
Drug Repurposing Using Gene Co-Expression and Module Preservation Analysis in Acute Respiratory Distress Syndrome (ARDS), Systemic Inflammatory Response Syndrome (SIRS), Sepsis, and COVID-19

Abstract
:Simple Summary
Abstract
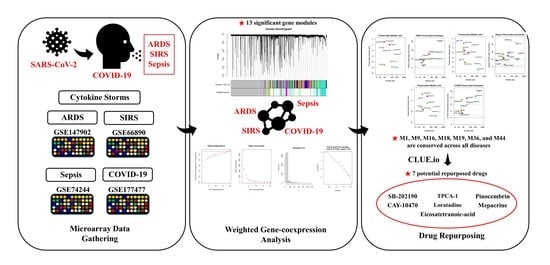
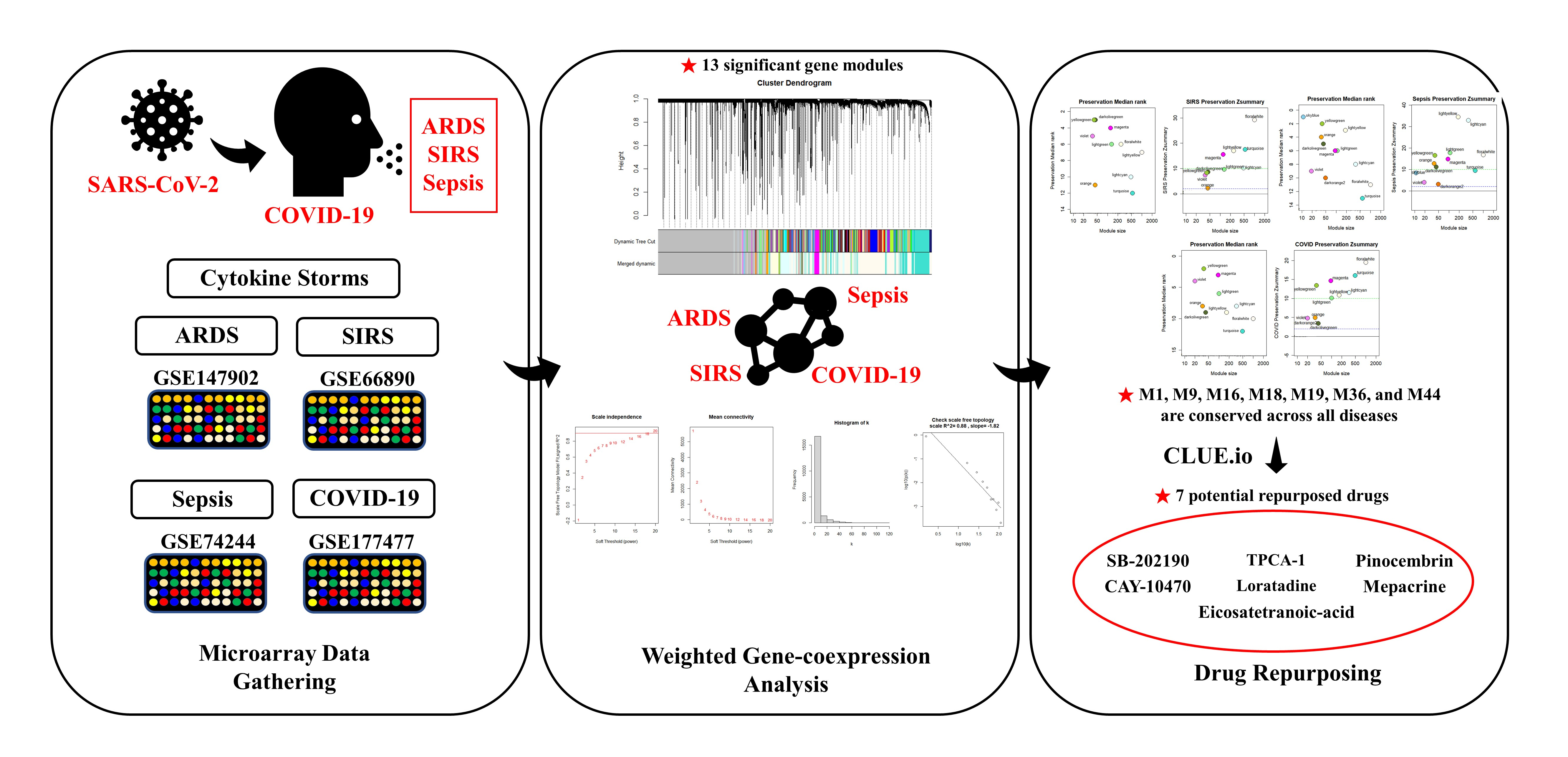
1. Introduction
2. Materials and Methods
2.1. Data Acquisition
Gathering and Preparation of Microarray Datasets
2.2. Weighted Gene Co-Expression Network Analysis (WGCNA)
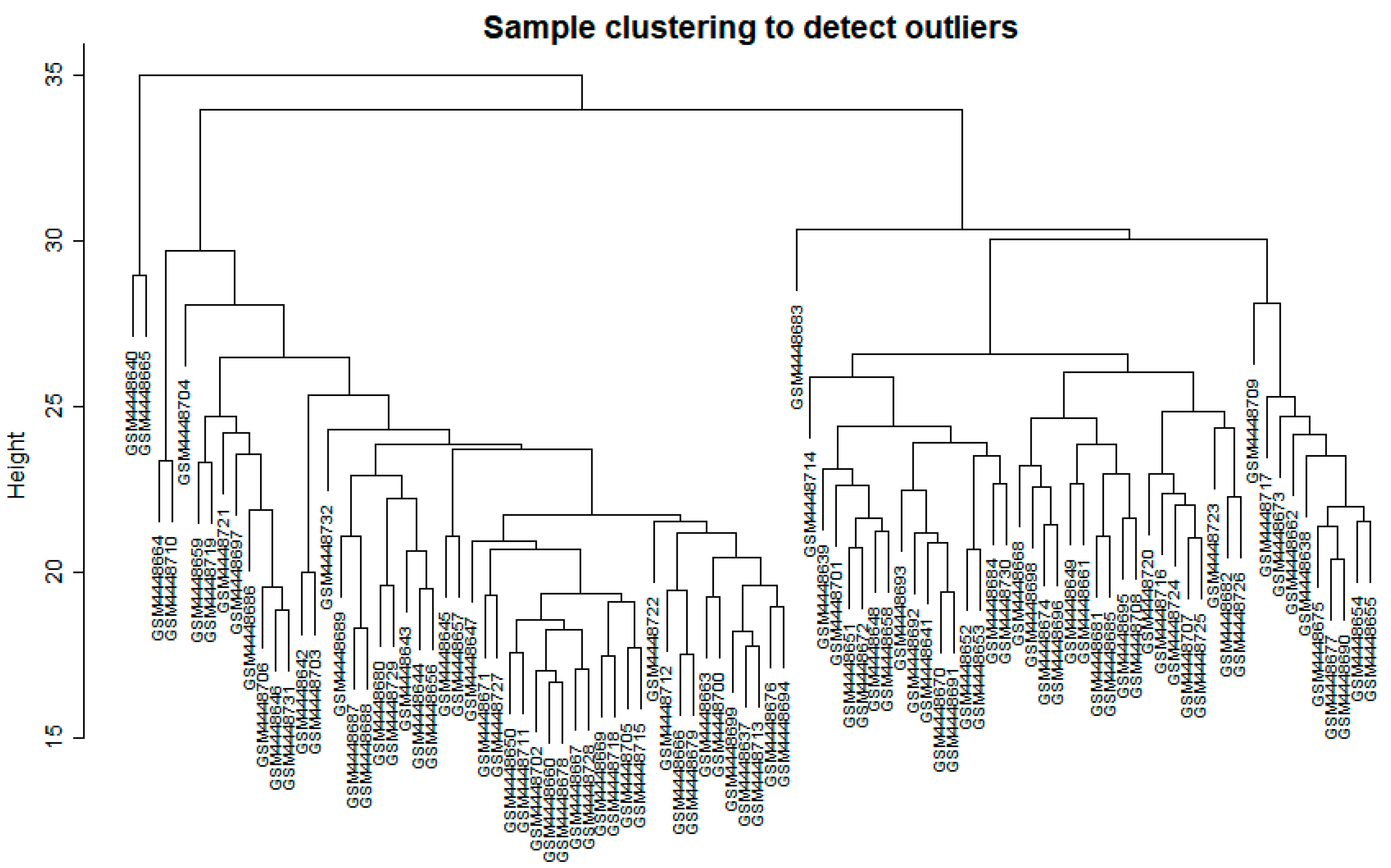
2.2.1. Data Input, Cleaning, and Pre-Processing
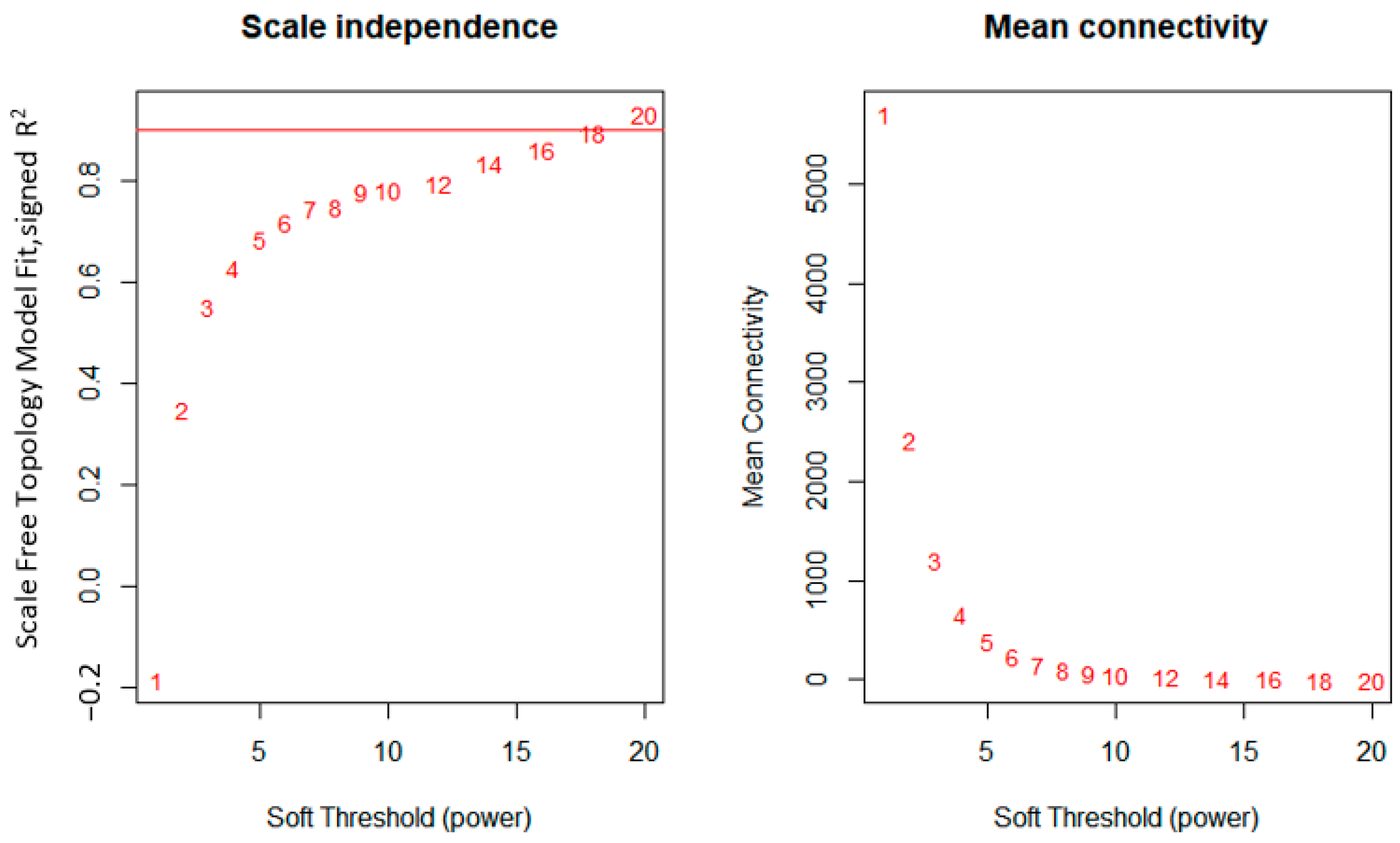
2.2.2. Network Construction
2.2.3. Module Preservation Analysis
2.3. GO Enrichment and KEGG Pathway Analysis of Module Genes
Identification of Hub Genes within Modules
2.4. Screening of Possible Repurposed Drug Candidates
Connectivity Map Analysis
3. Results
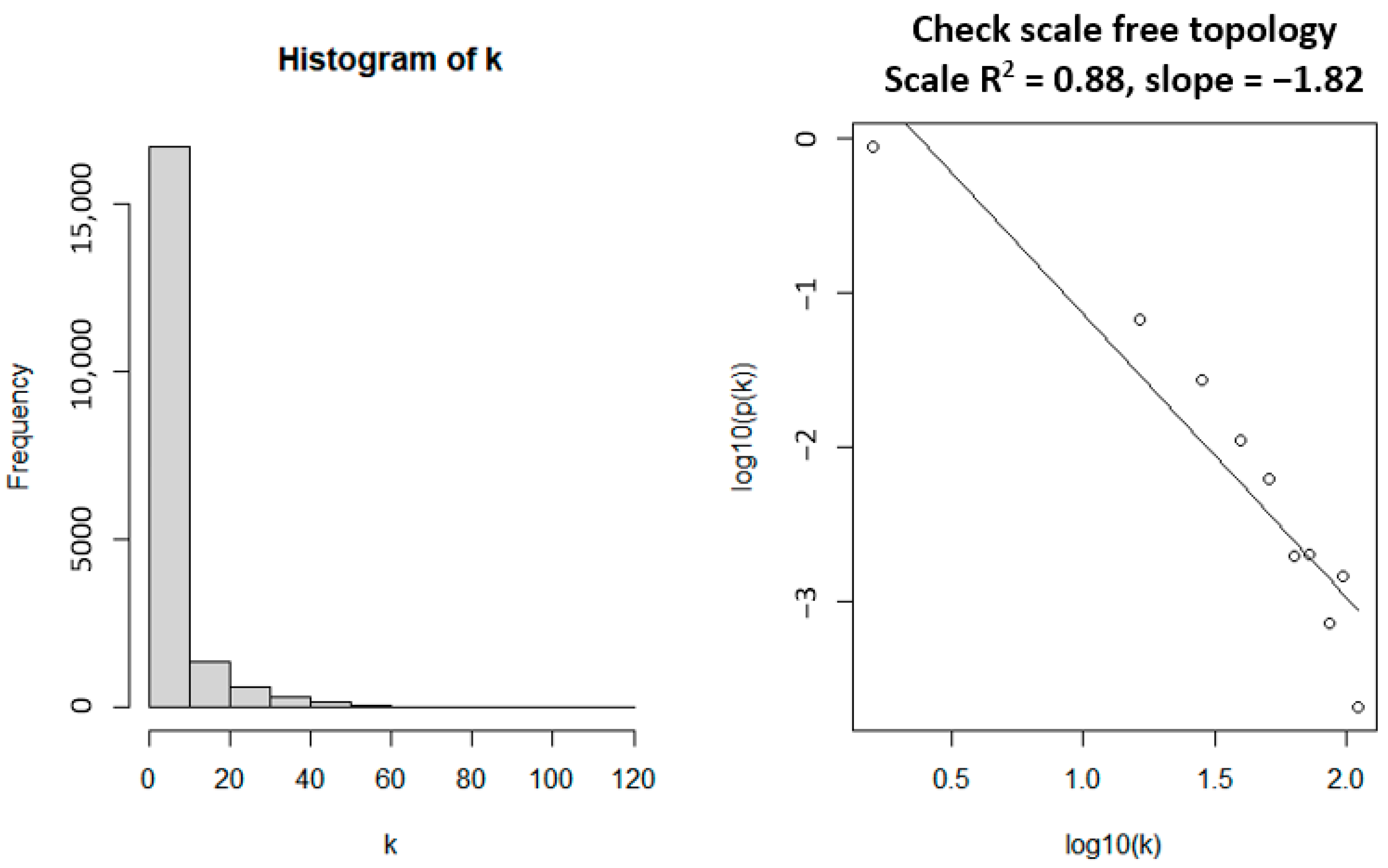
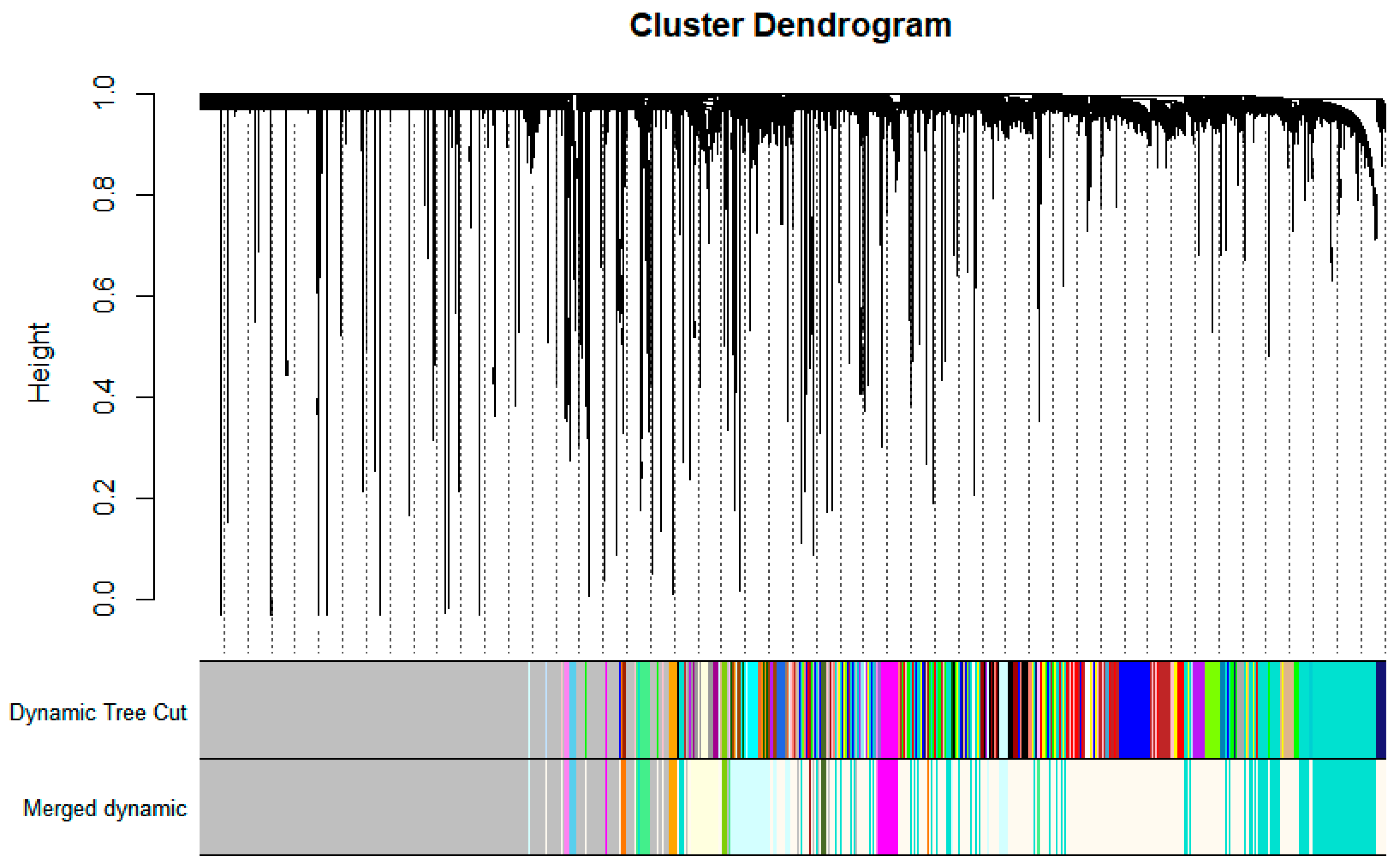
3.1. WGCNA Network Construction
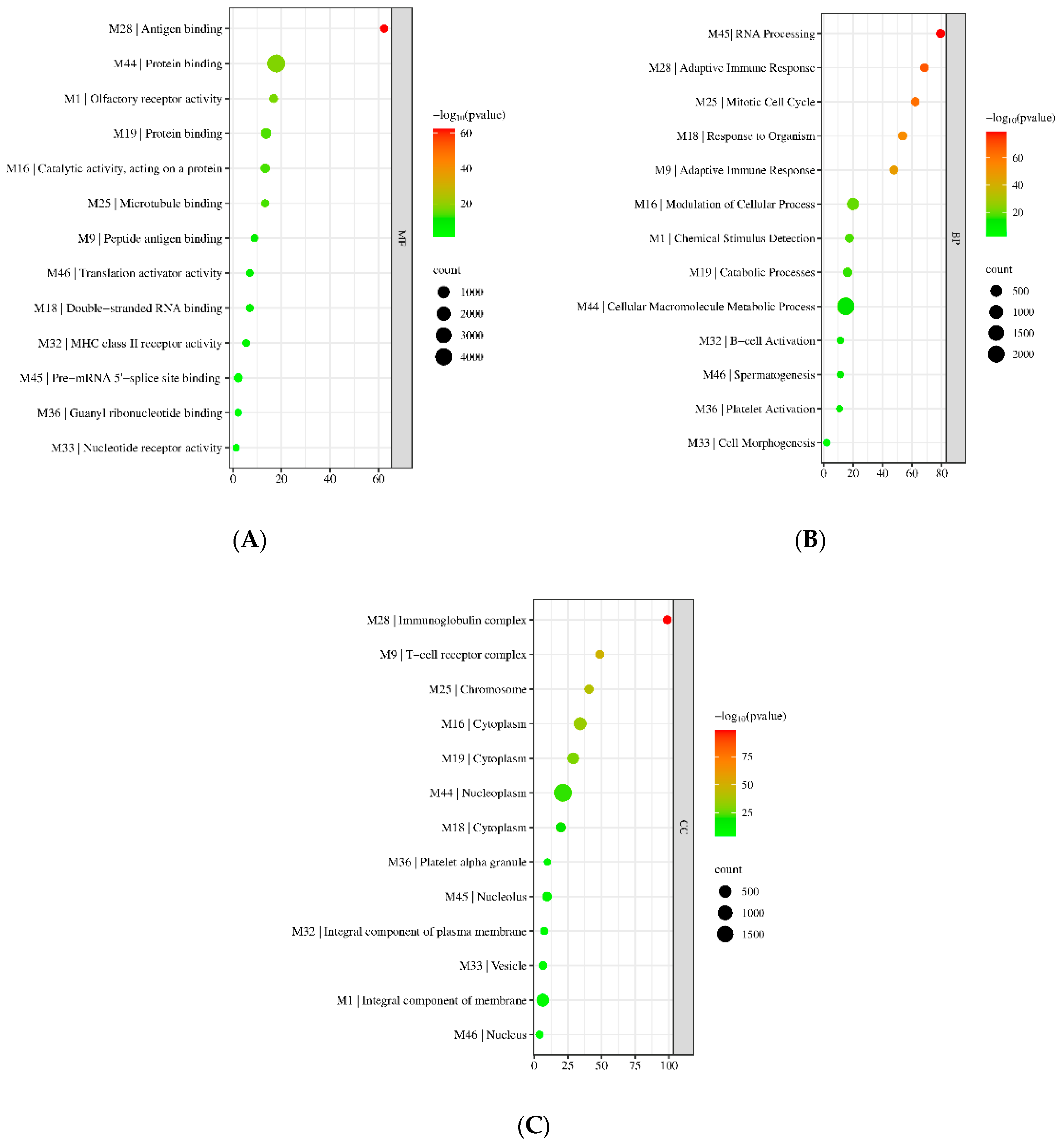
3.2. Gene Ontology (GO) Enrichment Results
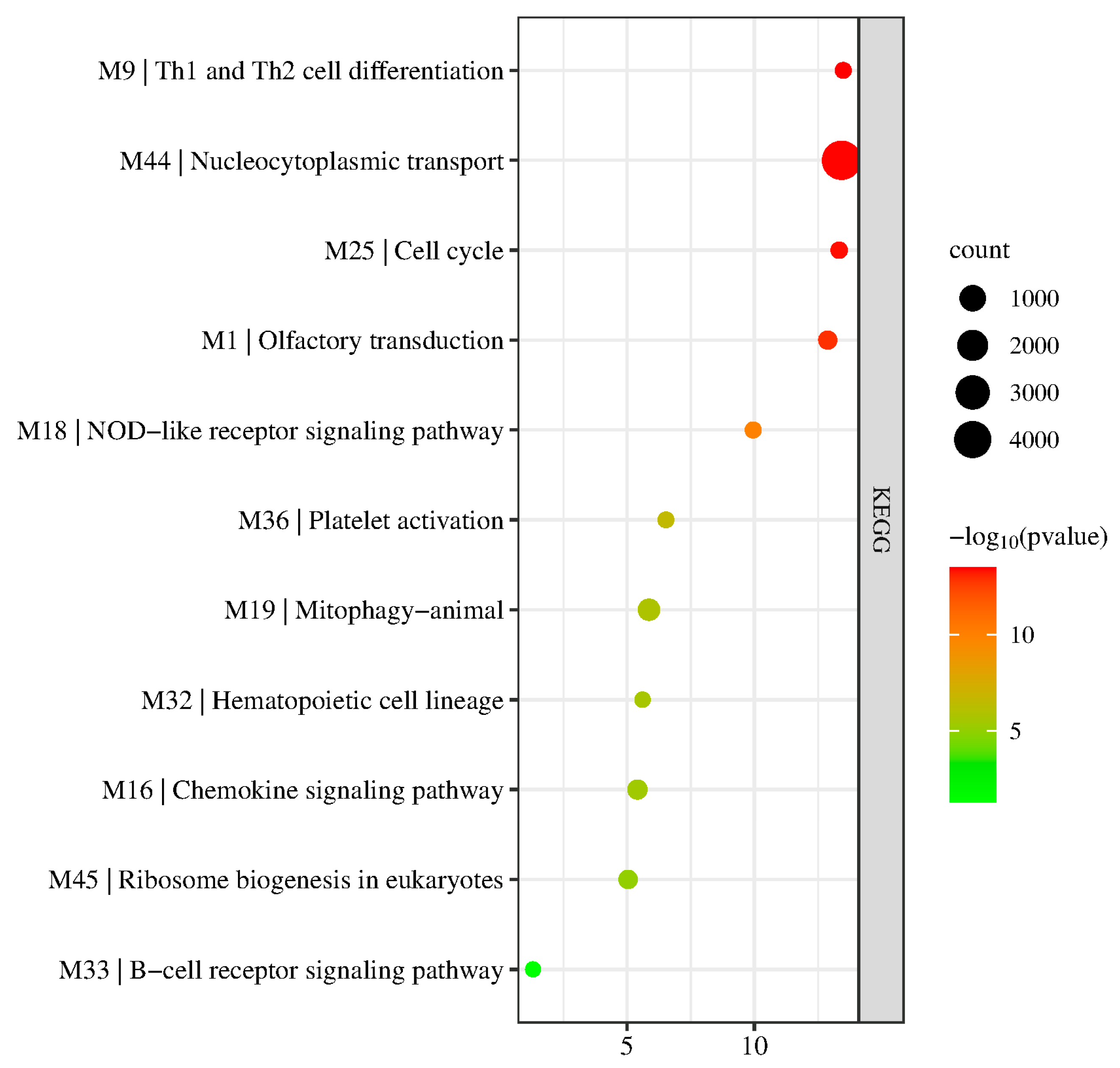
3.3. KEGG Pathway Enrichment
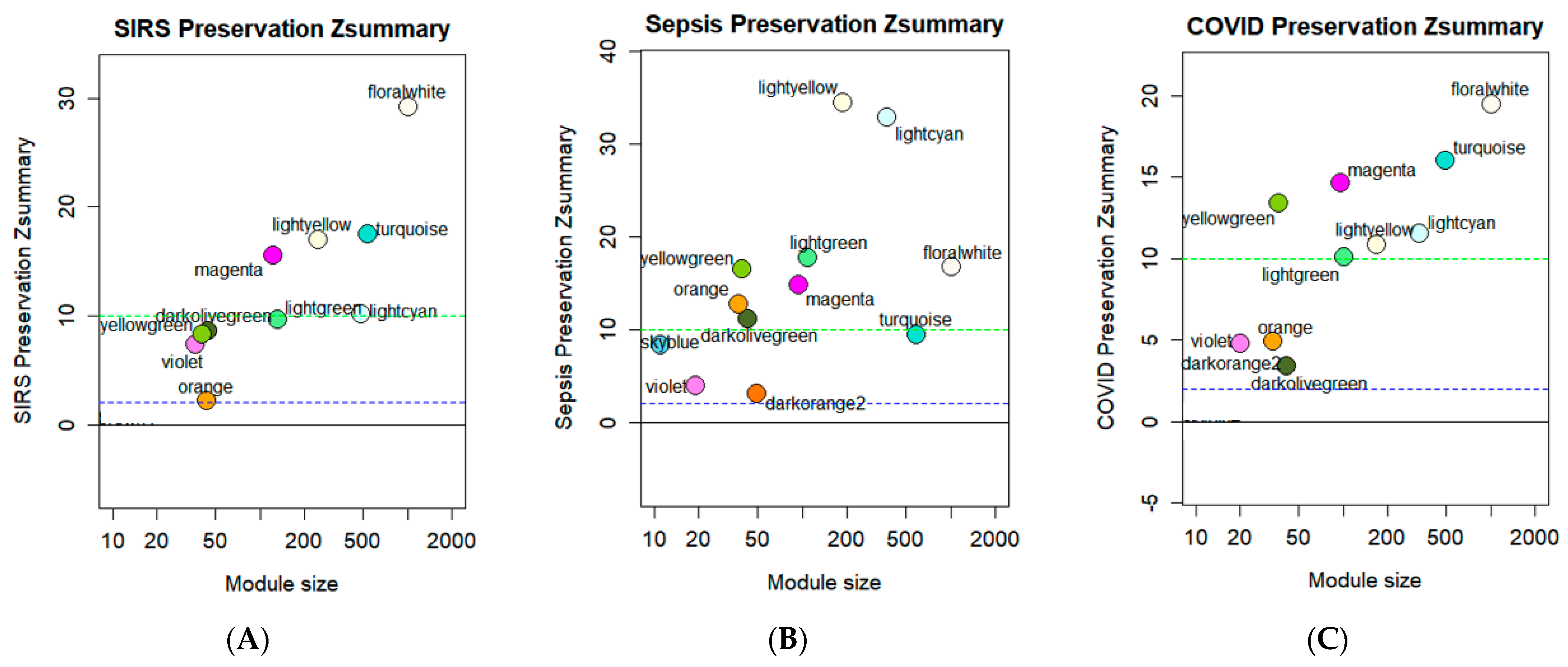
3.4. Module Preservation Analysis
3.5. Hub Gene Identification
3.6. Candidate Drug Identification
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and Immunological Features of Severe and Moderate Coronavirus Disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.-C.; Shih, T.-P.; Ko, W.-C.; Tang, H.-J.; Hsueh, P.-R. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) and Coronavirus Disease-2019 (COVID-19): The Epidemic and the Challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aslan, A.; Aslan, C.; Zolbanin, N.M.; Jafari, R. Acute Respiratory Distress Syndrome in COVID-19: Possible Mechanisms and Therapeutic Management. Pneumonia 2021, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Matthay, M.A.; Calfee, C.S. Is a “Cytokine Storm” Relevant to COVID-19? JAMA Intern. Med. 2020, 180, 1152–1154. [Google Scholar] [CrossRef] [PubMed]
- Nayak, R.R.; Kearns, M.; Spielman, R.S.; Cheung, V.G. Coexpression Network Based on Natural Variation in Human Gene Expression Reveals Gene Interactions and Functions. Genome Res. 2009, 19, 1953–1962. [Google Scholar] [CrossRef] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Darzi, M.; Gorgin, S.; Majidzadeh, A.K.; Esmaeili, R. Gene Co-Expression Network Analysis Reveals Immune Cell Infiltration as a Favorable Prognostic Marker in Non-Uterine Leiomyosarcoma. Sci. Rep. 2021, 11, 2339. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef]
- Li, G.; Ruan, S.; Zhao, X.; Liu, Q.; Dou, Y.; Mao, F. Transcriptomic Signatures and Repurposing Drugs for COVID-19 Patients: Findings of Bioinformatics Analyses. Comput. Struct. Biotechnol. J. 2021, 19, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Huo, J.; Wang, L.; Tian, Y.; Sun, W.; Zhang, G.; Zhang, Y.; Liu, Y.; Zhang, J.; Yang, X.; Liu, Y. Gene Co-Expression Analysis Identified Preserved and Survival-Related Modules in Severe Blunt Trauma, Burns, Sepsis, and Systemic Inflammatory Response Syndrome. Int. J. Gen. Med. 2021, 14, 7065–7076. [Google Scholar] [CrossRef] [PubMed]
- Masi, P.; Hékimian, G.; Lejeune, M.; Chommeloux, J.; Desnos, C.; Pineton De Chambrun, M.; Martin-Toutain, I.; Nieszkowska, A.; Lebreton, G.; Bréchot, N.; et al. Systemic Inflammatory Response Syndrome Is a Major Contributor to COVID-19–Associated Coagulopathy. Circulation 2020, 142, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Ramaswamy, S.; Harilal, D.; Uddin, M.; Loney, T.; Nowotny, N.; Alsuwaidi, H.; Varghese, R.; Deesi, Z.; Alkhajeh, A.; et al. Host Transcriptomic Profiling of COVID-19 Patients with Mild, Moderate, and Severe Clinical Outcomes. Comput. Struct. Biotechnol. J. 2021, 19, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Bhattacharya, M.; Zayed, H.; Lee, S.-S. Understanding Gene Expression and Transcriptome Profiling of COVID-19: An Initiative Towards the Mapping of Protective Immunity Genes Against SARS-CoV-2 Infection. Front. Immunol. 2021, 12, 724936. [Google Scholar] [CrossRef]
- Sun, J.; Ye, F.; Wu, A.; Yang, R.; Pan, M.; Sheng, J.; Zhu, W.; Mao, L.; Wang, M.; Xia, Z.; et al. Comparative Transcriptome Analysis Reveals the Intensive Early Stage Responses of Host Cells to SARS-CoV-2 Infection. Front. Microbiol. 2020, 11, 593857. [Google Scholar] [CrossRef]
- Yehya, N.; Varisco, B.M.; Thomas, N.J.; Wong, H.R.; Christie, J.D.; Feng, R. Peripheral Blood Transcriptomic Sub-Phenotypes of Pediatric Acute Respiratory Distress Syndrome. Crit. Care 2020, 24, 681. [Google Scholar] [CrossRef]
- Kangelaris, K.N.; Prakash, A.; Liu, K.D.; Aouizerat, B.; Woodruff, P.G.; Erle, D.J.; Rogers, A.; Seeley, E.J.; Chu, J.; Liu, T.; et al. Increased Expression of Neutrophil-Related Genes in Patients with Early Sepsis-Induced ARDS. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L1102–L1113. [Google Scholar] [CrossRef] [Green Version]
- McHugh, L.; Seldon, T.A.; Brandon, R.A.; Kirk, J.T.; Rapisarda, A.; Sutherland, A.J.; Presneill, J.J.; Venter, D.J.; Lipman, J.; Thomas, M.R.; et al. A Molecular Host Response Assay to Discriminate Between Sepsis and Infection-Negative Systemic Inflammation in Critically Ill Patients: Discovery and Validation in Independent Cohorts. PLoS Med. 2015, 12, e1001916. [Google Scholar] [CrossRef] [Green Version]
- Masood, K.I.; Yameen, M.; Ashraf, J.; Shahid, S.; Mahmood, S.F.; Nasir, A.; Nasir, N.; Jamil, B.; Ghanchi, N.K.; Khanum, I.; et al. Upregulated Type I Interferon Responses in Asymptomatic COVID-19 Infection Are Associated with Improved Clinical Outcome. Sci. Rep. 2021, 11, 22958. [Google Scholar] [CrossRef]
- Langfelder, P.; Zhang, B.; Horvath, S. Defining Clusters from a Hierarchical Cluster Tree: The Dynamic Tree Cut Package for R. Bioinformatics 2008, 24, 719–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded Annotation Database and Novel Algorithms to Better Extract Biology from Large Gene Lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.-L.; Ideker, T. Cytoscape 2.8: New Features for Data Integration and Network Visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siskind, S.; Brenner, M.; Wang, P. TREM-1 Modulation Strategies for Sepsis. Front. Immunol. 2022, 13, 2944. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Syed, M.; Panchal, D.; Joo, M.; Bedi, C.; Lim, S.; Onyuksel, H.; Rubinstein, I.; Colonna, M.; Sadikot, R.T. Translational Research in Acute Lung Injury and Pulmonary Fibrosis: TREM-1-Accentuated Lung Injury via MiR-155 Is Inhibited by LP17 Nanomedicine. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L426. [Google Scholar] [CrossRef] [Green Version]
- Roe, K.; Gibot, S.; Verma, S. Triggering Receptor Expressed on Myeloid Cells-1 (TREM-1): A New Player in Antiviral Immunity? Front. Microbiol. 2014, 5, 627. [Google Scholar] [CrossRef] [Green Version]
- Denning, N.L.; Aziz, M.; Murao, A.; Gurien, S.D.; Ochani, M.; Prince, J.M.; Wang, P. Extracellular CIRP as an Endogenous TREM-1 Ligand to Fuel Inflammation in Sepsis. JCI Insight 2020, 5, e134172. [Google Scholar] [CrossRef]
- Matsuyama, T.; Kubli, S.P.; Yoshinaga, S.K.; Pfeffer, K.; Mak, T.W. An Aberrant STAT Pathway Is Central to COVID-19. Cell Death Differ. 2020, 27, 3209–3225. [Google Scholar] [CrossRef]
- Li, H.; You, J.; Yang, X.; Wei, Y.; Zheng, L.; Zhao, Y.; Huang, Y.; Jin, Z.; Yi, C. Glycyrrhetinic Acid: A Potential Drug for the Treatment of COVID-19 Cytokine Storm. Phytomedicine 2022, 102, 154153. [Google Scholar] [CrossRef] [PubMed]
- Heissig, B.; Salama, Y.; Takahashi, S.; Osada, T.; Hattori, K. The Multifaceted Role of Plasminogen in Inflammation. Cell Signal. 2020, 75, 109761. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Yan, H. Methyltransferase like 7B Is Upregulated in Sepsis and Modulates Lipopolysaccharide-Induced Inflammatory Response and Macrophage Polarization. Bioengineered 2022, 13, 11753–11766. [Google Scholar] [CrossRef] [PubMed]
- CORDIS; European Commission. Final Report Summary—SNORD104 MICRORNA (SNORD104 Gene-Encoded MicroRNA and Its Role in Immune Homeostasis)|FP7|. Available online: https://cordis.europa.eu/project/id/334079/reporting (accessed on 18 July 2022).
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e17. [Google Scholar] [CrossRef] [PubMed]
- Gil-Etayo, F.J.; Garcinuño, S.; Utrero-Rico, A.; Cabrera-Marante, O.; Arroyo-Sanchez, D.; Mancebo, E.; Pleguezuelo, D.E.; Rodríguez-Frías, E.; Allende, L.M.; Morales-Pérez, P.; et al. An Early Th1 Response Is a Key Factor for a Favorable COVID-19 Evolution. Biomedicines 2022, 10, 296. [Google Scholar] [CrossRef]
- Pavel, A.B.; Glickman, J.W.; Michels, J.R.; Kim-Schulze, S.; Miller, R.L.; Guttman-Yassky, E. Th2/Th1 Cytokine Imbalance Is Associated With Higher COVID-19 Risk Mortality. Front. Genet. 2021, 12, 1273. [Google Scholar] [CrossRef]
- Grimes, J.M.; Grimes, K.v. P38 MAPK Inhibition: A Promising Therapeutic Approach for COVID-19. J. Mol. Cell Cardiol. 2020, 144, 63–65. [Google Scholar] [CrossRef]
- Mailem, R.C.; Tayo, L.L. Identification of Hub Genes and Key Pathways in TNF-α and IFN-γ Induced Cytokine Storms via Bioinformatics. In Proceedings of the 2022 10th International Conference on Bioinformatics and Computational Biology, ICBCB, Hangzhou, China, 13–15 May 2022. [Google Scholar] [CrossRef]
- García De Acilu, M.; Leal, S.; Caralt, B.; Roca, O.; Sabater, J.; Masclans, J.R. The Role of Omega-3 Polyunsaturated Fatty Acids in the Treatment of Patients with Acute Respiratory Distress Syndrome: A Clinical Review. Biomed. Res. Int. 2015, 2015, 653750. [Google Scholar] [CrossRef] [Green Version]
- Eldanasory, O.A.; Eljaaly, K.; Memish, Z.A.; Al-Tawfiq, J.A. Histamine Release Theory and Roles of Antihistamine in the Treatment of Cytokines Storm of COVID-19. Travel Med. Infect. Dis. 2020, 37, 101874. [Google Scholar] [CrossRef]
- Mura, C.; Preissner, S.; Nahles, S.; Heiland, M.; Bourne, P.E.; Preissner, R. Real-World Evidence for Improved Outcomes with Histamine Antagonists and Aspirin in 22,560 COVID-19 Patients. Signal Transduct. Target. Ther. 2021, 6, 1–3. [Google Scholar] [CrossRef]
- Bhatti, F.U.R.; Hasty, K.A.; Cho, H. Anti-Inflammatory Role of TPCA-1 Encapsulated Nanosomes in Porcine Chondrocytes against TNF-α Stimulation. Inflammopharmacology 2019, 27, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Bai, S.; Wang, J.; Ren, N.; Xie, R.; Cheng, G.; Yu, Y. TPCA-1 Negatively Regulates Inflammation Mediated by NF-ΚB Pathway in Mouse Chronic Periodontitis Model. Mol. Oral. Microbiol. 2021, 36, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Soromou, L.W.; Chu, X.; Jiang, L.; Wei, M.; Huo, M.; Chen, N.; Guan, S.; Yang, X.; Chen, C.; Feng, H.; et al. In Vitro and in Vivo Protection Provided by Pinocembrin against Lipopolysaccharide-Induced Inflammatory Responses. Int. Immunopharmacol. 2012, 14, 66–74. [Google Scholar] [CrossRef]
- Tobe, M.; Isobe, Y.; Tomizawa, H.; Nagasaki, T.; Takahashi, H.; Fukazawa, T.; Hayashi, H. Discovery of Quinazolines as a Novel Structural Class of Potent Inhibitors of NF-Kappa B Activation. Bioorg. Med. Chem. 2003, 11, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Fan, Q.; Xu, J.; Bai, J.; Han, X.; Dong, Z.; Zhou, X.; Liu, Z.; Gu, Z.; Wang, C. Calming Cytokine Storm in Pneumonia by Targeted Delivery of TPCA-1 Using Platelet-Derived Extracellular Vesicles. Matter 2020, 3, 287–301. [Google Scholar] [CrossRef]
- Ram, A.; Mabalirajan, U.; Singh, S.K.; Singh, V.P.; Ghosh, B. Mepacrine Alleviates Airway Hyperresponsiveness and Airway Inflammation in a Mouse Model of Asthma. Int. Immunopharmacol. 2008, 8, 893–899. [Google Scholar] [CrossRef]
- Krishna, S.; Augustin, Y.; Wang, J.; Xu, C.; Staines, H.M.; Platteeuw, H.; Kamarulzaman, A.; Sall, A.; Kremsner, P. Repurposing Antimalarials to Tackle the COVID-19 Pandemic. Trends Parasitol. 2021, 37, 8. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2020 | 2015 | 2015 | 2021 | |
|---|---|---|---|---|
| GSE147902 | GSE66890 | GSE74224 | GSE177477 | |
| Type | Expression profiling by array | |||
| Condition | ARDS | Sepsis | SIRS | COVID-19 |
| Platform | Affymetrix Human Gene 2.1 ST Array | Affymetrix Human Gene 1.0 ST Array | Affymetrix Clariom S Assay | |
| Source | Whole Blood RNA | |||
| No. of samples | 96 | 58 | 105 | 47 |
| Module | Color | Top 1 | Top 2 | Top3 |
|---|---|---|---|---|
| 1 | Turquoise | GAPDH | AKT1 | ALB |
| 9 | Magenta | CD8A | CTLA4 | LCK |
| 16 | Light Cyan | CTNNB1 | TLR4 | STAT3 |
| 18 | Light Green | STAT1 | IRF7 | IFIH1 |
| 19 | Light Yellow | UBB | UBA52 | TFRC |
| 36 | Yellow Green | GP6 | PF4 | ITGB3 |
| 44 | Floral White | HDAC1 | HSPA8 | RPS3 |
| Disease | Rank | Name | Connectivity Score | Function |
|---|---|---|---|---|
| COVID-19 | 1 | SB-202190 | −99.44 | P38 MAPK inhibitor |
| 2 | Eicosatetraenoic-acid | −99.40 | COX inhibitor | |
| 3 | Loratadine | −99.37 | Histamine receptor antagonist | |
| 4 | TPCA-1 | −99.12 | IKK inhibitor | |
| 5 | Pinocembrin | −98.58 | CYP1B1 inhibitor | |
| 6 | Mepacrine | −97.22 | Cytokine production inhibitor | |
| 7 | CAY-10470 | −96.80 | NFkβ pathway inhibitor |
| Hub Gene | Function | Reference |
|---|---|---|
| TRIM49D2 | Protein-coding gene predicted to play a role in the innate immune response. | |
| TRAJ12 | Plays a role in joining the two subunits of the T-cell receptor. | |
| ACAP2 | Enables GTPase activator activity. | |
| STAT2 | Induces IFN immune response and has been observed to be aberrant in COVID-19 cytokine storms. | [30] |
| SLC1A5 | Solute carrier which affects ferroptosis, a potential mechanism for tissue damage during cytokine storms. | |
| TOP2A | DNA topoisomerase that has been highlighted in gene expression studies of COVID-19. | [31] |
| IGKV1-6 | Variable domain of immunoglobulin light chains that facilitates antigen recognition. | |
| AFF3 | Transcriptional activator found primarily in lymphoid tissue. | |
| LRP1 | Facilitates cellular movement; controls cellular cytokine signaling. | [32] |
| METTL23 | Methyl transferase that has been reported to be upregulated in sepsis. | [33] |
| SNORD104 | Involved in immune homeostasis. | [34] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mailem, R.C.; Tayo, L.L. Drug Repurposing Using Gene Co-Expression and Module Preservation Analysis in Acute Respiratory Distress Syndrome (ARDS), Systemic Inflammatory Response Syndrome (SIRS), Sepsis, and COVID-19. Biology 2022, 11, 1827. https://doi.org/10.3390/biology11121827
Mailem RC, Tayo LL. Drug Repurposing Using Gene Co-Expression and Module Preservation Analysis in Acute Respiratory Distress Syndrome (ARDS), Systemic Inflammatory Response Syndrome (SIRS), Sepsis, and COVID-19. Biology. 2022; 11(12):1827. https://doi.org/10.3390/biology11121827
Chicago/Turabian StyleMailem, Ryan Christian, and Lemmuel L. Tayo. 2022. "Drug Repurposing Using Gene Co-Expression and Module Preservation Analysis in Acute Respiratory Distress Syndrome (ARDS), Systemic Inflammatory Response Syndrome (SIRS), Sepsis, and COVID-19" Biology 11, no. 12: 1827. https://doi.org/10.3390/biology11121827