
Computationally Designed Anti-LuxP DNA Aptamer Suppressed Flagellar Assembly- and Quorum Sensing-Related Gene Expression in Vibrio parahaemolyticus
 , , , , , , and
, , , , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Molecular Docking and Dynamics Simulation
2.2. Production and Purification of the Recombinant LuxP Receptor Protein
2.3. Protein Identification
2.4. Isothermal Titration Calorimetry
2.5. Comparative Transcriptome Analysis of V. parahaemolyticus Treated with the Aptamer Candidate
3. Results
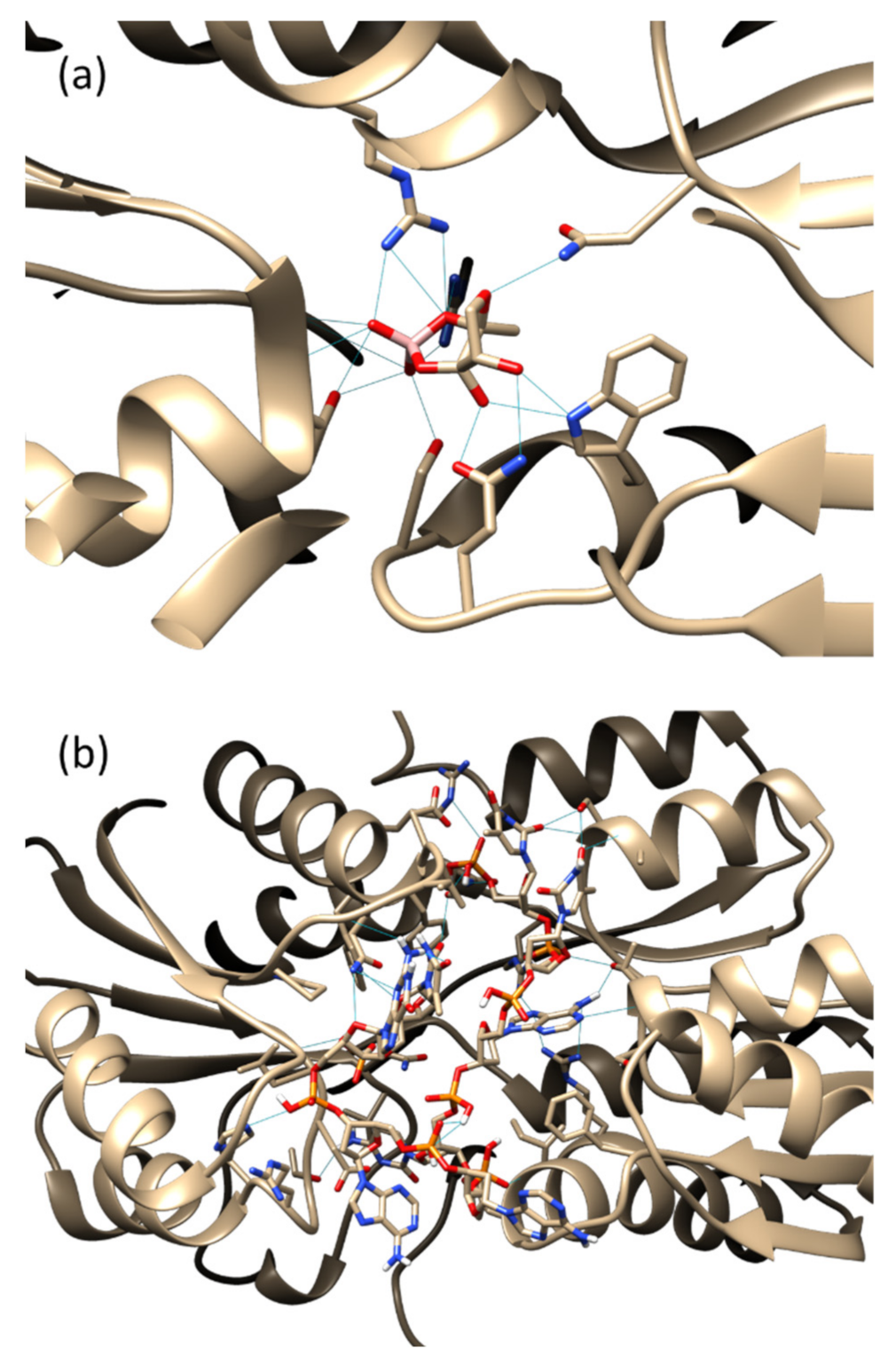
3.1. Molecular Docking
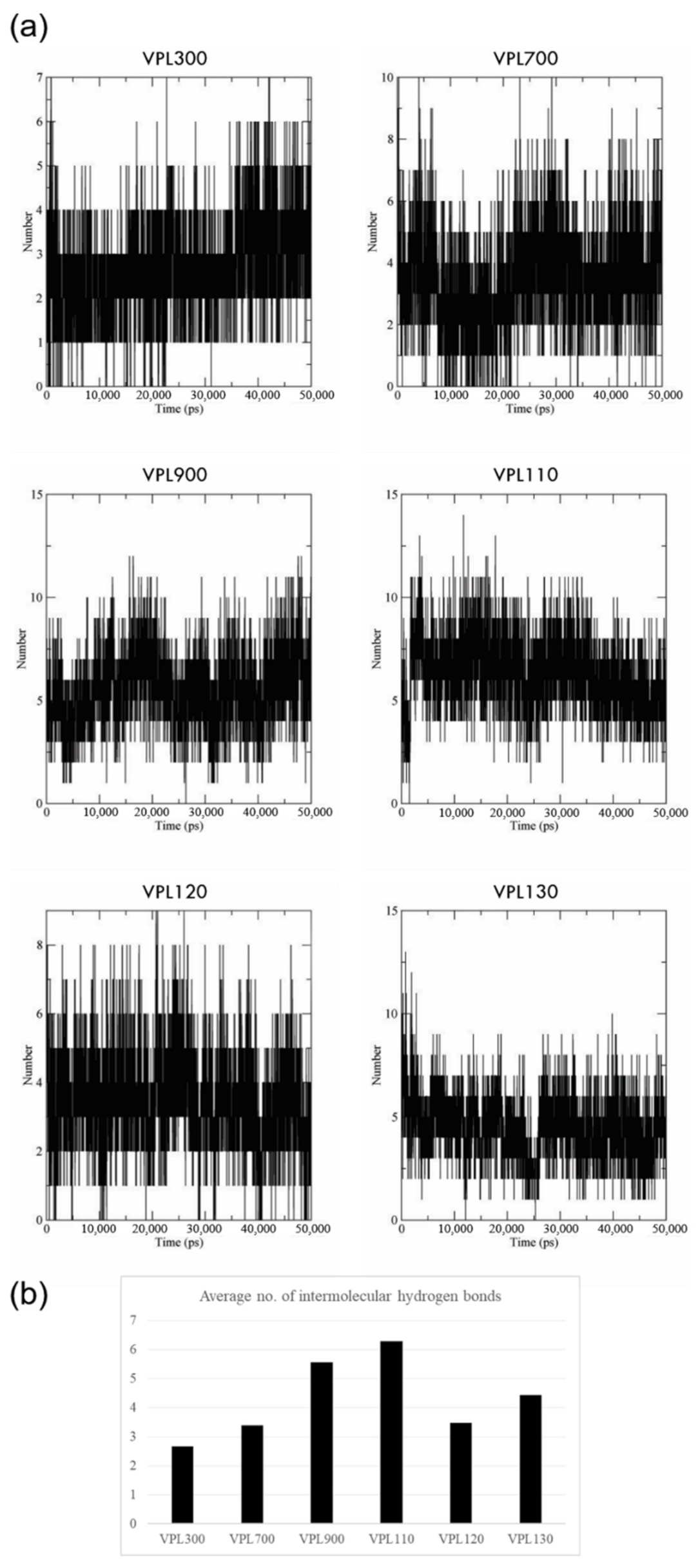
3.2. Molecular Dynamics Simulation
3.3. Isothermal Titration Calorimetry
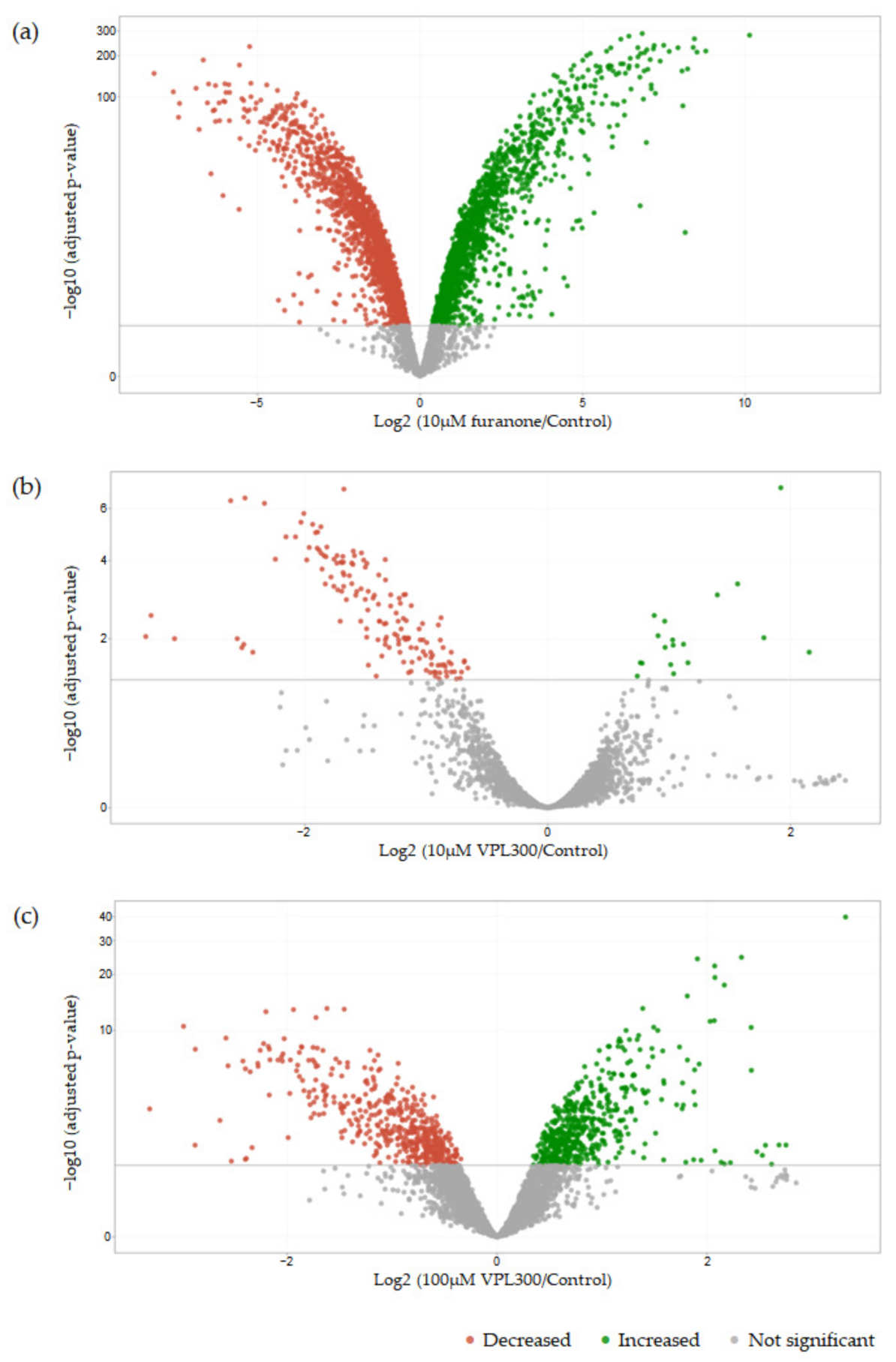
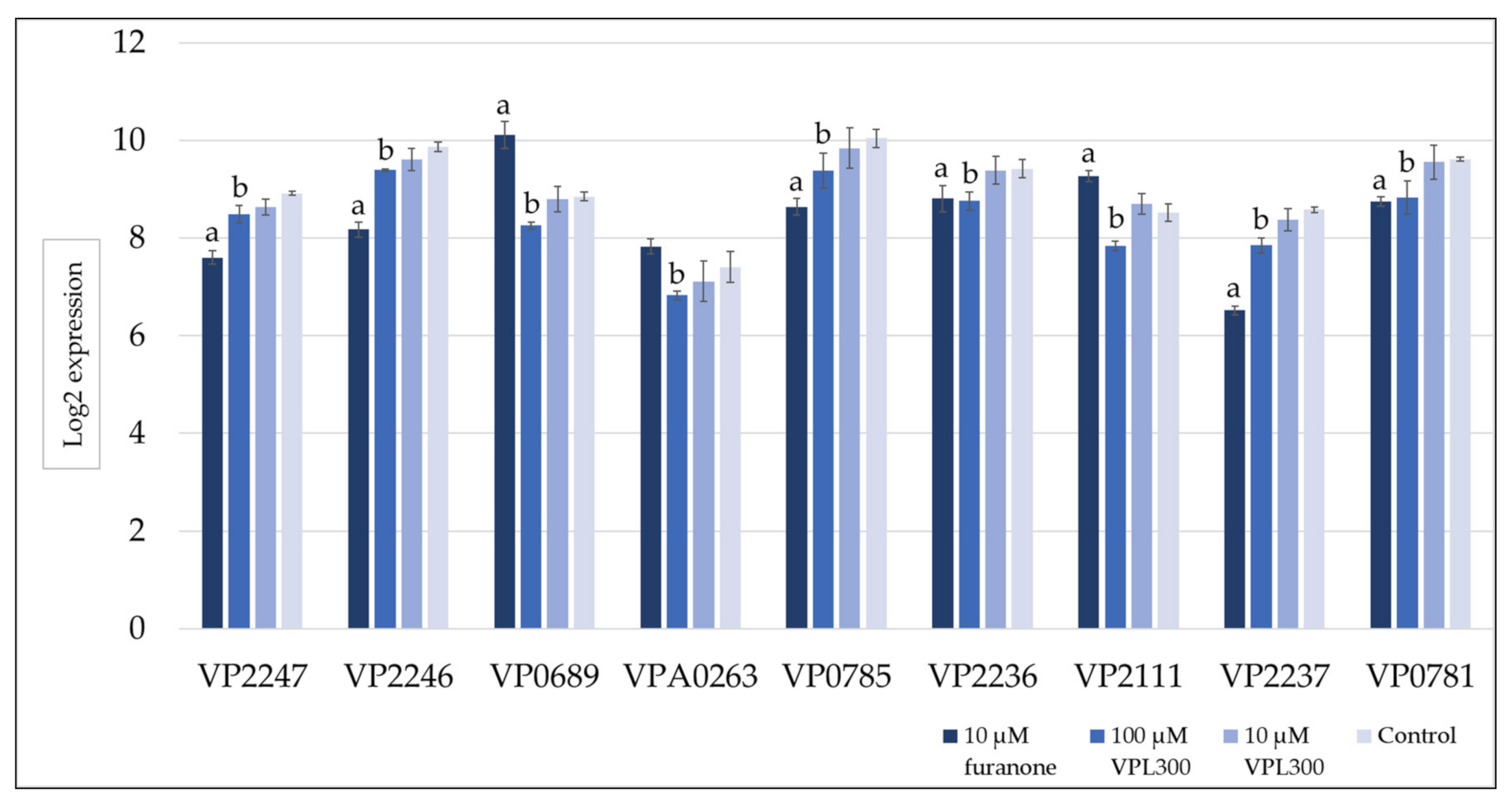
3.4. Comparative Transcriptome Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
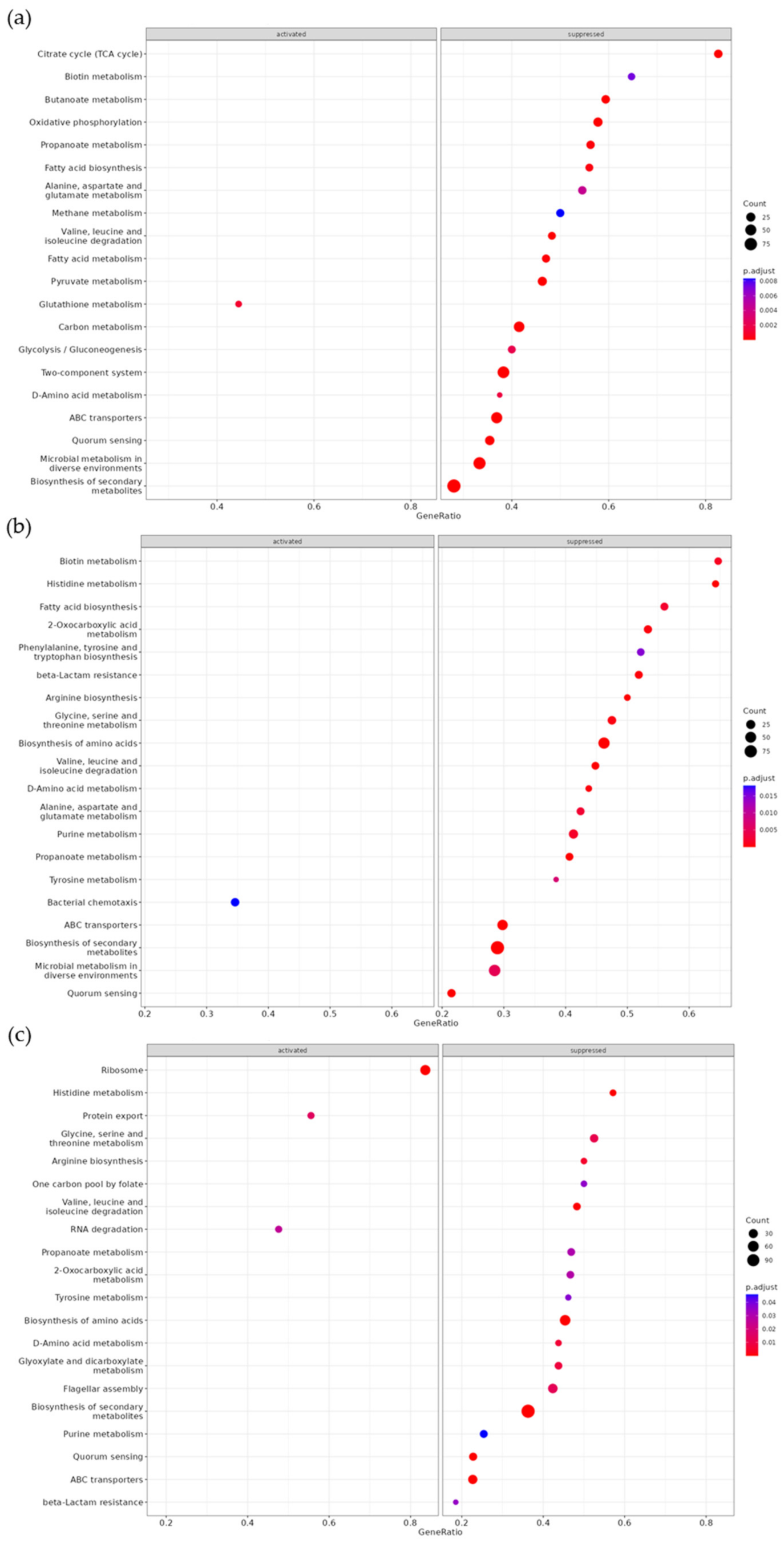
| KEGG | Set Size | Enrichment Score | NES | p Value | q Value |
|---|---|---|---|---|---|
| 10 µM furanone treatment/control | |||||
| Oxidative phosphorylation | 45 | −0.73 | −2.56 | 1.00 × 10−10 | 2.74 × 10−9 |
| Butanoate metabolism | 32 | −0.84 | −2.80 | 1.00 × 10−10 | 2.74 × 10−9 |
| Two-component system | 162 | −0.46 | −2.01 | 5.03 × 10−8 | 9.17 × 10−7 |
| Citrate cycle (TCA cycle) | 23 | −0.78 | −2.36 | 3.86 × 10−7 | 5.28 × 10−6 |
| Valine, leucine and isoleucine degradation | 29 | −0.72 | −2.31 | 1.31 × 10−6 | 1.43 × 10−5 |
| ABC transporters | 141 | −0.44 | −1.91 | 2.27 × 10−6 | 2.07 × 10−5 |
| Quorum sensing | 79 | −0.52 | −2.04 | 4.49 × 10−6 | 3.48 × 10−5 |
| Microbial metabolism in diverse environments | 207 | −0.39 | −1.77 | 5.08 × 10−6 | 3.48 × 10−5 |
| Pyruvate metabolism | 54 | −0.58 | −2.10 | 9.90 × 10−6 | 6.02 × 10−5 |
| Biosynthesis of secondary metabolites | 328 | −0.34 | −1.63 | 1.22 × 10−5 | 6.69 × 10−5 |
| Carbon metabolism | 106 | −0.46 | −1.90 | 2.06 × 10−5 | 1.02 × 10−4 |
| Fatty acid metabolism | 34 | −0.62 | −2.04 | 9.46 × 10−5 | 4.31 × 10−4 |
| Propanoate metabolism | 32 | −0.62 | −2.06 | 1.62 × 10−4 | 6.82 × 10−4 |
| Fatty acid biosynthesis | 25 | −0.65 | −2.01 | 2.78 × 10−4 | 1.09 × 10−3 |
| Glutathione metabolism | 18 | 0.70 | 1.87 | 1.08 × 10−3 | 3.95 × 10−3 |
| D-Amino acid metabolism | 16 | −0.67 | −1.86 | 1.60 × 10−3 | 5.47 × 10−3 |
| Glycolysis/gluconeogenesis | 35 | −0.54 | −1.81 | 2.03 × 10−3 | 6.55 × 10−3 |
| Alanine, aspartate, and glutamate metabolism | 33 | −0.53 | −1.76 | 4.42 × 10−3 | 0.01 |
| Biotin metabolism | 17 | −0.64 | −1.81 | 7.21 × 10−3 | 0.02 |
| Methane metabolism | 32 | −0.52 | −1.72 | 8.33 × 10−3 | 0.02 |
| Tyrosine metabolism | 13 | −0.66 | −1.74 | 8.89 × 10−3 | 0.02 |
| Starch and sucrose metabolism | 26 | −0.54 | −1.69 | 9.55 × 10−3 | 0.02 |
| Fatty acid degradation | 16 | −0.61 | −1.69 | 0.01 | 0.03 |
| Arginine and proline metabolism | 22 | −0.56 | −1.67 | 0.01 | 0.03 |
| 10 µM VPL300 treatment/control | |||||
| Biosynthesis of secondary metabolites | 328 | −0.51 | −1.98 | 1.00 × 10−10 | 5.58 × 10−9 |
| Biosynthesis of amino acids | 119 | −0.62 | −2.16 | 3.11 × 10−9 | 7.65 × 10−8 |
| ABC transporters | 141 | −0.59 | −2.11 | 4.11 × 10−9 | 7.65 × 10−8 |
| Valine, leucine and isoleucine degradation | 29 | −0.83 | −2.31 | 8.29 × 10−9 | 1.16 × 10−7 |
| Arginine biosynthesis | 14 | −0.87 | −2.07 | 3.42 × 10−6 | 3.81 × 10−5 |
| Histidine metabolism | 14 | −0.86 | −2.04 | 8.33 × 10−6 | 7.75 × 10−5 |
| Propanoate metabolism | 32 | −0.72 | −2.07 | 1.84 × 10−5 | 1.29 × 10−4 |
| Quorum sensing | 79 | −0.58 | −1.92 | 1.86 × 10−5 | 1.29 × 10−4 |
| beta-Lactam resistance | 27 | −0.70 | −1.94 | 3.67 × 10−4 | 2.27 × 10−3 |
| D-Amino acid metabolism | 16 | −0.78 | −1.90 | 4.14 × 10−4 | 2.31 × 10−3 |
| 2-Oxocarboxylic acid metabolism | 30 | −0.67 | −1.89 | 4.86 × 10−4 | 2.46 × 10−3 |
| Glycine, serine, and threonine metabolism | 40 | −0.62 | −1.83 | 6.14 × 10−4 | 2.86 × 10−3 |
| Biotin metabolism | 17 | −0.74 | −1.83 | 1.42 × 10−3 | 6.08 × 10−3 |
| Purine metabolism | 63 | −0.53 | −1.69 | 1.83 × 10−3 | 7.29 × 10−3 |
| Fatty acid biosynthesis | 25 | −0.67 | −1.83 | 2.24 × 10−3 | 8.35 × 10−3 |
| Alanine, aspartate, and glutamate metabolism | 33 | −0.63 | −1.83 | 2.58 × 10−3 | 9.01 × 10−3 |
| Microbial metabolism in diverse environments | 207 | −0.38 | −1.45 | 4.81 × 10−3 | 0.02 |
| Tyrosine metabolism | 13 | −0.75 | −1.74 | 7.30 × 10−3 | 0.02 |
| Phenylalanine, tyrosine, and tryptophan biosynthesis | 23 | −0.62 | −1.65 | 0.01 | 0.04 |
| 100 µM VPL300 treatment/control | |||||
| Ribosome | 55 | 0.70 | 2.53 | 2.51 × 10−10 | 1.59 × 10−8 |
| ABC transporters | 141 | −0.53 | −2.19 | 7.50 × 10−10 | 2.37 × 10−8 |
| Biosynthesis of secondary metabolites | 328 | −0.38 | −1.72 | 2.67 × 10−6 | 5.63 × 10−5 |
| Quorum sensing | 79 | −0.55 | −2.05 | 5.37 × 10−6 | 8.48 × 10−5 |
| Histidine metabolism | 14 | −0.81 | −2.07 | 4.61 × 10−5 | 5.53 × 10−4 |
| Biosynthesis of amino acids | 119 | −0.45 | −1.82 | 5.26 × 10−5 | 5.53 × 10−4 |
| Valine, leucine, and isoleucine degradation | 29 | −0.64 | −1.97 | 1.70 × 10−4 | 1.53 × 10−3 |
| Arginine biosynthesis | 14 | −0.70 | −1.78 | 5.47 × 10−3 | 0.04 |


References
- Broberg, C.A.; Calder, T.J.; Orth, K. Vibrio parahaemolyticus cell biology and pathogenicity determinants. Microbes Infect. 2011, 13, 992–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccarelli, D.; Hasan, N.A.; Huq, A.; Colwell, R.R. Distribution and dynamics of epidemic and pandemic Vibrio parahaemolyticus virulence factors. Front. Cell. Infect. Microbiol. 2013, 3, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Hu, L.; Qiu, Y.; Osei-Adjei, G.; Tang, H.; Zhang, Y.; Zhang, R.; Sheng, X.; Xu, S.; Yang, W.; et al. QsvR integrates into quorum sensing circuit to control Vibrio parahaemolyticus virulence. Environ. Microbiol. 2019, 21, 1054–1067. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Lu, R.; Liu, F.; Ye, J.; Zhao, J.; Wang, F.; Yang, M. Identification of LuxR family regulators that integrate into quorum sensing circuit in Vibrio parahaemolyticus. Front. Microbiol. 2021, 12, 691842. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Osei-Adjei, G.; Huang, X.; Zhang, Y. Role and regulation of the orphan AphA protein of quorum sensing in pathogenic Vibrios. Future Microbiol. 2018, 13, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Freeman, J.A.; Bassler, B.L. A genetic analysis of the function of LuxO, a two-component response regulator involved in quorum sensing in Vibrio harveyi. Mol. Microbiol. 1999, 31, 665–677. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiu, Y.; Tan, Y.; Guo, Z.; Yang, R.; Zhou, D. Transcriptional regulation of opaR, qrr2-4 and aphA by the master quorum-sensing regulator opaR in Vibrio parahaemolyticus. PLoS ONE 2012, 7, e0034622. [Google Scholar] [CrossRef]
- Guo, M.; Fang, Z.; Sun, L.; Sun, D.; Wang, Y.; Li, C.; Wang, R.; Liu, Y.; Hu, H.; Liu, Y.; et al. Regulation of Thermostable Direct Hemolysin and Biofilm Formation of Vibrio parahaemolyticus by Quorum-Sensing Genes luxM and luxS. Curr. Microbiol. 2018, 75, 1190–1197. [Google Scholar] [CrossRef]
- Henke, J.M.; Bassler, B.L. Quorum sensing regulates type III secretion in Vibrio harveyi and Vibrio parahaemolyticus. J. Bacteriol. 2004, 186, 3794–3805. [Google Scholar] [CrossRef] [Green Version]
- Waters, C.M.; Wu, J.T.; Ramsey, M.E.; Harris, R.C.; Bassler, B.L. Control of the type 3 secretion system in Vibrio harveyi by quorum sensing through repression of exsA. Appl. Environ. Microbiol. 2010, 76, 4996–5004. [Google Scholar] [CrossRef]
- Noor, N.M.; Defoirdt, T.; Alipiah, N.; Karim, M.; Daud, H.; Natrah, I. Quorum sensing is required for full virulence of Vibrio campbellii towards tiger grouper (Epinephelus fuscoguttatus) larvae. J. Fish Dis. 2019, 42, 489–495. [Google Scholar] [CrossRef]
- Defoirdt, T.; Sorgeloos, P. Monitoring of Vibrio harveyi quorum sensing activity in real time during infection of brine shrimp larvae. ISME J. 2012, 6, 2314–2319. [Google Scholar] [CrossRef]
- Chen, X.; Schauder, S.; Potier, N.; van Dorsselaer, A.; Pelczer, I.; Bassler, B.L.; Hughson, F.M. Structural identification of a bacterial quorum-sensing signal containing boron. Nature 2002, 415, 545–549. [Google Scholar] [CrossRef]
- Neiditch, M.B.; Federle, M.J.; Miller, S.T.; Bassler, B.L.; Hughson, F.M. Regulation of LuxPQ receptor activity by the quorum-sensing signal autoinducer-2. Mol. Cell 2005, 18, 507–518. [Google Scholar] [CrossRef]
- Bognár, Z.; Gyurcsányi, R.E. Aptamers against immunoglobulins: Design, selection and bioanalytical applications. Int. J. Mol. Sci. 2020, 21, 5748. [Google Scholar] [CrossRef]
- Torres-Vázquez, B.; de Lucas, A.M.; Crespo, C.; Mart, J.A.; Fragoso, A.; Fernández-Algar, M.; Perales, C.; Domingo, E.; Morenom, M.; Briones, C. In vitro selection of high affinity DNA and RNA aptamers that detect hepatitis C virus core protein of genotypes 1 to 4 and inhibit virus production in cell culture. J. Mol. Biol. 2022, 434, 167501. [Google Scholar] [CrossRef]
- Shangguan, D.; Cao, Z.; Meng, L.; Mallikaratchy, P.; Sefah, K.; Wang, H.; Li, Y.; Tan, W. Cell-specific aptamer probes for membrane protein elucidation in cancer cells. J. Proteome Res. 2008, 7, 2133–2139. [Google Scholar] [CrossRef] [Green Version]
- Van Simaeys, D.; Turek, D.; Champanhac, C.; Vaizer, J.; Sefah, K.; Zhen, J.; Sutphen, R.; Tan, W. Identification of cell membrane protein stress-induced phosphoprotein 1 as a potential ovarian cancer biomarker using aptamers selected by cell systematic evolution of ligands by exponential enrichment. Anal. Chem. 2014, 86, 4521–4527. [Google Scholar] [CrossRef]
- Chong, C.; Low, C. Synthetic antibody: Prospects in aquaculture biosecurity. Fish Shellfish Immunol. 2019, 86, 361–367. [Google Scholar] [CrossRef]
- Nimjee, S.M.; White, R.R.; Becker, R.C.; Sullenger, B.A. Aptamers as therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 61. [Google Scholar] [CrossRef]
- Punnarak, P.; Santos, M.D.; Hwang, S.D.; Kondo, H.; Hirono, I.; Kikuchi, Y.; Aoki, T. RNA aptamers inhibit the growth of the fish pathogen viral hemorrhagic septicemia virus (VHSV). Mar. Biotechnol. 2012, 14, 752–761. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Yan, Y.; Wei, S.; Wei, J.; Gao, R.; Huang, X.; Huang, Y.; Jiang, G.; Qin, Q. Isolation and characterization of a new class of DNA aptamers specific binding to Singapore grouper iridovirus (SGIV) with antiviral activities. Virus Res. 2014, 188, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, P.; Yang, M.; Yu, Y.; Huang, Y.; Wei, J.; Wei, S.; Qin, Q. Generation and characterization of novel DNA aptamers against coat protein of grouper nervous necrosis virus (GNNV) with antiviral activities and delivery potential in grouper cells. Antiviral Res. 2016, 129, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Z.; Yu, Y.; Wang, M.; Li, J.; Zhang, Z.; Liu, J.; Wu, X.; Lu, A.; Zhang, G.; Zhang, B. Recent advances in SELEX technology and aptamer applications in biomedicine. Int. J. Mol. Sci. 2017, 18, 2142. [Google Scholar] [CrossRef] [Green Version]
- Bayat, P.; Nosrati, R.; Alibolandi, M.; Rafatpanah, H.; Abnous, K.; Khedri, M.; Ramezani, M. SELEX methods on the road to protein targeting with nucleic acid aptamers. Biochimie 2018, 154, 132–155. [Google Scholar] [CrossRef]
- Kalra, P.; Dhiman, A.; Cho, W.C.; Bruno, J.G.; Sharma, T.K. Simple methods and rational design for enhancing aptamer sensitivity and specificity. Front. Mol. Biosci. 2018, 5, 41. [Google Scholar] [CrossRef]
- Bell, D.R.; Weber, J.K.; Yin, W.; Huynh, T.; Duan, W.; Zhou, R. In silico design and validation of high-affinity RNA aptamers targeting epithelial cellular adhesion molecule dimers. Proc. Natl. Acad. Sci. USA 2020, 117, 8486–8493. [Google Scholar] [CrossRef]
- Pinzi, L.; Rastelli, G. Molecular docking: Shifting paradigms in drug discovery. Int. J. Mol. Sci. 2019, 20, 4331. [Google Scholar] [CrossRef] [Green Version]
- Bavi, R.; Liu, Z.; Han, Z.; Zhang, H.; Gu, Y. In silico designed RNA aptamer against epithelial cell adhesion molecule for cancer cell imaging. Biochem. Biophys. Res. Commun. 2019, 509, 937–942. [Google Scholar] [CrossRef]
- Mousivand, M.; Anfossi, L.; Bagherzadeh, K.; Barbero, N.; Mirzadi-Gohari, A.; Javan-Nikkhah, M. In silico maturation of affinity and selectivity of DNA aptamers against aflatoxin B1 for biosensor development. Anal. Chim. Acta 2020, 1105, 178–186. [Google Scholar] [CrossRef]
- Buglak, A.A.; Samokhvalov, A.V.; Zherdev, A.V.; Dzantiev, B.B. Methods and applications of in silico aptamer design and modeling. Int. J. Mol. Sci. 2020, 21, 8420. [Google Scholar] [CrossRef]
- Chen, J.; Wang, J.; Zhu, W. Binding modes of three inhibitors 8CA, F8A and I4A to A-FABP studied based on molecular dynamics simulation. PLoS ONE 2014, 9, e99862. [Google Scholar] [CrossRef] [Green Version]
- Turner, P.J. XMGRACE, version 5.1.25; Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Rizzo, R.C.; Aynechi, T.; Case, D.A.; Kuntz, I.D. Estimation of absolute free energies of hydration using continuum methods: Accuracy of partial charge models and optimization of nonpolar contributions. J. Chem. Theory Comput. 2006, 2, 128–139. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Sabri, M.Z.; Hamid, A.A.A.; Hitam, S.M.S.; Rahim, M.Z.A. In-silico selection of aptamer: A review on the revolutionary approach to understand the aptamer design and interaction through computational chemistry. Mater. Today Proc. 2019, 19, 1572–1581. [Google Scholar] [CrossRef]
- Hasegawa, H.; Savory, N.; Abe, K.; Ikebukuro, K. Methods for improving aptamer binding affinity. Molecules 2016, 21, 421. [Google Scholar] [CrossRef]
- Navien, T.N.; Thevendran, R.; Hamdani, H.Y.; Tang, T.-H.; Citartan, M. In silico molecular docking in DNA aptamer development. Biochimie 2021, 180, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.; Melquiond, A.S.J.; Karaca, E.; Trellet, M.; van Dijk, M.; van Zundert, G.C.P.; Schmitz, C.; de Vries, S.J.; Bordogna, A.; Bonati, L.; et al. Defining the limits of homology modeling in information-driven protein docking. Proteins 2013, 81, 2119–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational protein--ligand docking and virtual drug screening with the AutoDock suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanchuk, V.Y.; Tanin, V.O.; Vovk, A.I.; Poda, G. A new, improved hybrid scoring function for molecular docking and scoring based on AutoDock and AutoDock Vina. Chem. Biol. Drug Des. 2016, 87, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- De Vries, S.J.; van Dijk, M.; Bonvin, A.M.J.J. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijk, M.; van Dijk, A.D.J.; Hsu, V.; Boelens, R.; Bonvin, A.M.J.J. Information-driven protein--DNA docking using HADDOCK: It is a matter of flexibility. Nucleic Acids Res. 2006, 34, 3317–3325. [Google Scholar] [CrossRef] [Green Version]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.-H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein--protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.-L.; Cui, H.-F.; Du, J.-F.; Lv, Q.-Y.; Song, X. In silico post-SELEX screening and experimental characterizations for acquisition of high affinity DNA aptamers against carcinoembryonic antigen. RSC Adv. 2019, 9, 6328–6334. [Google Scholar] [CrossRef]
- Bjerregaard, N.; Andreasen, P.A.; Dupont, D.M. Expected and unexpected features of protein-binding RNA aptamers. Wiley Interdiscip. Rev. RNA 2016, 7, 744–757. [Google Scholar] [CrossRef]
- Minagawa, H.; Onodera, K.; Fujita, H.; Sakamoto, T.; Akitomi, J.; Kaneko, N.; Shiratori, I.; Kuwahara, M.; Horii, K.; Waga, I. Selection, characterization and application of artificial DNA aptamer containing appended bases with sub-nanomolar affinity for a salivary biomarker. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Szalewicz, K. Hydrogen Bond. In Encyclopedia of Physical Science and Technology, 3rd ed.; Meyers, R.A., Ed.; Academic Press: New York, NY, USA, 2003; pp. 505–538. Available online: https://www.sciencedirect.com/science/article/pii/B0122274105003227 (accessed on 15 September 2021).
- Aeksiri, N.; Songtawee, N.; Gleeson, M.P.; Hannongbua, S.; Choowongkomon, K. Insight into HIV-1 reverse transcriptase-aptamer interaction from molecular dynamics simulations. J. Mol. Model. 2014, 20, 1–10. [Google Scholar] [CrossRef]
- Wang, C.; Greene, D.; Xiao, L.; Qi, R.; Luo, R. Recent developments and applications of the MMPBSA method. Front. Mol. Biosci. 2018, 4, 87. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.; Yuan, X.; Li, J.; Lin, S.; Yang, B.; Chen, C.; Zhao, J.; Zheng, W.; Liao, H.; Yang, Z.; et al. Assessing the performance of the g_mmpbsa tools to simulate the inhibition of oseltamivir to influenza virus neuraminidase by molecular mechanics Poisson-Boltzmann surface area methods. J. Chin. Chem. Soc. 2020, 67, 46–53. [Google Scholar] [CrossRef]
- Basit, A.; Mishra, R.K.; Bandyopadhyay, P. Calcium ion binding to calmodulin: Binding free energy calculation using the molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) method by incorporating implicit polarization. J. Biomol. Struct. Dyn. 2021, 39, 7213–7222. [Google Scholar] [CrossRef]
- Wang, C.; Nguyen, P.H.; Pham, K.; Huynh, D.; Le, T.-B.N.; Wang, H.; Ren, P.; Luo, R. Calculating protein-ligand binding affinities with MMPBSA: Method and error analysis. J. Comput. Chem. 2016, 37, 2436–2446. [Google Scholar] [CrossRef] [Green Version]
- Shcherbinin, D.S.; Gnedenko, O.V.; Khmeleva, S.A.; Usanov, S.A.; Gilep, A.A.; Yantsevich, A.V.; Shkel, T.V.; Yushkevich, I.V.; Radko, S.P.; Ivanov, A.S.; et al. Computer-aided design of aptamers for cytochrome p450. J. Struct. Biol. 2015, 191, 112–119. [Google Scholar] [CrossRef]
- Xie, Y.; Wu, S.; Chen, Z.; Jiang, J.; Sun, J. Rapid nanomolar detection of methamphetamine in biofluids via a reagentless electrochemical aptamer-based biosensor. Anal. Chim. Acta 2022, 1207, 339742. [Google Scholar] [CrossRef]
- Minagawa, H.; Kataoka, Y.; Fujita, H.; Kuwahara, M.; Horii, K.; Shiratori, I.; Waga, I. Modified DNA aptamers for C-reactive protein and lactate dehydrogenase-5 with sub-nanomolar affinities. Int. J. Mol. Sci. 2020, 21, 2683. [Google Scholar] [CrossRef]
- Akke, M. Conformational dynamics and thermodynamics of protein-ligand binding studied by NMR relaxation. Biochem. Soc. Trans. 2012, 40, 419–423. [Google Scholar] [CrossRef]
- Low, C.-F.; Shamsir, M.S.; Mohamed-Hussein, Z.-A.; Baharum, S.N. Evaluation of potential molecular interaction between quorum sensing receptor, LuxP and grouper fatty acids: In-silico screening and simulation. PeerJ 2019, 7, e6568. [Google Scholar] [CrossRef]
- Federle, M.J. Autoinducer-2-based chemical communication in bacteria: Complexities of interspecies signaling. Contrib. Microbiol. 2009, 16, 18–32. [Google Scholar]
- Rajamani, S.; Sayre, R. Biosensors for the detection and quantification of AI-2 class quorum-sensing compounds. In Quorum Sensing; Springer: Berlin/Heidelberg, Germany, 2018; pp. 73–88. [Google Scholar]
- Byeon, J.-Y.; Sim, J.; Ryu, E.-J.; Sim, J.; Lee, H.; Cho, K.-H.; Choi, B.-K.; Lee, J. In Silico Development of Quorum-Sensing Inhibitors. Bull. Korean Chem. Soc. 2017, 38, 728–734. [Google Scholar] [CrossRef]
- Neiditch, M.B.; Federle, M.J.; Pompeani, A.J.; Kelly, R.C.; Swem, D.L.; Jeffrey, P.D.; Bassler, B.L.; Hughson, F.M. Ligand-induced asymmetry in histidine sensor kinase complex regulates quorum sensing. Cell 2006, 126, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Theisen, F.F.; Staby, L.; Tidemand, F.G.; O’Shea, C.; Prestel, A.; Willemoës, M.; Kragelund, B.B.; Skriver, K. Quantification of Conformational Entropy Unravels Effect of Disordered Flanking Region in Coupled Folding and Binding. J. Am. Chem. Soc. 2021, 143, 14540–14550. [Google Scholar] [CrossRef]
- Defoirdt, T.; Miyamoto, C.M.; Wood, T.K.; Meighen, E.A.; Sorgeloos, P.; Verstraete, W.; Bossier, P. The natural furanone (5Z)-4-bromo-5-(bromomethylene)-3-butyl-2 (5H)-furanone disrupts quorum sensing-regulated gene expression in Vibrio harveyi by decreasing the DNA-binding activity of the transcriptional regulator protein LuxR. Environ. Microbiol. 2007, 9, 2486–2495. [Google Scholar] [CrossRef] [Green Version]
- De Nys, R.; Givskov, M.; Kumar, N.; Kjelleberg, S.; Steinberg, P.D. Furanones. In Antifouling Compounds; Springer: Berlin/Heidelberg, Germany, 2006; pp. 55–86. [Google Scholar]
- Ren, D.; Zuo, R.; Wood, T.K. Quorum-sensing antagonist (5Z)-4-bromo-5-(bromomethylene)-3-butyl-2 (5H)-furanone influences siderophore biosynthesis in Pseudomonas putida and Pseudomonas aeruginosa. Appl. Microbiol. Biotechnol. 2005, 66, 689–695. [Google Scholar] [CrossRef]
- Gregory, G.J.; Morreale, D.P.; Carpenter, M.R.; Kalburge, S.S.; Boyd, E.F. Quorum sensing regulators AphA and OpaR control expression of the operon responsible for biosynthesis of the compatible solute ectoine. Appl. Environ. Microbiol. 2019, 85, e01543-19. [Google Scholar] [CrossRef]
- Chang, S.-C.; Lee, C.-Y. Quorum-Sensing regulator OpaR directly represses seven protease genes in Vibrio parahaemolyticus. Front. Microbiol. 2020, 11, 534692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, L.; Hou, S.; Huang, X.; Sun, F.; Gao, H. The master quorum-sensing regulator OpaR is activated indirectly by H-NS in Vibrio parahaemolyticus. Curr. Microbiol. 2016, 73, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.K.; Guthrie, L.T.C.; Modise, T.; Cormier, G.; Jensen, R.V.; McCarter, L.L.; Stevens, A.M. OpaR controls a network of downstream transcription factors in Vibrio parahaemolyticus BB22OP. PLoS ONE 2015, 10, e0121863. [Google Scholar]
- Gode-Potratz, C.J.; McCarter, L.L. Quorum sensing and silencing in Vibrio parahaemolyticus. J. Bacteriol. 2011, 193, 4224–4237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haiko, J.; Westerlund-Wikström, B. The role of the bacterial flagellum in adhesion and virulence. Biology 2013, 2, 1242–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, K.; Kashimoto, T.; Morita, M.; Kado, T.; Matsuda, K.; Yamasaki, M.; Ueno, S. Identification of in vivo essential genes of Vibrio vulnificus for establishment of wound infection by signature-tagged mutagenesis. Front. Microbiol. 2019, 10, 123. [Google Scholar] [CrossRef]
- Kim, S.Y.; Thanh, X.T.T.; Jeong, K.; Kim, S.B.; Pan, S.O.; Jung, C.H.; Hong, S.H.; Lee, S.E.; Rhee, J.H. Contribution of six flagellin genes to the flagellum biogenesis of Vibrio vulnificus and in vivo invasion. Infect. Immun. 2014, 82, 29–42. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Defoirdt, T. Quorum sensing positively regulates flagellar motility in pathogenic V ibrio harveyi. Environ. Microbiol. 2015, 17, 960–968. [Google Scholar] [CrossRef]
- Echazarreta, M.A.; Klose, K.E. Vibrio flagellar synthesis. Front. Cell. Infect. Microbiol. 2019, 9, 131. [Google Scholar] [CrossRef]
- Lu, R.; Sun, J.; Qiu, Y.; Zhang, M.; Xue, X.; Li, X.; Yang, W.; Zhou, D.; Hu, L. The quorum sensing regulator OpaR is a repressor of polar flagellum genes in Vibrio parahaemolyticus. J. Microbiol. 2021, 59, 651–657. [Google Scholar] [CrossRef]
- Komulainen, H.; Huuskonen, H.; Kosma, V.-M.; Lötjönen, S.; Vartiainen, T. Toxic effects and excretion in urine of 3-chloro-4-(dichloromethyl)-5-hydroxy-2 (5H)-furanone (MX) in the rat after a single oral dose. Arch. Toxicol. 1994, 68, 398–400. [Google Scholar] [CrossRef]
- Komulainen, H.; Vaittinen, S.-L.; Vartiainen, T.; Tuomisto, J.; Kosma, V.-M.; Kaliste-Korhonen, E.; Lötjönen, S.; Tuominen, R.K. Carcinogenicity of the drinking water mutagen 3-chloro-4-(dichloromethyl)-5-hydroxy-2 (5H)-furanone in the rat. J. Natl. Cancer Inst. 1997, 89, 848–856. [Google Scholar] [CrossRef]








| Oligonucleotides | Affinity (kcal/mol) | Amino Acids Involved in Hydrogen Bonding | |
|---|---|---|---|
| VPL300 | ATAA GTGT | −7.5 | Asn159; Ser293; Ala294; Arg310; Arg215; Ile211 |
| VPL700 | ATGG GTGG | −7.7 | Gln116; Thr162; Ile211; Arg215; Asp136; Ser207 |
| VPL900 | GTGG AGCA | −8.4 | Lys238; Ser207; Asp267; Ser293; Thr266; Ser265; Ala239; Asn159; Thr138; Thr137; Asp136; Tyr28; Trp82; Ala294 |
| VPL110 | AGCA TTAC | −8.1 | Thr138; Ser207; Asn159; Arg215; Thr266; Gln77; Ser79; Pro109; Asp136; Tyr81 |
| VPL120 | TTAC AGCA | −8.0 | Ile211; Tyr210; Asp136; Pro109; Thr138; Thr107; Thr162; Ser79; Asn159; Arg310 |
| VPL130 | TTAC AAGT | −9.2 | Trp82; Thr134; His140; Ile211; Arg215; Thr137; Arg310; Thr266; Ala294; Arg84; Ser293; Asn312; Ser79; Gln77; Gln76; Gln116 |
| Oligonucleotides | MMPBSA Binding Energy (kJ/mol) | |
|---|---|---|
| VPL300 | ATAA GTGT | −251.615 ± 44.305 |
| VPL700 | ATGG GTGG | −265.427 ± 24.302 |
| VPL900 | GTGG AGCA | −326.981 ± 38.291 |
| VPL110 | AGCA TTAC | −260.524 ± 21.035 |
| VPL120 | TTAC AGCA | −301.524 ± 24.673 |
| VPL130 | TTAC AAGT | −293.980 ± 25.769 |
| Aptamer Candidates | Kd (M) | Stoichiometry Value, n | ΔH (kJ/mol) | Calculated ΔS (kJ/mol.K) | Calculated ΔG (kJ/mol) |
|---|---|---|---|---|---|
| VPL300 | 2.11 × 10−7 ± 2.03 × 10−7 | 0.928 ± 0.083 | −84.37 ± 11.89 | −0.1552 | −38.12 |
| VPL130 | 5.64 × 10−7 ± 5.29 × 10−7 | 0.746 ± 0.107 | −51.91 ± 9.24 | −0.05449 | −35.67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yusof, N.A.M.; Razali, S.A.; Mohd Padzil, A.; Lau, B.Y.C.; Baharum, S.N.; Nor Muhammad, N.A.; Raston, N.H.A.; Chong, C.M.; Ikhsan, N.F.M.; Situmorang, M.L.; et al. Computationally Designed Anti-LuxP DNA Aptamer Suppressed Flagellar Assembly- and Quorum Sensing-Related Gene Expression in Vibrio parahaemolyticus. Biology 2022, 11, 1600. https://doi.org/10.3390/biology11111600
Yusof NAM, Razali SA, Mohd Padzil A, Lau BYC, Baharum SN, Nor Muhammad NA, Raston NHA, Chong CM, Ikhsan NFM, Situmorang ML, et al. Computationally Designed Anti-LuxP DNA Aptamer Suppressed Flagellar Assembly- and Quorum Sensing-Related Gene Expression in Vibrio parahaemolyticus. Biology. 2022; 11(11):1600. https://doi.org/10.3390/biology11111600
Chicago/Turabian StyleYusof, Nur Afiqah Md, Siti Aisyah Razali, Azyyati Mohd Padzil, Benjamin Yii Chung Lau, Syarul Nataqain Baharum, Nor Azlan Nor Muhammad, Nurul Hanun Ahmad Raston, Chou Min Chong, Natrah Fatin Mohd Ikhsan, Magdalena Lenny Situmorang, and et al. 2022. "Computationally Designed Anti-LuxP DNA Aptamer Suppressed Flagellar Assembly- and Quorum Sensing-Related Gene Expression in Vibrio parahaemolyticus" Biology 11, no. 11: 1600. https://doi.org/10.3390/biology11111600