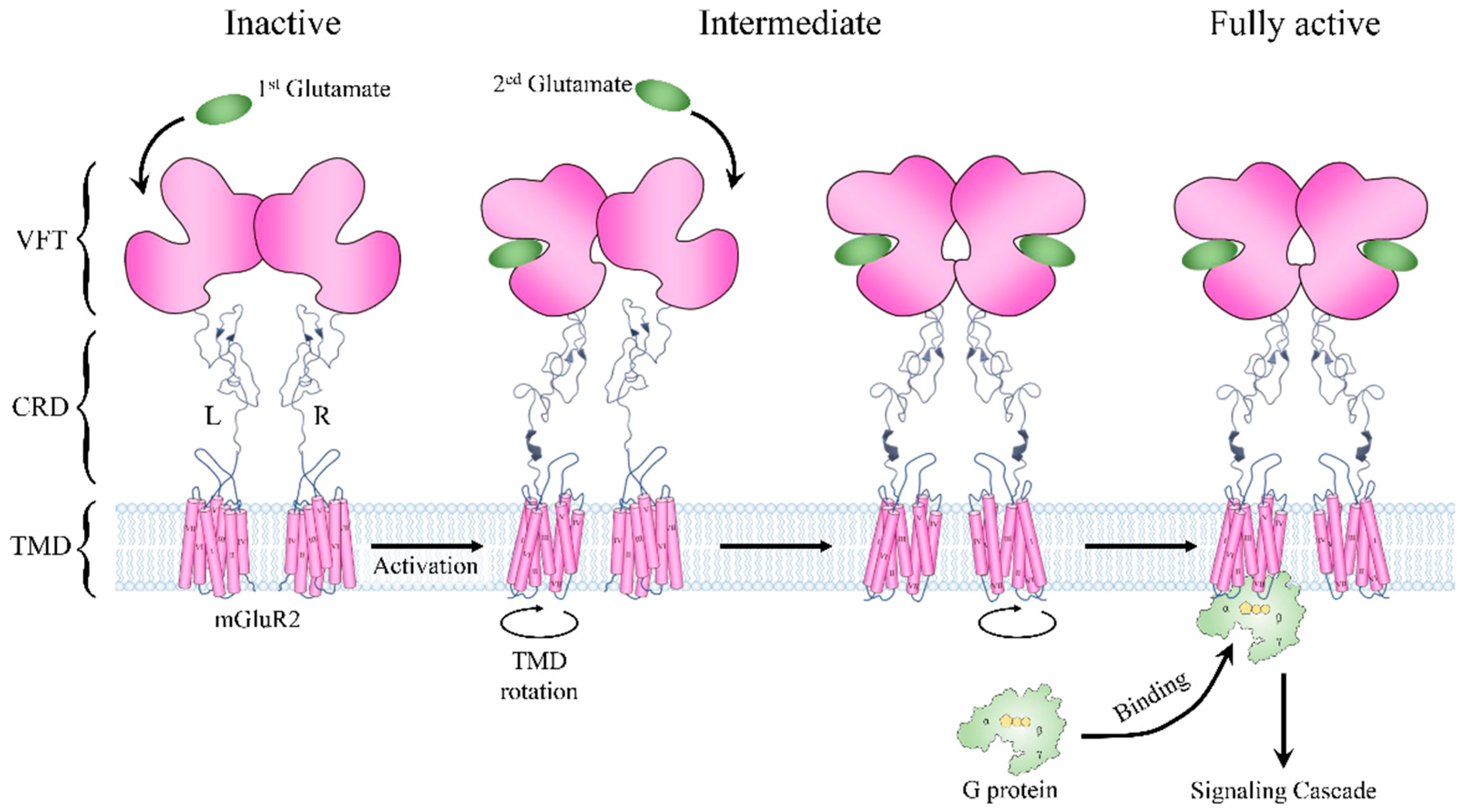
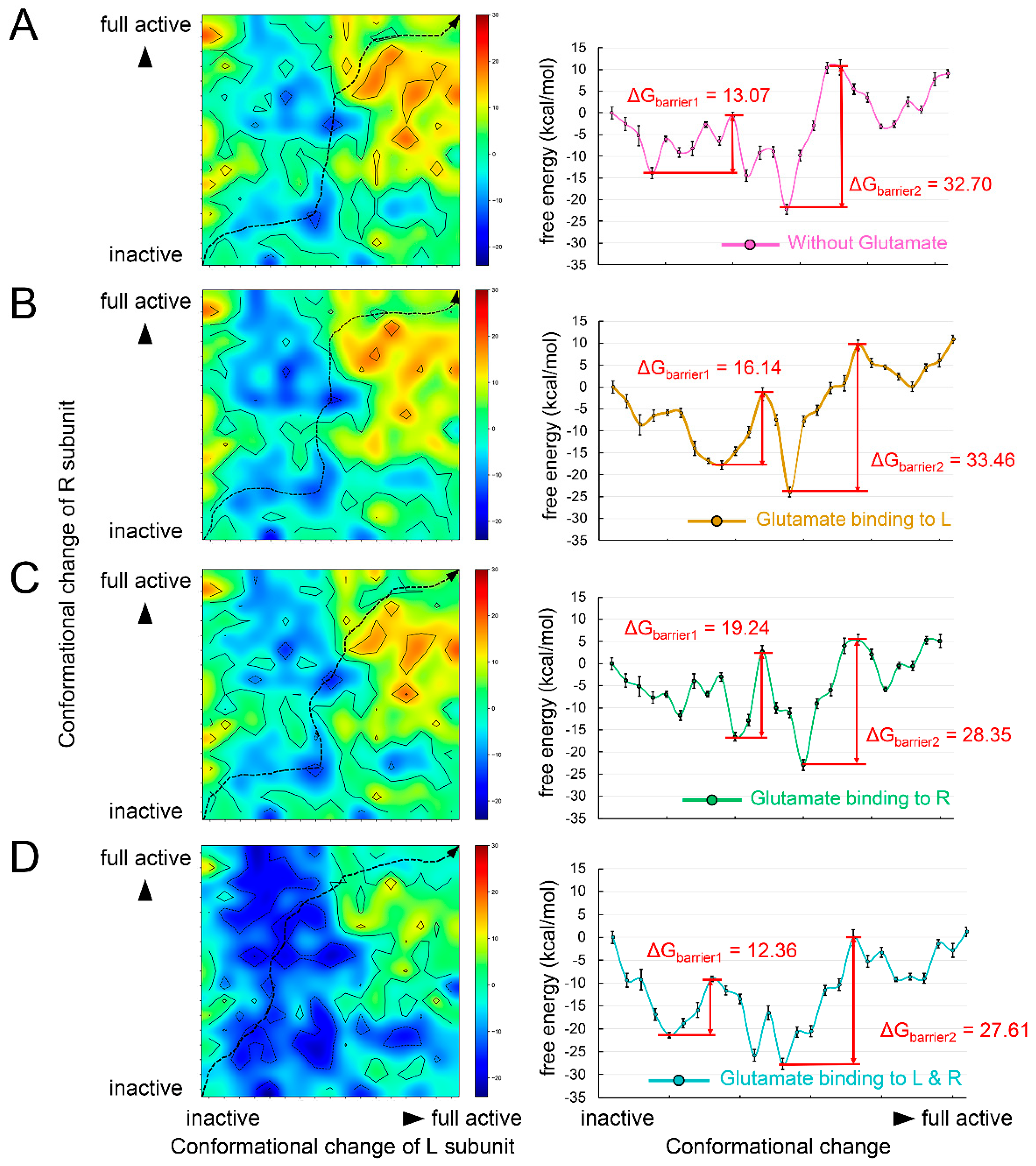
3.3. Two Agonists Are Required for the Activation of Metabotropic Glutamate Receptor 2
4. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Congreve, M.; de Graaf, C.; Swain, N.A.; Tate, C.G. Impact of GPCR Structures on Drug Discovery. Cell 2020, 181, 81–91. [Google Scholar] [CrossRef]
- Yang, D.; Zhou, Q.; Labroska, V.; Qin, S.; Darbalaei, S.; Wu, Y.; Yuliantie, E.; Xie, L.; Tao, H.; Cheng, J.; et al. G protein-coupled receptors: Structure- and function-based drug discovery. Signal Transduct. Target. Ther. 2021, 6, 7. [Google Scholar] [CrossRef]
- Gusach, A.; Maslov, I.; Luginina, A.; Borshchevskiy, V.; Mishin, A.; Cherezov, V. Beyond structure: Emerging approaches to study GPCR dynamics. Curr. Opin. Struct. Biol. 2020, 63, 18–25. [Google Scholar] [CrossRef]
- Lefkowitz, R.J. A brief history of G-protein coupled receptors (Nobel Lecture). Angew. Chem. Int. Ed. 2013, 52, 6366–6378. [Google Scholar] [CrossRef]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef]
- Shimada, I.; Ueda, T.; Kofuku, Y.; Eddy, M.T.; Wuthrich, K. GPCR drug discovery: Integrating solution NMR data with crystal and cryo-EM structures. Nat. Rev. Drug Discov. 2019, 18, 59–82. [Google Scholar] [CrossRef]
- Bissantz, C. Conformational changes of G protein-coupled receptors during their activation by agonist binding. J. Recept. Signal Transduct Res. 2003, 23, 123–153. [Google Scholar] [CrossRef]
- Manglik, A.; Kruse, A.C. Structural Basis for G Protein-Coupled Receptor Activation. Biochemistry 2017, 56, 5628–5634. [Google Scholar] [CrossRef]
- Latorraca, N.R.; Venkatakrishnan, A.J.; Dror, R.O. GPCR Dynamics: Structures in Motion. Chem. Rev. 2017, 117, 139–155. [Google Scholar] [CrossRef]
- Hilger, D.; Kumar, K.K.; Hu, H.; Pedersen, M.F.; O’Brien, E.S.; Giehm, L.; Jennings, C.; Eskici, G.; Inoue, A.; Lerch, M.; et al. Structural insights into differences in G protein activation by family A and family B GPCRs. Science 2020, 369, eaba3373. [Google Scholar] [CrossRef]
- Zhang, D.; Zhao, Q.; Wu, B. Structural Studies of G Protein-Coupled Receptors. Mol. Cells 2015, 38, 836–842. [Google Scholar] [CrossRef] [Green Version]
- Kooistra, A.J.; Mordalski, S.; Pandy-Szekeres, G.; Esguerra, M.; Mamyrbekov, A.; Munk, C.; Keseru, G.M.; Gloriam, D.E. GPCRdb in 2021: Integrating GPCR sequence, structure and function. Nucleic Acids Res. 2021, 49, D335–D343. [Google Scholar] [CrossRef]
- Hanson, M.A.; Orencia, M.C. Ten Years of GPCR Structures. GPCRs Ther. Targets 2022, 1, 299–345. [Google Scholar]
- Sutkeviciute, I.; Vilardaga, J.P. Structural insights into emergent signaling modes of G protein-coupled receptors. J. Biol. Chem. 2020, 295, 11626–11642. [Google Scholar] [CrossRef]
- Deupi, X.; Kobilka, B.K. Energy landscapes as a tool to integrate GPCR structure, dynamics, and function. Physiology 2010, 25, 293–303. [Google Scholar] [CrossRef] [Green Version]
- Kamerlin, S.C.; Vicatos, S.; Dryga, A.; Warshel, A. Coarse-grained (multiscale) simulations in studies of biophysical and chemical systems. Annu. Rev. Phys. Chem. 2011, 62, 41–64. [Google Scholar] [CrossRef]
- Vicatos, S.; Rychkova, A.; Mukherjee, S.; Warshel, A. An effective coarse-grained model for biological simulations: Recent refinements and validations. Proteins 2014, 82, 1168–1185. [Google Scholar] [CrossRef]
- Vorobyov, I.; Kim, I.; Chu, Z.T.; Warshel, A. Refining the treatment of membrane proteins by coarse-grained models. Proteins 2016, 84, 92–117. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Kolev, V.; Warshel, A. Validating a Coarse-Grained Voltage Activation Model by Comparing Its Performance to the Results of Monte Carlo Simulations. J. Phys. Chem. B 2017, 121, 11284–11291. [Google Scholar] [CrossRef]
- Shi, D.; An, K.; Zhang, H.; Xu, P.; Bai, C. Application of Coarse-Grained (CG) Models to Explore Conformational Pathway of Large-Scale Protein Machines. Entropy 2022, 24, 620. [Google Scholar] [CrossRef]
- Bai, C.; Warshel, A. Revisiting the protomotive vectorial motion of F0-ATPase. Proc. Natl. Acad. Sci. USA 2019, 116, 19484–19489. [Google Scholar] [CrossRef]
- Bai, C.; Asadi, M.; Warshel, A. The catalytic dwell in ATPases is not crucial for movement against applied torque. Nat. Chem. 2020, 12, 1187–1192. [Google Scholar] [CrossRef]
- Bai, C.; Warshel, A. Critical Differences between the Binding Features of the Spike Proteins of SARS-CoV-2 and SARS-CoV. J. Phys. Chem. B 2020, 124, 5907–5912. [Google Scholar] [CrossRef]
- Bai, C.; Wang, J.; Chen, G.; Zhang, H.; An, K.; Xu, P.; Du, Y.; Ye, R.D.; Saha, A.; Zhang, A.; et al. Predicting Mutational Effects on Receptor Binding of the Spike Protein of SARS-CoV-2 Variants. J. Am. Chem. Soc. 2021, 143, 17646–17654. [Google Scholar] [CrossRef]
- Alhadeff, R.; Vorobyov, I.; Yoon, H.W.; Warshel, A. Exploring the free-energy landscape of GPCR activation. Proc. Natl. Acad. Sci. USA 2018, 115, 10327–10332. [Google Scholar] [CrossRef] [Green Version]
- Alhadeff, R.; Warshel, A. A free-energy landscape for the glucagon-like peptide 1 receptor GLP1R. Proteins 2020, 88, 127–134. [Google Scholar] [CrossRef]
- Bai, C.; Wang, J.; Mondal, D.; Du, Y.; Ye, R.D.; Warshel, A. Exploring the Activation Process of the beta2AR-Gs Complex. J. Am. Chem. Soc. 2021, 143, 11044–11051. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Schlitter, J.; Engels, M.; Krüger, P. Targeted molecular dynamics: A new approach for searching pathways of conformational transitions. J. Mol. Graph. 1994, 12, 84–88. [Google Scholar] [CrossRef]
- Lee, F.S.; Chu, Z.T.; Warshel, A. Microscopic and semimicroscopic calculations of electrostatic energies in proteins by the POLARIS and ENZYMIX programs. J. Comput. Chem. 1993, 14, 161–185. [Google Scholar]
- Yang, F.; Ling, S.; Zhou, Y.; Zhang, Y.; Lv, P.; Liu, S.; Fang, W.; Sun, W.; Hu, L.A.; Zhang, L. Different conformational responses of the β2-adrenergic receptor-Gs complex upon binding of the partial agonist salbutamol or the full agonist isoprenaline. Natl. Sci. Rev. 2021, 8, nwaa284. [Google Scholar]
- Du, J.; Wang, D.; Fan, H.; Xu, C.; Tai, L.; Lin, S.; Han, S.; Tan, Q.; Wang, X.; Xu, T.; et al. Structures of human mGlu2 and mGlu7 homo- and heterodimers. Nature 2021, 594, 589–593. [Google Scholar] [CrossRef]
- Muegge, I.; Tao, H.; Warshel, A. A fast estimate of electrostatic group contributions to the free energy of protein-inhibitor binding. Protein Eng. Des. Sel. 1997, 10, 1363–1372. [Google Scholar] [CrossRef]
- Schutz, C.N.; Warshel, A. What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins 2001, 44, 400–417. [Google Scholar] [CrossRef]
- Singh, N.; Warshel, A. Absolute binding free energy calculations: On the accuracy of computational scoring of protein-ligand interactions. Proteins 2010, 78, 1705–1723. [Google Scholar] [CrossRef] [Green Version]
- Cherezov, V.; Rosenbaum, D.M.; Hanson, M.A.; Rasmussen, S.G.; Thian, F.S.; Kobilka, T.S.; Choi, H.J.; Kuhn, P.; Weis, W.I.; Kobilka, B.K.; et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science 2007, 318, 1258–1265. [Google Scholar] [CrossRef] [Green Version]
- Patel, C.B.; Noor, N.; Rockman, H.A. Functional selectivity in adrenergic and angiotensin signaling systems. Mol. Pharmacol. 2010, 78, 983–992. [Google Scholar] [CrossRef] [Green Version]
- Plazinska, A.; Plazinski, W.; Jozwiak, K. Agonist binding by the beta2-adrenergic receptor: An effect of receptor conformation on ligand association-dissociation characteristics. Eur. Biophys. J. 2015, 44, 149–163. [Google Scholar] [CrossRef] [Green Version]
- Chan, H.C.; Filipek, S.; Yuan, S. The Principles of Ligand Specificity on beta-2-adrenergic receptor. Sci. Rep. 2016, 6, 34736. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yang, F.; Ling, S.; Lv, P.; Zhou, Y.; Fang, W.; Sun, W.; Zhang, L.; Shi, P.; Tian, C. Single-particle cryo-EM structural studies of the beta(2)AR-Gs complex bound with a full agonist formoterol. Cell Discov. 2020, 6, 45. [Google Scholar] [CrossRef]
- Mayo, K.E.; Miller, L.J.; Bataille, D.; Dalle, S.; Goke, B.; Thorens, B.; Drucker, D.J. International Union of Pharmacology. XXXV. The glucagon receptor family. Pharmacol. Rev. 2003, 55, 167–194. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Chen, Y.; Shenoy, S.K. Agonist-activated glucagon receptors are deubiquitinated at early endosomes by two distinct deubiquitinases to facilitate Rab4a-dependent recycling. J. Biol. Chem. 2020, 295, 16630–16642. [Google Scholar] [CrossRef]
- Zhang, H.; Qiao, A.; Yang, L.; Van Eps, N.; Frederiksen, K.S.; Yang, D.; Dai, A.; Cai, X.; Zhang, H.; Yi, C.; et al. Structure of the glucagon receptor in complex with a glucagon analogue. Nature 2018, 553, 106–110. [Google Scholar] [CrossRef]
- Sloop, K.W.; Michael, M.D.; Moyers, J.S. Glucagon as a target for the treatment of Type 2 diabetes. Expert Opin. Ther. Targets 2005, 9, 593–600. [Google Scholar] [CrossRef]
- Bagger, J.I.; Knop, F.K.; Holst, J.J.; Vilsboll, T. Glucagon antagonism as a potential therapeutic target in type 2 diabetes. Diabetes Obes. Metab. 2011, 13, 965–971. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: Physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [Green Version]
- Pin, J.P.; Kniazeff, J.; Liu, J.; Binet, V.; Goudet, C.; Rondard, P.; Prezeau, L. Allosteric functioning of dimeric class C G-protein-coupled receptors. FEBS J. 2005, 272, 2947–2955. [Google Scholar] [CrossRef]
- Kunishima, N.; Shimada, Y.; Tsuji, Y.; Sato, T.; Yamamoto, M.; Kumasaka, T.; Nakanishi, S.; Jingami, H.; Morikawa, K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature 2000, 407, 971–977. [Google Scholar] [CrossRef]
- Hlavackova, V.; Zabel, U.; Frankova, D.; Batz, J.; Hoffmann, C.; Prezeau, L.; Pin, J.P.; Blahos, J.; Lohse, M.J. Sequential inter- and intrasubunit rearrangements during activation of dimeric metabotropic glutamate receptor 1. Sci. Signal. 2012, 5, ra59. [Google Scholar] [CrossRef]
- Kniazeff, J.; Bessis, A.S.; Maurel, D.; Ansanay, H.; Prezeau, L.; Pin, J.P. Closed state of both binding domains of homodimeric mGlu receptors is required for full activity. Nat. Struct. Mol. Biol. 2004, 11, 706–713. [Google Scholar] [CrossRef]
- Hlavackova, V.; Goudet, C.; Kniazeff, J.; Zikova, A.; Maurel, D.; Vol, C.; Trojanova, J.; Prezeau, L.; Pin, J.P.; Blahos, J. Evidence for a single heptahelical domain being turned on upon activation of a dimeric GPCR. EMBO J. 2005, 24, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.; Han, S.; Cai, X.; Tan, Q.; Zhou, K.; Wang, D.; Wang, X.; Du, J.; Yi, C.; Chu, X.; et al. Structures of Gi-bound metabotropic glutamate receptors mGlu2 and mGlu4. Nature 2021, 594, 583–588. [Google Scholar] [CrossRef]
- Liauw, B.W.; Afsari, H.S.; Vafabakhsh, R. Conformational rearrangement during activation of a metabotropic glutamate receptor. Nat. Chem. Biol. 2021, 17, 291–297. [Google Scholar] [CrossRef]
- Manglik, A.; Kim, T.H.; Masureel, M.; Altenbach, C.; Yang, Z.; Hilger, D.; Lerch, M.T.; Kobilka, T.S.; Thian, F.S.; Hubbell, W.L.; et al. Structural Insights into the Dynamic Process of beta2-Adrenergic Receptor Signaling. Cell 2015, 161, 1101–1111. [Google Scholar] [CrossRef] [Green Version]
- Koehl, A.; Hu, H.; Feng, D.; Sun, B.; Zhang, Y.; Robertson, M.J.; Chu, M.; Kobilka, T.S.; Laeremans, T.; Steyaert, J.; et al. Structural insights into the activation of metabotropic glutamate receptors. Nature 2019, 566, 79–84. [Google Scholar] [CrossRef]
- Mao, C.; Shen, C.; Li, C.; Shen, D.D.; Xu, C.; Zhang, S.; Zhou, R.; Shen, Q.; Chen, L.N.; Jiang, Z.; et al. Cryo-EM structures of inactive and active GABA(B) receptor. Cell Res. 2020, 30, 564–573. [Google Scholar] [CrossRef]
- Shaye, H.; Ishchenko, A.; Lam, J.H.; Han, G.W.; Xue, L.; Rondard, P.; Pin, J.P.; Katritch, V.; Gati, C.; Cherezov, V. Structural basis of the activation of a metabotropic GABA receptor. Nature 2020, 584, 298–303. [Google Scholar] [CrossRef]
- Isaikina, P.; Tsai, C.J.; Dietz, N.; Pamula, F.; Grahl, A.; Goldie, K.N.; Guixa-Gonzalez, R.; Branco, C.; Paolini-Bertrand, M.; Calo, N.; et al. Structural basis of the activation of the CC chemokine receptor 5 by a chemokine agonist. Sci. Adv. 2021, 7, eabg8685. [Google Scholar] [CrossRef]
- Kiss, D.L.; Longden, J.; Fechner, G.A.; Avery, V.M. The functional antagonist Met-RANTES: A modified agonist that induces differential CCR5 trafficking. Cell. Mol. Biol. Lett. 2009, 14, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Costa, T.; Cotecchia, S. Historical review: Negative efficacy and the constitutive activity of G-protein-coupled receptors. Trends Pharmacol. Sci. 2005, 26, 618–624. [Google Scholar] [CrossRef]
- Smith, J.S.; Lefkowitz, R.J.; Rajagopal, S. Biased signalling: From simple switches to allosteric microprocessors. Nat. Rev. Drug Discov. 2018, 17, 243–260. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, K.; Zhu, X.; Bai, C. The Nature of Functional Features of Different Classes of G-Protein-Coupled Receptors. Biology 2022, 11, 1839. https://doi.org/10.3390/biology11121839
An K, Zhu X, Bai C. The Nature of Functional Features of Different Classes of G-Protein-Coupled Receptors. Biology. 2022; 11(12):1839. https://doi.org/10.3390/biology11121839
Chicago/Turabian StyleAn, Ke, Xiaohong Zhu, and Chen Bai. 2022. "The Nature of Functional Features of Different Classes of G-Protein-Coupled Receptors" Biology 11, no. 12: 1839. https://doi.org/10.3390/biology11121839