
Re-Enlightenment of Fulminant Type 1 Diabetes under the COVID-19 Pandemic

Abstract
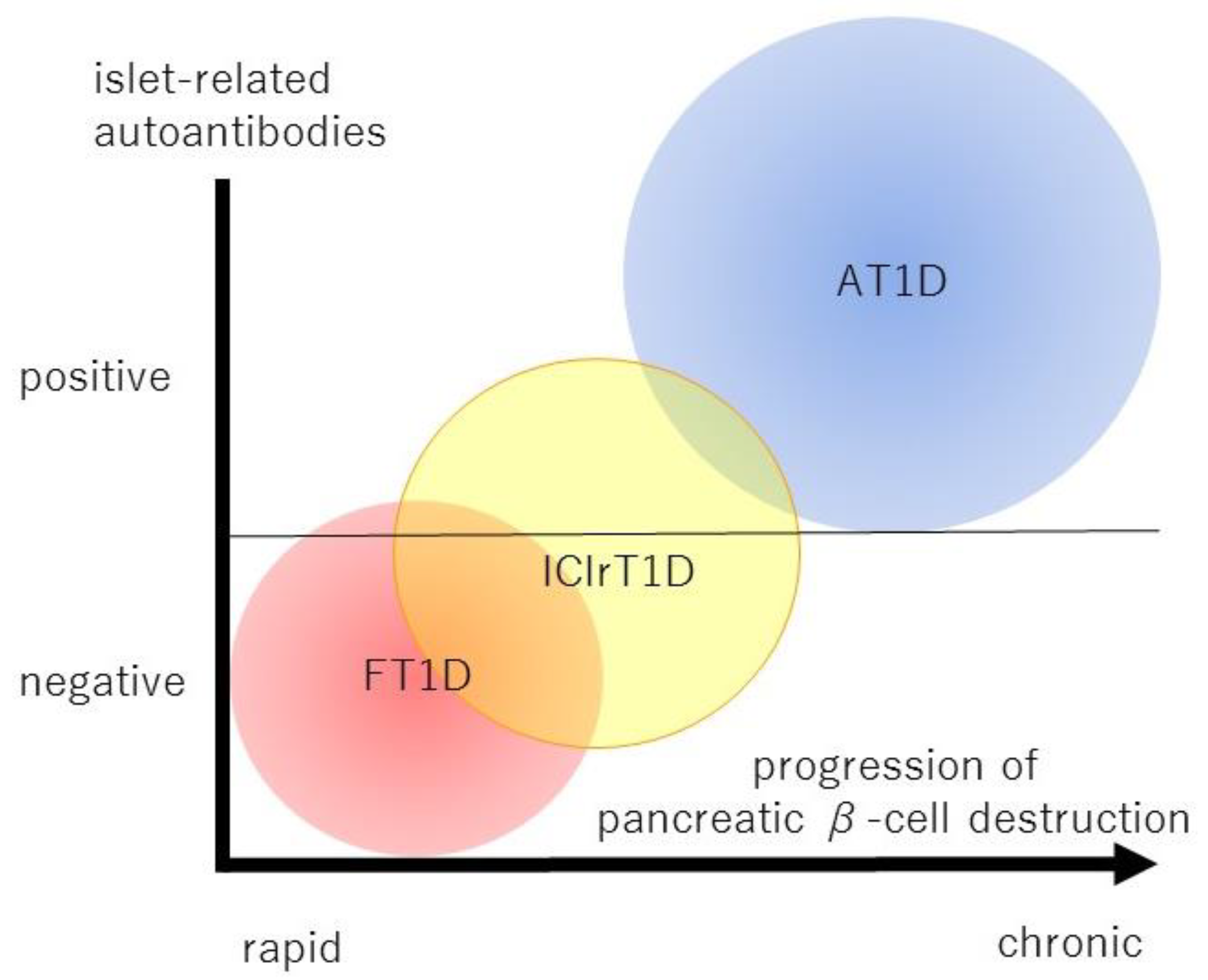
:Simple Summary
Abstract
1. Introduction
2. Clinical Characteristics
3. Treatment of FT1D
4. Etiology
4.1. Disease-Susceptibility Genes
4.2. Viruses and FT1D
4.3. Drug-Induced Hypersensitivity Syndrome (DIHS) and FT1D
4.4. COVID-19 and FT1D
4.5. Mouse Diabetes Model Induced by Encephalomyocarditis Virus (EMCV)
4.6. Pregnancy-Related FT1D (PF)
4.7. Immune Checkpoint Inhibitor (ICI)-Related FT1D
5. Immune Mechanism
5.1. Innate Immunity and FT1D
5.2. Acquired Immunity and FT1D
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Imagawa, A.; Hanafusa, T.; Miyagawa, J.; Matsuzawa, Y.; for the Osaka IDDM Study Group. A Novel Subtype of Type 1 Diabetes Mellitus Characterized by a Rapid Onset and an Absence of Diabetes-Related Antibodies. N. Engl. J. Med. 2000, 342, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, S.; Imagawa, A.; Tauriainen, S.; Iino, M.; Oikarinen, M.; Abiru, H.; Tamaki, K.; Seino, H.; Nishi, K.; Takase, I.; et al. Expression of Toll-Like Receptors in the Pancreas of Recent-Onset Fulminant type 1 Diabetes. Endocr. J. 2010, 57, 211–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imagawa, A.; Hanafusa, T.; Uchigata, Y.; Kanatsuka, A.; Kawasaki, E.; Kobayashi, T.; Shimada, A.; Shimizu, I.; Toyoda, T.; Maruyama, T.; et al. Fulminant type 1 Diabetes: A Nationwide Survey in Japan. Diabetes Care 2003, 26, 2345–2352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.M.; Kim, J.T.; Ko, K.S.; Koo, B.K.; Yang, S.W.; Park, M.H.; Lee, H.K.; Park, K.S. Fulminant type 1 diabetes in Korea: High prevalence among patients with adult-onset type 1 diabetes. Diabetologia 2007, 50, 2276–2279. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Mao, J.; Lu, Z.; Yan, X.; Ye, Y.; Jiang, F. Clinical analysis of fulminant type 1 diabetes in China and comparison with a nationwide survey in Japan. Ann. Endocrinol. 2013, 74, 36–39. [Google Scholar] [CrossRef]
- Murao, S.; Makino, H.; Kaino, Y.; Konoue, E.; Ohashi, J.; Kida, K.; Fujii, Y.; Shimizu, I.; Kawasaki, E.; Fujiyama, M.; et al. Differences in the contribution of HLA-DR and -DQ haplotypes to susceptibility to adult-onset and childhood-onset type 1 diabetes in Japanese patients. Diabetes 2004, 53, 2684–2690. [Google Scholar] [CrossRef] [Green Version]
- Shibasaki, S.; Imagawa, A.; Hanafusa, T. Fulminant Type 1 Diabetes Mellitus: A New Class of Type 1 Diabetes. Adv. Exp. Med. Biol. 2012, 771, 20–23. [Google Scholar] [CrossRef]
- Imagawa, A.; Hanafusa, T. Fulminant Type 1 Diabetes Mellitus. Endocr. J. 2006, 53, 577–584. [Google Scholar] [CrossRef] [Green Version]
- Sekine, N.; Motokura, T.; Oki, T.; Umeda, Y.; Sasaki, N.; Hayashi, M.; Sato, H.; Fujita, T.; Kaneko, T.; Asano, Y.; et al. Rapid Loss of Insulin Secretion in a Patient with Fulminant Type 1 Diabetes Mellitus and Carbamazepine Hypersensitivity Syndrome. JAMA 2001, 285, 1153–1154. [Google Scholar] [CrossRef]
- Hirota, H.; Tsutsumi, C.; Kimata, H.; Watanabe, D.; Imbe, H.; Takamoto, S.; Shibasaki, S.; Sano, H.; Onishi, M.; Furukawa, K.; et al. A Case of Fulminant Type 1 Diabetes Patient Accompanied by Hyperinsulinemic Hypoglycemia Prior to Clinical Diagnosis of Diabetes. J. Jpn. Diabetes Soc. 2016, 59, 210–217. [Google Scholar]
- Imagawa, A.; Hanafusa, T.; Awata, T.; Ikegami, H.; Uchigata, Y.; Osawa, H.; Kawasaki, E.; Kawabata, Y.; Kobayashi, T.; Shimada, A.; et al. Report of the Committee of the Japan Diabetes Society on the Research of Fulminant and Acute-Onset Type 1 Diabetes Mellitus: New Diagnostic Criteria of Fulminant Type 1 Diabetes Mellitus. J. Diabetes Investig. 2012, 3, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Albandar, H.J.; Fuqua, J.; Albandar, J.M.; Safi, S.; Merrill, S.A.; Ma, P.C. Immune-Related Adverse Events (IrAE) in Cancer Immune Checkpoint Inhibitors (ICI) and Survival Outcomes Correlation: To Rechallenge or Not? Cancers 2021, 13, 989. [Google Scholar] [CrossRef] [PubMed]
- Endo, T.; Takizawa, S.; Tanaka, S.; Takahashi, M.; Fujii, H.; Kamisawa, T.; Kobayashi, T. Amylase alpha-2A Autoantibodies: Novel Marker of Autoimmune Pancreatitis and Fulminant type 1 Diabetes. Diabetes 2009, 58, 732–737. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, S.; Endo, T.; Wanjia, X.; Tanaka, S.; Takahashi, M.; Kobayashi, T. HSP 10 Is a New Autoantigen in Both Autoimmune Pancreatitis and Fulminant type 1 Diabetes. Biochem. Biophys. Res. Commun. 2009, 386, 192–196. [Google Scholar] [CrossRef]
- Haseda, F.; Imagawa, A.; Nishikawa, H.; Mitsui, S.; Tsutsumi, C.; Fujisawa, R.; Sano, H.; Murase-Mishiba, Y.; Terasaki, J.; Sakaguchi, S.; et al. Antibody to CMRF35-Like Molecule 2, CD300e A Novel Biomarker Detected in Patients with Fulminant Type 1 Diabetes. PLoS ONE 2016, 11, e0160576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oram, R.A.; Jones, A.G.; Besser, R.E.J.; Knight, B.A.; Shields, B.M.; Brown, R.J.; Hattersley, A.T.; McDonald, T.J. Erratum to: The Majority of Patients with Long-Duration Type 1 Diabetes Are Insulin Microsecretors and have Functioning Beta Cells. Diabetologia 2014, 57, 262. [Google Scholar] [CrossRef] [Green Version]
- Infante, M.; Ricordi, C. Editorial—Moving Forward on the Pathway of Targeted Immunotherapies for Type 1 Diabetes: The Importance of Disease Heterogeneity. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8702–8704. [Google Scholar]
- Murase, Y.; Imagawa, A.; Hanafusa, T.; Iwahashi, H.; Uchigata, Y.; Kanatsuka, A.; Kawasaki, E.; Kobayashi, T.; Shimada, A.; Shimizu, I.; et al. Fulminant Type 1 Diabetes as a High Risk Group for Diabetic Microangiopathy—A Nationwide 5-Year-Study in Japan. Diabetologia 2007, 50, 531–537. [Google Scholar] [CrossRef] [Green Version]
- Takaike, H.; Uchigata, Y.; Nakagami, T.; Iwamoto, Y. Incidence and Development of Diabetic Microangiopathy of Fulminant Type 1 Diabetes—Comparison with Non-Fulminant Type 1 Diabetes. Intern. Med. 2010, 49, 1079–1083. [Google Scholar] [CrossRef] [Green Version]
- Beik, P.; Ciesielska, M.; Kucza, M.; Kurczewska, A.; Kuźmińska, J.; Maćkowiak, B.; Niechciał, E. Prevention of Type 1 Diabetes: Past Experiences and Future Opportunities. J. Clin. Med. 2020, 9, 2805. [Google Scholar] [CrossRef]
- Bougnères, P.F.; Landais, P.; Boisson, C.; Carel, J.C.; Frament, N.; Boitard, C.; Chaussain, J.L.; Bach, J.F. Limited Duration of Remission of Insulin Dependency in Children with Recent Overt Type I Diabetes Treated with Low-Dose Cyclosporin. Diabetes 1990, 39, 1264–1272. [Google Scholar] [CrossRef] [PubMed]
- Keymeulen, B.; Vandemeulebroucke, E.; Ziegler, A.G.; Mathieu, C.; Kaufman, L.; Hale, G.; Gorus, F.; Goldman, M.; Walter, M.; Candon, S.; et al. Insulin Needs after CD3-Antibody Therapy in New-Onset Type 1 Diabetes. N. Engl. J. Med. 2005, 352, 2598–2608. [Google Scholar] [CrossRef]
- Keymeulen, B.; Walter, M.; Mathieu, C.; Kaufman, L.; Gorus, F.; Hilbrands, R.; Vandemeulebroucke, E.; Van De Velde, U.; Crenier, L.; De Block, C.; et al. Four-year metabolic outcome of a randomised controlled CD3-antibody trial in recent-onset type 1 diabetic patients depends on their age and baseline residual beta cell mass. Diabetologia 2010, 53, 614–623. [Google Scholar] [CrossRef] [PubMed]
- Sherry, N.; Hagopian, W.; Ludvigsson, J.; Jain, S.M.; Wahlen, J.; Ferry, R.J.; Bode, B.; Aronoff, S.; Holland, C.; Carlin, D.; et al. Teplizumab for Treatment of Type 1 Diabetes (Protégé study): 1-year Results from a Randomised, Placebo-controlled Trial. Lancet 2011, 378, 487–497. [Google Scholar] [CrossRef] [Green Version]
- Orban, T.; Bundy, B.; Becker, D.J.; DiMeglio, L.A.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Greenbaum, C.J.; Marks, J.B.; Monzavi, R.; et al. Co-stimulation Modulation with Abatacept in Patients with Recent-onset Type 1 Diabetes: A randomised, double-blind, placebo-controlled trial. Lancet 2011, 378, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Pescovitz, M.D.; Greenbaum, C.J.; Krause-Steinrauf, H.; Becker, D.J.; Gitelman, S.E.; Goland, R.; Gottlieb, P.A.; Marks, J.B.; McGee, P.F.; Moran, A.M.; et al. Rituximab, B-Lymphocyte Depletion, and Preservation of Beta-Cell Function. N. Engl. J. Med. 2009, 361, 2143–2152. [Google Scholar] [CrossRef]
- Ludvigsson, J.; Faresjö, M.; Hjorth, M.; Axelsson, S.; Chéramy, M.; Pihl, M.; Vaarala, O.; Forsander, G.; Ivarsson, S.; Johansson, C.; et al. GAD Treatment and Insulin Secretion in Recent-Onset Type 1 Diabetes. N. Engl. J. Med. 2008, 359, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Herold, K.C.; Bundy, B.N.; Long, S.A.; Bluestone, J.A.; DiMeglio, L.A.; Dufort, M.J.; Gitelman, S.E.; Gottlieb, P.A.; Krischer, J.P.; Linsley, P.S.; et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N. Engl. J. Med. 2019, 381, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Stone, V.M.; Hankaniemi, M.M.; Svedin, E.; Sioofy-Khojine, A.; Oikarinen, S.; Hyöty, H.; Laitinen, O.H.; Hytönen, V.P.; Flodström-Tullberg, M. A Coxsackievirus B Vaccine Protects against Virus-Induced Diabetes in an Experimental Mouse Model of Type 1 Diabetes. Diabetologia 2018, 61, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Imagawa, A.; Hanafusa, T.; Uchigata, Y.; Kanatsuka, A.; Kawasaki, E.; Kobayashi, T.; Shimada, A.; Shimizu, I.; Maruyama, T.; Makino, H. Different Contribution of Class II HLA in Fulminant and Typical Autoimmune Type 1 Diabetes Mellitus. Diabetologia 2005, 48, 294–300. [Google Scholar] [CrossRef]
- Tsutsumi, C.; Imagawa, A.; Ikegami, H.; Makino, H.; Kobayashi, T.; Hanafusa, T.; Japan Diabetes Society Committee on Type 1 Diabetes Mellitus Research. Class II HLA Genotype in Fulminant type 1 Diabetes: A Nationwide Survey with Reference to Glutamic Acid Decarboxylase Antibodies. J. Diabetes Investig. 2012, 3, 62–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawabata, Y.; Nishida, N.; Awata, T.; Kawasaki, E.; Imagawa, A.; Shimada, A.; Osawa, H.; Tanaka, S.; Takahashi, K.; Nagata, M.; et al. Genome-Wide Association Study Confirming a Strong Effect of HLA and Identifying Variants in CSAD/lnc-ITGB7-1 on Chromosome 12q13.13 Associated with Susceptibility to Fulminant Type 1 Diabetes. Diabetes 2019, 68, 665–675. [Google Scholar] [CrossRef]
- Arany, E.; Strutt, B.; Romanus, P.; Remacle, C.; Reusens, B.; Hill, D.J. Taurine Supplement in Early Life Altered Islet Morphology, Decreased Insulitis and Delayed the Onset of Diabetes in Non-Obese Diabetic Mice. Diabetologia 2004, 47, 1831–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berlin, C.; Berg, E.L.; Briskin, M.J.; Andrew, D.P.; Kilshaw, P.J.; Holzmann, B.; Weissman, I.L.; Hamann, A.; Butcher, E.C. Alpha 4 Beta 7 Integrin Mediates Lymphocyte Binding to the Mucosal Vascular Addressin MAdCAM-1. Cell 1993, 74, 185–195. [Google Scholar] [CrossRef]
- Hanafusa, T.; Imagawa, A.; Iwahashi, H.; Uchigata, Y.; Kanatsuka, A.; Kawasaki, E.; Kobayashi, T.; Shimada, A.; Shimizu, I.; Maruyama, T.; et al. Report of the Japan Diabetes Society’s Committee on Research on Fulminant Type 1 Diabetes Mellitus: Analysis of Antiviral Antibodies at Disease Onset. J. Jpn. Diab. Soc. 2008, 51, 531–536. [Google Scholar]
- Hanafusa, T.; Imagawa, A. Fulminant type 1 Diabetes: A Novel Clinical Entity Requiring Special Attention by All Medical Practitioners. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 36–45. [Google Scholar] [CrossRef]
- Imagawa, A.; Hanafusa, T. Fulminant type 1 Diabetes--an Important Subtype in East Asia. Diabetes Metab. Res. Rev. 2011, 27, 959–964. [Google Scholar] [CrossRef]
- Goto, A.; Takahashi, Y.; Kishimoto, M.; Nakajima, Y.; Nakanishi, K.; Kajio, H.; Noda, M. A Case of Fulminant Type 1 Diabetes Associated with Significant Elevation of Mumps Titers. Endocr. J. 2008, 55, 561–564. [Google Scholar] [CrossRef] [Green Version]
- Akatsuka, H.; Yano, Y.; Gabazza, E.C.; Morser, J.; Sasaki, R.; Suzuki, T.; Fujiwara, R.; Katsuki, A.; Takei, Y.; Sumida, Y. A Case of Fulminant type 1 Diabetes with Coxsackie B4 Virus Infection Diagnosed by Elevated Serum Levels of Neutralizing Antibody. Diabetes Res. Clin. Pract. 2009, 84, e50–e52. [Google Scholar] [CrossRef]
- Sano, H.; Terasaki, J.; Tsutsumi, C.; Imagawa, A.; Hanafusa, T. A Case of Fulminant Type 1 Diabetes Mellitus After Influenza B Infection. Diabetes Res. Clin. Pract. 2008, 79, e8–e9. [Google Scholar] [CrossRef]
- Kawasaki, S.; Kuroiwa, S.; Mikami, T.; Shibata, H.; Misawa, H.; Tamura, Y.; Yamamoto, S.; Terauchi, Y. A Case of Fulminant Type 1 Diabetes Mellitus Which was Estimated to Triggered by Enterovirus Infection after 25 Years of the Vogt-Koyanagi-Harada (VKH) Disease Onset. J. Jpn. Diab. Soc. 2016, 59, 768–774. [Google Scholar]
- Ohta, M.; Miura, J.; Ohsato, A.; Maruyama, S.; Ishii, A.; Iizuka, J.; Tanabe, K.; Iwamoto, Y. A Case of Fulminant Type 1 Diabetes Mellitus Undergoing Immunosuppression Therapy after Kidney Transplantation with Cytomegalovirus Antigenemia Elevation. J. Jpn. Diab. Soc. 2009, 52, 919–925. [Google Scholar]
- Ohara, N.; Kaneko, M.; Nishibori, T.; Sato, K.; Furukawa, T.; Koike, T.; Sone, H.; Kaneko, K.; Kamoi, K. Fulminant Type 1 Diabetes Mellitus Associated with Coxsackie Virus Type A2 Infection: A Case Report and Literature Review. Intern. Med. 2016, 55, 643–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirtipal, N.; Bharadwaj, S.; Kang, S.G. From SARS to SARS-CoV-2, Insights on Structure, Pathogenicity and Immunity Aspects of Pandemic Human Coronaviruses. Infect. Genet. Evol. 2020, 85, 104502. [Google Scholar] [CrossRef]
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.-L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.; et al. Angiotensin-converting Enzyme 2 (ACE2), SARS-CoV-2 and the Pathophysiology of Coronavirus Disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248. [Google Scholar] [CrossRef]
- Imagawa, A.; Hanafusa, T.; Makino, H.; Miyagawa, J.I.; Juto, P. High Titres of IgA Antibodies to Enterovirus in Fulminant type-1 Diabetes. Diabetologia 2005, 48, 290–293. [Google Scholar] [CrossRef] [Green Version]
- Pak, C.Y.; Eun, H.M.; Mcarthur, R.G.; Yoon, J.W. Association of Cytomegalovirus Infection with Autoimmune Type 1 Diabe-Tes. Lancet 1988, 2, 1–4. [Google Scholar] [CrossRef]
- Honeyman, M.C.; Stone, N.L.; Harrison, L.C. T-Cell Epitopes in Type 1 Diabetes Autoantigen Tyrosine Phosphatase IA-2: Potential for Mimicry with Rotavirus and Other Environmental Agents. Mol. Med. 1998, 4, 231–239. [Google Scholar] [CrossRef] [Green Version]
- Filippi, C.M.; von Herrath, M.G. Viral Trigger for Type 1 Diabetes. Diabetes 2008, 57, 2863–2871. [Google Scholar] [CrossRef] [Green Version]
- Oikarinen, M.; Tauriainen, S.; Honkanen, T.; Oikarinen, S.; Vuori, K.; Kaukinen, K.; Rantala, I.; Maki, M.; Hyoty, H. Detection of Enteroviruses in the Intestine of Type 1 Diabetic Patients. Clin. Exp. Immunol. 2008, 151, 71–75. [Google Scholar] [CrossRef]
- Krogvold, L.; Edwin, B.; Buanes, T.; Frisk, G.; Skog, O.; Anagandula, M.; Korsgren, O.; Undlien, D.; Eike, M.C.; Richardson, S.J. Detection of a Low-grade Enteroviral Infection in the Islets of Langerhans of Living Patients Newly Diagnosed with Type 1 Diabetes. Diabetes 2015, 64, 1682–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geravandi, S.; Richardson, S.; Pugliese, A.; Maedler, K. Localization of Enteroviral RNA within the Pancreas in Donors with T1D and T1D-Associated Autoantibodies. Cell Rep. Med. 2021, 2, 100371. [Google Scholar] [CrossRef] [PubMed]
- Insel, R.A.; Dunne, J.L.; Atkinson, M.A.; Chiang, J.L.; Dabelea, D.; Gottlieb, P.A.; Greenbaum, C.J.; Herold, K.C.; Krischer, J.P.; Lernmark, Å.; et al. Staging Presymptomatic Type 1 Diabetes: A Scientific Statement of JDRF, the Endocrine Society, and the American Diabetes Association. Diabetes Care 2015, 38, 1964–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, S.J.; Morgan, N.G. Enteroviral Infections in the Pathogenesis of Type 1 Diabetes: New Insights for Therapeutic Intervention. Curr. Opin. Pharmacol. 2018, 43, 11. [Google Scholar] [CrossRef] [PubMed]
- Elshebani, A.; Olsson, A.; Westman, J.; Tuvemo, T.; Korsgren, O.; Frisk, G. Effects on Isolated Human Pancreatic Islet Cells after Infection with Strains of Enterovirus Isolated at Clinical Presentation of Type 1 Diabetes. Virus Res. 2007, 124, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Shiohara, T.; Kano, Y.; Hirahara, K.; Aoyama, Y. Prediction and Management of Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS). Expert Opin. Drug Metab. Toxicol. 2017, 13, 701–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiohara, T.; Mizukawa, Y. Drug-Induced Hypersensitivity Syndrome (DiHS)/Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS): An Update in 2019. Allergol. Int. 2019, 68, 301–308. [Google Scholar] [CrossRef]
- Kano, Y.; Inaoka, M.; Shiohara, T. Association between Anticonvulsant Hypersensitivity Syndrome and Human Herpesvirus 6 Reactivation and Hypogammaglobulinemia. Arch. Dermatol. 2004, 140, 183–188. [Google Scholar] [CrossRef] [Green Version]
- Sugita, K.; Tohyama, M.; Watanabe, H.; Otsuka, A.; Nakajima, S.; Iijima, M.; Hashimoto, K.; Tokura, Y.; Miyachi, Y.; Kabashima, K. Fluctuation of Blood and Skin Plasmacytoid Dendritic Cells in Drug-Induced Hypersensitivity Syndrome. J. Allergy Clin. Immunol. 2010, 126, 408–410. [Google Scholar] [CrossRef]
- Kano, Y.; Hiraharas, K.; Sakuma, K.; Shiohara, T. Several Herpesviruses Can Reactivate in a Severe Drug-Induced Multiorgan Reaction in the Same Sequential Order as in Graft-Versus-Host Disease. Br. J. Dermatol. 2006, 155, 301–306. [Google Scholar] [CrossRef]
- Yoneda, S.; Imagawa, A.; Fukui, K.; Uno, S.; Kozawa, J.; Sakai, M.; Yumioka, T.; Iwahashi, H.; Shimomura, I. A Histological Study of Fulminant Type 1 Diabetes Mellitus Related to Human Cytomegalovirus Reactivation. J. Clin. Endocrinol. Metab. 2017, 102, 2394–2400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onuma, H.; Tohyama, M.; Imagawa, A.; Hanafusa, T.; Kobayashi, T.; Kano, Y.; Ohashi, J.; Hashimoto, K.; Osawa, H.; Makino, H.; et al. High Frequency of HLA B62 in Fulminant Type 1 Diabetes with the Drug-Induced Hypersensitivity Syndrome. J. Clin. Endocrinol. Metab. 2012, 97, E2277–E2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, J.A.; Groß, R.; Conzelmann, C.; Krüger, J.; Merle, U.; Steinhart, J.; Weil, T.; Koepke, L.; Bozzo, C.P.; Read, C.; et al. SARS-CoV-2 Infects and Replicates in Cells of the Human Endocrine and Exocrine Pancreas. Nature Metab. 2021, 3, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, J. Pancreatic β-cell fate in subjects with COVID-19. J. Diabetes Investig. 2021, 12, 2126–2128. [Google Scholar] [CrossRef]
- Yang, L.; Han, Y.; Nilsson-Payant, B.E.; Gupta, V.; Wang, P.; Duan, X.; Tang, X.; Zhu, J.; Zhao, Z.; Jaffré, F.; et al. A Human Pluripotent Stem Cell-Based Platform to Study SARS-CoV-2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem Cell. 2020, 27, 125–136. [Google Scholar] [CrossRef]
- Geravandi, S.; Mahmoudi-aznaveh, A.; Azizi, Z.; Maedler, K.; Ardestani, A. SARS-CoV-2 and Pancreas: A Potential Pathological Interaction? Trends Endocrinol. Metab. 2021, 32, 842–845. [Google Scholar] [CrossRef]
- Wu, C.-T.; Lidsky, P.V.; Xiao, Y.; Lee, I.T.; Cheng, R.; Nakayama, T.; Jiang, S.; Demeter, J.; Bevacqua, R.J.; Chang, C.A.; et al. SARS-CoV-2 Infects Human Pancreatic β Cells and Elicits β Cell Impairment. Cell Metab. 2021, 33, 1565–1576. [Google Scholar] [CrossRef]
- Tang, X.; Uhl, S.; Zhang, T.; Xue, D.; Li, B.; Vandana, J.J.; Acklin, J.A.; Bonnycastle, L.L.; Narisu, N.; Erdos, M.R.; et al. SARS-CoV-2 infection induces beta cell transdifferentiation. Cell Metab. 2021, 33, 1577–1591.e7. [Google Scholar] [CrossRef]
- Rathmann, W.; Kuss, O.; Kostev, K. Incidence of Newly Diagnosed Diabetes after COVID-19. Diabetologia 2022, 65, 949–954. [Google Scholar] [CrossRef]
- Kuchay, M.S.; Reddy, P.K.; Gagneja, S.; Mathew, A.; Mishra, S.K. Short Term Follow-up of Patients Presenting with Acute Onset Diabetes and Diabetic Ketoacidosis during an Episode of COVID-19. Diabetes Metab. Syndr. 2020, 14, 2039–2041. [Google Scholar] [CrossRef]
- Yang, J.-K.; Lin, S.-S.; Ji, X.-J.; Guo, L.-M. Binding of SARS Coronavirus to Its Receptor Damages Islets and Causes Acute Diabetes. Acta Diabetol. 2010, 47, 193–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.K.; Kuchay, M.S.; Mehta, Y.; Mishra, S.K. Diabetic Ketoacidosis Precipitated by COVID-19: A Report of Two Cases and Review of Literature. Diabetes Metab. Syndr. 2020, 14, 1459–1462. [Google Scholar] [CrossRef] [PubMed]
- Paengsai, N.; Jourdain, G.; Salvadori, N.; Tantraworasin, A.; Mary, J.Y.; Cressey, T.R.; Chaiwarith, R.; Bowonwatanuwong, C.; Bhakeecheep, S.; Kosachunhanun, N. Recommended First-Line Antiretroviral Therapy Regimens and Risk of Diabetes Mellitus in HIV-Infected Adults in Resource-Limited Settings. Open Forum Infect. Dis. 2019, 6, ofz298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, F.; Gao, D.; Ma, X.; Guo, Y.; Wang, R.; Jiang, W.; Gong, S. Corticosteroids in diabetes patients infected with COVID-19. Ir. J. Med. Sci. 2021, 190, 29–31. [Google Scholar] [CrossRef]
- Montefusco, L.; Ben Nasr, M.; D’Addio, F.; Loretelli, C.; Rossi, A.; Pastore, I.; Daniele, G.; Abdelsalam, A.; Maestroni, A.; Dell’Acqua, M.; et al. Acute and Long-Term Disruption of Glycometabolic Control after SARS-CoV-2 Infection. Nat. Metab. 2021, 3, 774–785. [Google Scholar] [CrossRef]
- Sefik, E.; Qu, R.; Junqueira, C.; Kaffe, E.; Mirza, H.; Zhao, J.; Brewer, J.R.; Han, A.; Steach, H.R.; Israelow, B.; et al. Inflammasome activation in infected macrophages drives COVID-19 pathology. Nature 2022, 606, 585–593. [Google Scholar] [CrossRef]
- Unsworth, R.; Wallace, S.; Oliver, N.S.; Yeung, S.; Kshirsagar, A.; Naidu, H.; Kwong, R.M.W.; Kumar, P.; Logan, K.M. New-Onset Type 1 Diabetes in Children during COVID-19: Multicenter Regional Findings in the U.K. Diabetes Care 2020, 43, e170–e171. [Google Scholar] [CrossRef]
- Tittel, S.R.; Rosenbauer, J.; Kamrath, C.; Ziegler, J.; Reschke, F.; Hammersen, J.; Mönkemöller, K.; Pappa, A.; Kapellen, T.; Holl, R.W.; et al. Did the COVID-19 Lockdown Affect the Incidence of Pediatric Type 1 Diabetes in Germany? Diabetes Care 2020, 43, e172–e173. [Google Scholar] [CrossRef]
- Qeadan, F.; Tingey, B.; Egbert, J.; Pezzolesi, M.G.; Burge, M.R.; Peterson, K.A.; Honda, T. The associations between COVID-19 diagnosis, type 1 diabetes, and the risk of diabetic ketoacidosis: A nationwide cohort from the US using the Cerner Real-World Data. PLoS ONE 2022, 17, e0266809. [Google Scholar] [CrossRef]
- McGlacken-Byrne, S.M.; Drew, S.E.V.; Turner, K.; Peters, C.; Amin, R. The SARS-CoV-2 Pandemic Is Associated with Increased Severity of Presentation of Childhood Onset Type 1 Diabetes Mellitus: A Multi-Centre Study of the First COVID-19 Wave. Diabet. Med. 2021, 38, e14640. [Google Scholar] [CrossRef]
- Goldman, S.; Pinhas-Hamiel, O.; Weinberg, A.; Auerbach, A.; German, A.; Haim, A.; Zung, A.; Brener, A.; Strich, D.; Azoulay, E.; et al. Alarming Increase in Ketoacidosis in Children and Adolescents with Newly Diagnosed Type 1 Diabetes during the First Wave of the COVID-19 Pandemic in Israel. Pediatr. Diabetes 2022, 23, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Beliard, K.; Ebekozien, O.; Demeterco-Berggren, C.; Alonso, G.T.; Gallagher, M.P.; Clements, M.; Rapaport, R. Increased DKA at presentation among newly diagnosed type 1 diabetes patients with or without COVID-19: Data from a multi-site surveillance registry. J. Diabetes 2021, 13, 270–272. [Google Scholar] [CrossRef]
- Lazzeroni, P.; Bernardi, L.; Pecora, F.; Motta, M.; Bianchi, L.; Ruozi, M.B.; Giacometti, A.; Venezia, S.; Iovane, D.I. Diabetic Ke-Toacidosis at Type 1 Diabetes Onset: Indirect Impact of COVID-19 Pandemic. Acta Biomed. 2020, 91, e2020193. [Google Scholar] [PubMed]
- Dżygało, K.; Nowaczyk, J.; Szwilling, A.; Kowalska, A. Increased Frequency of Severe Diabetic Ketoacidosis at Type 1 Diabetes Onset among Children during COVID-19 Pandemic Lockdown: An Observational Cohort Study. Pediatr. Endocrinol. Diabetes Metab. 2020, 26, 167–175. [Google Scholar] [CrossRef]
- Alaqeel, A.; Aljuraibah, F.; Alsuhaibani, M.; Huneif, M.; Alsaheel, A.; Dubayee, M.A.; Alsaedi, A.; Bakkar, A.; Alnahari, A.; Taha, A.; et al. The Impact of COVID-19 Pandemic Lockdown on the Incidence of New-Onset Type 1 Diabetes and Ketoacidosis among Saudi Children. Front. Endocrinol. 2021, 12, 669302. [Google Scholar] [CrossRef] [PubMed]
- Salmi, H.; Heinonen, S.; Hästbacka, J.; Lääperi, M.; Rautiainen, P.; Miettinen, P.J.; Vapalahti, O.; Hepojoki, J.; Knip, M. New-Onset Type 1 Diabetes in Finnish Children during the COVID-19 Pandemic. Arch. Dis. Child. 2022, 107, 180–185. [Google Scholar] [CrossRef]
- Li, M.; Wang, H.; Tian, L.; Pang, Z.; Yang, Q.; Huang, T.; Fan, J.; Song, L.; Tong, Y.; Fan, H. COVID-19 vaccine development: Milestones, lessons and prospects. Signal Transduct. Target. Ther. 2022, 7, 146. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; He, B.; Liu, Z.; Zhou, Z.; Li, X. Fulminant type 1 diabetes after COVID-19 vaccination. Diabetes Metab. 2022, 48, 101324. [Google Scholar] [CrossRef] [PubMed]
- Ohuchi, K.; Amagai, R.; Tamabuchi, E.; Kambayashi, Y.; Fujimura, T. Fulminant type 1 diabetes mellitus triggered by coronavirus disease 2019 vaccination in an advanced melanoma patient given adjuvant nivolumab therapy. J. Dermatol. 2022, 49, e167–e168. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Morioka, T.; Okada, N.; Natsuki, Y.; Kakutani, Y.; Ochi, A.; Yamazaki, Y.; Shoji, T.; Ohmura, T.; Emoto, M. New-onset fulminant type 1 diabetes after severe acute respiratory syndrome coronavirus 2 vaccination: A case report. J. Diabetes Investig. 2022, 13, 1286–1289. [Google Scholar] [CrossRef]
- Toussirot, É.; Bereau, M. Vaccination and Induction of Autoimmune Diseases. Inflamm. Allergy Drug Targets 2015, 14, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, Z.; Wang, P.; Li, X.M.; Shuai, Z.W.; Ye, D.Q.; Pan, H.F. New-onset autoimmune phenomena post-COVID-19 vaccination. Immunology 2022, 165, 386–401. [Google Scholar] [CrossRef] [PubMed]
- Churilov, L.P.; Normatov, M.G.; Utekhin, V.J. Molecular Mimicry between SARS-CoV-2 and Human Endocrinocytes: A Prerequisite of Post-COVID-19 Endocrine Autoimmunity? Pathophysiology 2022, 29, 486–494. [Google Scholar] [CrossRef]
- Baek, H.S.; Yoon, J.W. Direct involvement of macrophages in destruction of beta-cells leading to development of diabetes in virus-infected mice. Diabetes 1991, 40, 1586–1597. [Google Scholar] [CrossRef]
- Shimada, A.; Maruyama, T. Encephalomyocarditis-virus-induced Diabetes Model Resembles “Fulminant” Type 1 Diabetes in Humans. Diabetologia 2004, 47, 1854–1855. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; McClintock, P.R.; Onodera, T.; Notkins, A.L. Virus-induced diabetes mellitus. XVIII. Inhibition by a Nondiabetogenic Variant of Encephalomyocarditis Virus. J. Exp. Med. 1980, 152, 878–892. [Google Scholar] [CrossRef] [Green Version]
- Sano, H.; Terasaki, J.; Mishiba, Y.; Imagawa, A.; Hanafusa, T. Exendin-4, a Glucagon-Like Peptide-1 Receptor Agonist, Suppresses Pancreatic β-Cell Destruction Induced by Encephalomyocarditis Virus. Biochem. Biophys. Res. Commun. 2011, 404, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.-W.; Onodera, T.; Notkins, A.L. Virus-Induced Diabetes Mellitus: VIII. Passage of Encephalomyocarditis Virus and Severity of Diabetes in Susceptible and Resistant Strains of Mice. J. Gen. Virol. 1977, 37, 225–232. [Google Scholar] [CrossRef]
- Izumi, K.; Mine, K.; Inoue, Y.; Teshima, M.; Ogawa, S.; Kai, Y.; Kurafuji, T.; Hirakawa, K.; Miyakawa, D.; Ikeda, H.; et al. Reduced Tyk2 Gene Expression in β-Cells Due to Natural Mutation Determines Susceptibility to Virus-Induced Diabetes. Nat. Commun. 2015, 6, 6748. [Google Scholar] [CrossRef] [Green Version]
- Kounoue, E.; Nagafuchi, S.; Nakamura, M.; Nakano, S.; Koga, T.; Nakayama, M.; Mituyama, M.; Niho, Y.; Takaki, R. Encephalomyocarditis (EMC) Virus-Induced Diabetes Mellitus Prevented by Corynebacterium parvum in Mice. Experientia 1987, 43, 430–431. [Google Scholar] [CrossRef]
- Baek, H.S.; Yoon, Y.W. Role of Macrophages in the Pathogenesis of Encephalomyocarditis Virus-induced Diabetes in Mice. J. Virol. 1990, 64, 5708–5715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, I.; Makino, H.; Osawa, H.; Kounoue, E.; Imagawa, A.; Hanafusa, T.; Kawasaki, E.; Fujii, Y. Association of Fulminant type 1 Diabetes with Pregnancy. Diabetes Res. Clin. Pract. 2003, 62, 33–38. [Google Scholar] [CrossRef]
- Shimizu, I.; Makino, H.; Imagawa, A.; Iwahashi, H.; Uchigata, Y.; Kanatsuka, A.; Kawasaki, E.; Kobayashi, T.; Shimada, A.; Maruyama, T.; et al. Clinical and Immunogenetic Characteristics of Fulminant type 1 Diabetes Associated with Pregnancy. J. Clin. Endocrinol. Metab. 2006, 91, 471–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baden, M.Y.; Imagawa, A.; Abiru, N.; Awata, T.; Ikegami, H.; Uchigata, Y.; Oikawa, Y.; Osawa, H.; Kajio, H.; Kawasaki, E.; et al. Characteristics and Clinical Course of Type 1 Diabetes Mellitus Related to Anti-Programmed Cell death-1 Therapy. Diabetol. Int. 2019, 10, 58–66. [Google Scholar] [CrossRef]
- Barroso-Sousa, R.; Barry, W.T.; Garrido-Castro, A.C.; Hodi, F.S.; Min, L.; Krop, I.E.; Tolaney, S.M. Incidence of endocrine dysfunction following the use of different immune checkpoint inhibitor regimens: A systematic review and meta-analysis. JAMA Oncol. 2018, 4, 173–182. [Google Scholar] [CrossRef]
- Stamatouli, A.M.; Quandt, Z.; Perdigoto, A.L.; Clark, P.L.; Kluger, H.; Weiss, S.A.; Gettinger, S.; Sznol, M.; Young, A.; Rushakoff, R.; et al. Collateral Damage: Insulin-Dependent Diabetes Induced with Checkpoint Inhibitors. Diabetes 2018, 67, 1471–1480. [Google Scholar] [CrossRef] [Green Version]
- Kotwal, A.; Haddox, C.; Block, M.; Kudva, Y.C. Immune checkpoint inhibitors: An emerging cause of insulin-dependent diabetes. BMJ Open Diabetes Res. Care 2019, 7, e000591. [Google Scholar] [CrossRef] [Green Version]
- Lo Preiato, V.; Salvagni, S.; Ricci, C.; Ardizzoni, A.; Pagotto, U.; Pelusi, C. Diabetes mellitus induced by immune checkpoint inhibitors: Type 1 diabetes variant or new clinical entity? Review of the literature. Rev. Endocr. Metab. Disord. 2021, 22, 337–349. [Google Scholar] [CrossRef]
- Kyriacou, A.; Melson, E.; Chen, W.; Kempegowda, P. Is Immune Checkpoint Inhibitor-Associated Diabetes the Same as Fulminant Type 1 Diabetes Mellitus? Clin. Med. 2020, 20, 417–423. [Google Scholar] [CrossRef]
- Clotman, K.; Janssens, K.; Specenier, P.; Weets, I.; De Block, C.E.M. Programmed Cell Death-1 Inhibitor-induced Type 1 Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2018, 103, 3144–3154. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, M.; Okamoto, M.; Gotoh, K.; Masaki, T.; Ozeki, Y.; Ando, H.; Anai, M.; Sato, A.; Yoshida, Y.; Ueda, S.; et al. Fulminant Type 1 Diabetes Mellitus with Anti-programmed Cell Death-1 therapy. J. Diabetes Investig. 2016, 7, 915–918. [Google Scholar] [CrossRef] [Green Version]
- Neu, A.; Hofer, S.E.; Karges, B.; Oeverink, R.; Rosenbauer, J.; Holl, R.W. Ketoacidosis at Diabetes Onset Is Still Frequent in Children and Adolescents. A Multicenter Analysis of 14,664 Patients from 106 Institutions. Diabetes Care 2009, 32, 1647–1648. [Google Scholar] [CrossRef] [Green Version]
- Grönberg, A.; Espes, D.; Carlsson, P.-O. Better HbA1c during the First Years after Diagnosis of Type 1 Diabetes Is Associated with Residual C Peptide 10 Years Later. BMJ Open Diabetes Res. Care 2020, 8, e000819. [Google Scholar] [CrossRef] [Green Version]
- Pihoker, C.; Gilliam, L.K.; Hampe, C.S.; Lernmark, A. Autoantibodies in diabetes. Diabetes 2005, 54 (Suppl. S2), S52–S61. [Google Scholar] [CrossRef] [Green Version]
- Marchand, L.; Thivolet, A.; Dalle, S.; Chikh, K.; Reffet, S.; Vouillarmet, J.; Fabien, N.; Cugnet-Anceau, C.; Thivolet, C. Diabetes Mellitus Induced by PD-1 and PD-L1 Inhibitors: Description of Pancreatic Endocrine and Exocrine Phenotype. Acta Diabetol. 2019, 56, 441–448. [Google Scholar] [CrossRef]
- Yadav, D. Nonspecific Hyperamylasemia and Hyperlipasemia in Diabetic Ketoacidosis: Incidence and Correlation with Biochemical Abnormalities. Am. J. Gastroenterol. 2000, 95, 3123–3128. [Google Scholar] [CrossRef]
- Campbell-Thompson, M.; Rodriguez-Calvo, T.; Battaglia, M. Abnormalities of the Exocrine Pancreas in Type 1 Diabetes. Curr. Diabetes Rep. 2015, 15, 79. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yoshida, T.; Nakaki, F.; Hiai, H.; Okazaki, T.; Honjo, T. Establishment of NOD-Pdcd1−/− Mice as an Efficient Animal Model of Type I Diabetes. Proc. Natl. Acad. Sci. USA 2005, 102, 11823–11828. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.J.; Salama, A.D.; Chitnis, T.; Smith, R.N.; Yagita, H.; Akiba, H.; Yamazaki, T.; Azuma, M.; Iwai, H.; Khoury, S.J.; et al. The programmed death-1 (PD-1) pathway regulates autoimmune diabetes in nonobese diabetic (NOD) mice. J. Exp. Med. 2003, 198, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.C.; Latchman, Y.E.; Buhlmann, J.E.; Tomczak, M.F.; Horwitz, B.H.; Freeman, G.J.; Sharpe, A.H. Regulation of PD-1, PD-L1, and PD-L2 Expression during Normal and Autoimmune Responses. Eur. J. Immunol. 2003, 33, 2706–2716. [Google Scholar] [CrossRef]
- Ni, R.; Ihara, K.; Miyako, K.; Kuromaru, R.; Inuo, M.; Kohno, H.; Hara, T. PD-1 Gene Haplotype is Associated with the Development of Type 1 Diabetes Mellitus in Japanese Children. Hum. Genet. 2007, 121, 223–232. [Google Scholar] [CrossRef]
- Gu, Y.; Xiao, L.; Gu, W.; Chen, S.; Feng, Y.; Wang, J.; Wang, Z.; Cai, Y.; Chen, H.; Xu, X.; et al. Rs2227982 and rs2227981 in PDCD1 Gene are Functional SNPs Associated with T1D Risk in East Asian. Acta Diabetol. 2018, 55, 813–819. [Google Scholar] [CrossRef]
- Nielsen, C.; Hansen, D.; Husby, S.; Jacobsen, B.B.; Lillevang, S.T. Association of a Putative Regulatory Polymorphism in the PD-1 Gene with Susceptibility to Type 1 Diabetes. Tissue Antigens 2003, 62, 492–497. [Google Scholar] [CrossRef]
- Tanaka, S.; Kobayashi, T.; Momotsu, T. A Novel Subtype of Type 1 Diabetes Mellitus. N. Engl. J. Med. 2000, 342, 1835–1837. [Google Scholar]
- Aida, K.; Nishida, Y.; Tanaka, S.; Maruyama, T.; Shimada, A.; Awata, T.; Suzuki, M.; Shimura, H.; Takizawa, S.; Ichijo, M.; et al. RIG-I− and MDA5-Initiated Innate Immunity Linked with Adaptive Immunity Accelerates Beta-Cell Death in Fulminant type 1 Diabetes. Diabetes 2011, 60, 884–889. [Google Scholar] [CrossRef] [Green Version]
- Nishida, Y.; Aida, K.; Kihara, M.; Kobayashi, T. Antibody-validated Proteins in Inflamed Islets of Fulminant Type 1 Diabetes Profiled by Laser-capture Microdissection Followed by Mass Spectrometry. PLoS ONE 2014, 9, e107664. [Google Scholar] [CrossRef]
- McCartney, S.A.; Vermi, W.; Lonardi, S.; Rossini, C.; Otero, K.; Calderon, B.; Gilfillan, S.; Diamond, M.S.; Unanue, E.R.; Colonna, M. RNA Sensor-Induced Type I IFN Prevents Diabetes Caused by a β Cell-Tropic Virus in Mice. J. Clin. Investig. 2011, 121, 1497–1507. [Google Scholar] [CrossRef]
- Nakata, S.; Imagawa, A.; Miyata, Y.; Yoshikawa, A.; Kozawa, J.; Okita, K.; Funahashi, T.; Nakamura, S.; Matsubara, K.; Iwahashi, H.; et al. Low Gene Expression Levels of Activating Receptors of Natural Killer Cells (NKG2E and CD94) in Patients with Fulminant type 1 Diabetes. Immunol. Lett. 2013, 156, 149–155. [Google Scholar] [CrossRef]
- Wang, Z.; Zheng, C.; Tan, Y.Y.; Li, Y.J.; Yang, L.; Huang, G.; Lin, J.; Zhou, Z.G. Gene Expression Changes in Patients with Fulminant type 1 Diabetes. Chin. Med. J. 2011, 124, 3613–3617. [Google Scholar]
- Roep, B.O.; Kleijwegt, F.S.; van Halteren, A.G.S.; Bonato, V.; Boggi, U.; Vendrame, F.; Marchetti, P.; Dotta, F. Islet Inflammation and CXCL10 in Recent-Onset Type 1 Diabetes. Clin. Exp. Immunol. 2010, 159, 338–343. [Google Scholar] [CrossRef]
- Berg, A.-K.; Korsgren, O.; Frisk, G. Induction of the Chemokine Interferon-γ-Inducible Protein-10 in Human Pancreatic Islets during Enterovirus Infection. Diabetologia 2006, 49, 2697–2703. [Google Scholar] [CrossRef]
- Nakagawa, Y.; Shimada, A.; Oikawa, Y.; Irie, J.-I.; Shigihara, T.; Tsumura, K.; Narumi, S.; Saruta, T. Two Cases of “Fulminant” Type 1 Diabetes Suggesting Involvement of Autoimmunity. Ann. N. Y. Acad. Sci. 2003, 1005, 359–361. [Google Scholar] [CrossRef]
- Tanaka, S.; Nishida, Y.; Aida, K.; Maruyama, T.; Shimada, A.; Suzuki, M.; Shimura, H.; Takizawa, S.; Takahashi, M.; Akiyama, D.; et al. Enterovirus Infection, CXC Chemokine Ligand 10 (CXCL10), and CXCR3 Circuit. Diabetes 2009, 58, 2285–2291. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, E.; Nakamura, K.; Kuriya, G.; Satoh, T.; Kobayashi, M.; Kuwahara, H.; Abiru, N.; Yamasaki, H.; Matsuura, N.; Miura, J.; et al. Zinc Transporter 8 Autoantibodies in Fulminant, Acute-Onset, and Slow-Onset Patients with Type 1 Diabetes. Diabetes Metab. Res. Rev. 2011, 27, 895–898. [Google Scholar] [CrossRef]
- Shimada, A.; Oikawa, Y.; Shigihara, T.; Senda, T.; Kodama, K. A Case of Fulminant type 1 Diabetes with Strong Evidence of Autoimmunity. Diabetes Care 2002, 25, 1482–1483. [Google Scholar] [CrossRef] [Green Version]
- Kotani, R.; Nagata, M.; Imagawa, A.; Moriyama, H.; Yasuda, H.; Miyagawa, J.; Hanafusa, T.; Yokono, K. T Lymphocyte Response against Pancreatic Beta Cell Antigens in Fulminant type 1 Diabetes. Diabetologia 2004, 47, 1285–1291. [Google Scholar] [CrossRef] [Green Version]
- Tada, A.; Shimada, A.; Yamada, T.; Oikawa, Y.; Yamada, Y.; Okubo, Y.; Irie, J.; Bluestone, J.A.; Itoh, H. A Mimic of Viral Double-Stranded RNA Triggers Fulminant Type 1 Diabetes-Like Syndrome in Regulatory T Cell-Deficient Autoimmune Diabetic Mouse. J. Immunol. 2011, 187, 4947–4953. [Google Scholar] [CrossRef]
- Haseda, F.; Imagawa, A.; Murase-Mishiba, Y.; Terasaki, J.; Hanafusa, T. CD4 + CD45RA − FoxP3 High Activated Regulatory T Cells Are Functionally Impaired and Related to Residual Insulin-Secreting Capacity in Patients with Type 1 Diabetes. Clin. Exp. Immunol. 2013, 173, 207–216. [Google Scholar] [CrossRef]
- Haseda, F.; Imagawa, A.; Murase-Mishiba, Y.; Sano, H.; Hirano-Kuwata, S.; Ueda, H.; Terasaki, J.; Hanafusa, T. Low CTLA-4 Expression in CD4+ Helper T-Cells in Patients with Fulminant type 1 Diabetes. Immunol. Lett. 2011, 139, 80–86. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| FT1D | AT1D | |
|---|---|---|
| Age at onset | Wide age range but mainly adult | Wide age range but the peak is in adolescence |
| Sex | Male ≒ female | Male < female |
| Onset (duration from onset to DKA) | Very rapid (within approximately 7 days) | Acute (within approximately 3 months) |
| HbA1c level at onset | ~8.7% | Very high |
| Insulin secretion | Depleted at onset | Slight residual for some time after onset |
| Flu-like symptoms | 71.7% | 26.9% |
| Elevation of pancreatic enzyme levels | Frequently | Rarely |
| Anti-GAD antibody | Mostly negative | Positive |
| Susceptible HLA haplotype in Japanese | DRB1*04:05-DQB1*04:01 DRB1*09:01-DQB1*03:03 | DRB1*09:01-DQB1*03:03 DRB1*04:05-DQB1*04:01 DRB1*08:02-DQB1*03:02 |
| Fulminant type 1 diabetes mellitus is confirmed when the following three findings are present: |
|
|
|
| Other findings in fulminant type 1 diabetes mellitus: |
|
|
|
|
|
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sano, H.; Imagawa, A. Re-Enlightenment of Fulminant Type 1 Diabetes under the COVID-19 Pandemic. Biology 2022, 11, 1662. https://doi.org/10.3390/biology11111662
Sano H, Imagawa A. Re-Enlightenment of Fulminant Type 1 Diabetes under the COVID-19 Pandemic. Biology. 2022; 11(11):1662. https://doi.org/10.3390/biology11111662
Chicago/Turabian StyleSano, Hiroyuki, and Akihisa Imagawa. 2022. "Re-Enlightenment of Fulminant Type 1 Diabetes under the COVID-19 Pandemic" Biology 11, no. 11: 1662. https://doi.org/10.3390/biology11111662