
Identification of NRAS Downstream Genes with CRISPR Activation Screening
 , , ,
, , ,
Abstract
:Simple Summary
Abstract
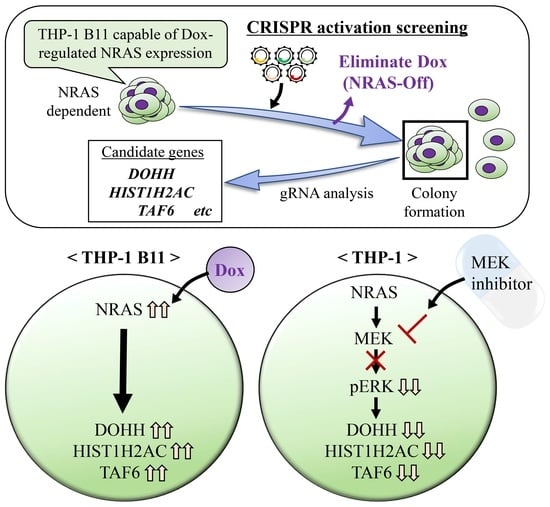
1. Introduction
2. Materials and Methods
2.1. Cell Culture
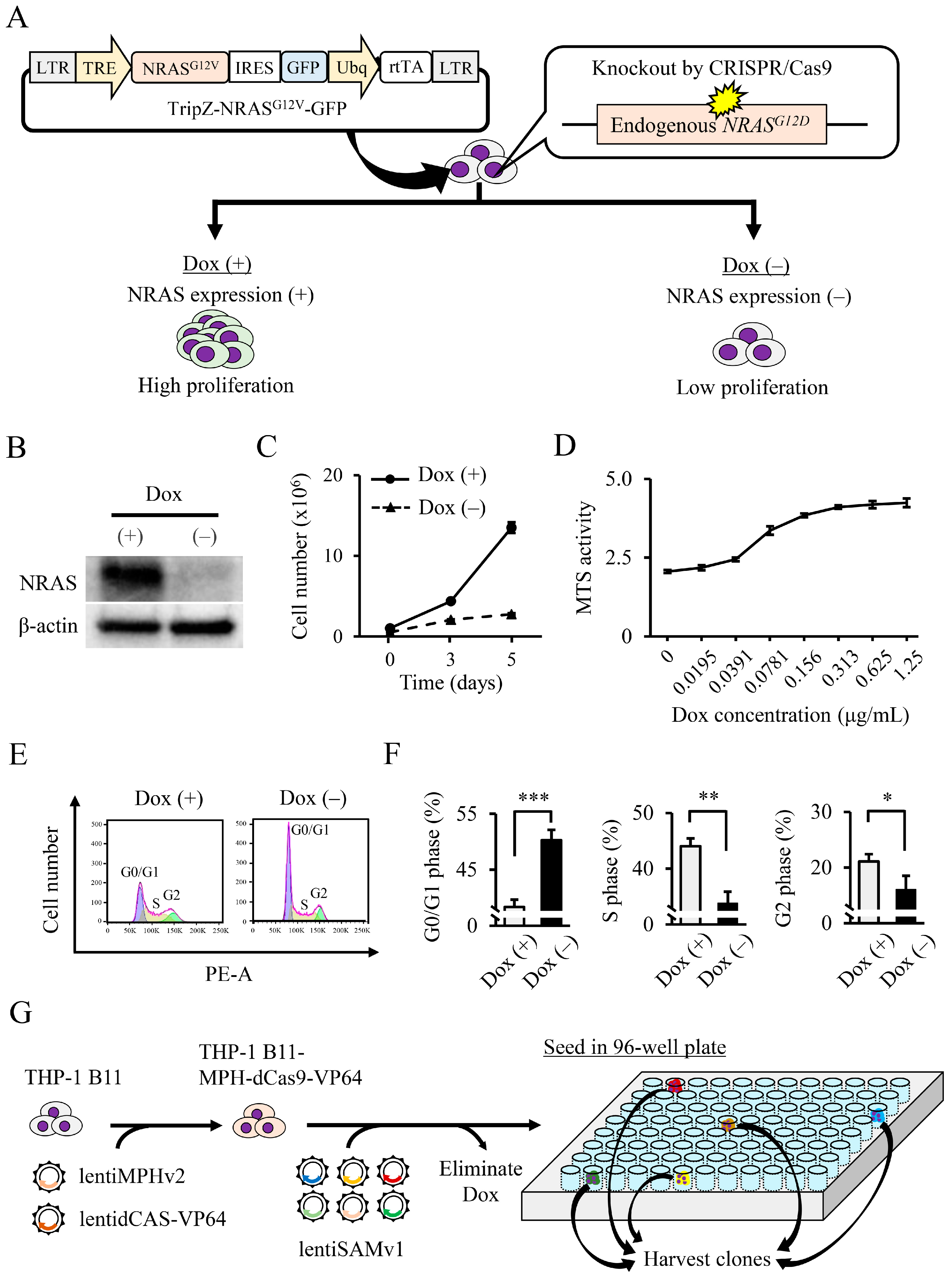
2.2. CRISPR Activation Screening and Plasmid Construction
2.3. Cell Proliferation
2.4. RT-qPCR
2.5. Western Blotting
2.6. Small Interfering RNA (siRNA)
2.7. Statistical Analysis
3. Results
3.1. Identification of NRAS Signaling-Related Growth Factors by Genome-Wide CRISPR Activation Screening
3.2. Validation of Candidate Genes Associated with NRAS Proliferation Signaling
3.3. DOHH, HIST1H2AC, and TAF6 Are Located Downstream of NRAS Signaling
3.4. DOHH, HIST1H2AC, and TAF6 Are Located Downstream of ERK Pathway
3.5. DOHH, HIST1H2AC, and TAF6 Contribute to Leukemia Cell Proliferation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vojtek, A.B.; Der, C.J. Increasing complexity of the Ras signaling pathway. J. Biol. Chem. 1998, 273, 19925–19928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosravi-Far, R.; Der, C.J. The Ras signal transduction pathway. Cancer Metastasis Rev. 1994, 13, 67–89. [Google Scholar] [CrossRef] [PubMed]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The frequency of Ras mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Couselo, E.; Adelantado, E.Z.; Ortiz, C.; Garcia, J.S.; Perez-Garcia, J. NRAS-mutant melanoma: Current challenges and future prospect. Onco Targets Ther. 2017, 10, 3941–3947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Zhang, Y.; Qian, L.; Wang, P. Emerging strategies to target RAS signaling in human cancer therapy. J. Hematol. Oncol. 2021, 14, 116. [Google Scholar] [CrossRef]
- Vu, H.L.; Aplin, A.E. Targeting mutant NRAS signaling pathways in melanoma. Pharmacol. Res. 2016, 107, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Caunt, C.J.; Sale, M.J.; Smith, P.D.; Cook, S.J. MEK1 and MEK2 inhibitors and cancer therapy: The long and winding road. Nat. Rev. Cancer 2015, 15, 577–592. [Google Scholar] [CrossRef]
- Kurata, M.; Yamamoto, K.; Moriarity, B.S.; Kitagawa, M.; Largaespada, D.A. CRISPR/Cas9 library screening for drug target discovery. J. Hum. Genet. 2018, 63, 179–186. [Google Scholar] [CrossRef]
- He, C.; Han, S.; Chang, Y.; Wu, M.; Zhao, Y.; Chen, C.; Chu, X. CRISPR screen in cancer: Status quo and future perspectives. Am. J. Cancer Res. 2021, 11, 1031–1050. [Google Scholar]
- Kurata, M.; Antony, M.L.; Noble-Orcutt, K.E.; Rathe, S.K.; Lee, Y.; Furuno, H.; Ishibash, S.; Ikeda, M.; Yamamoto, K.; Kitagawa, M.; et al. Proliferation and self-renewal are differentially sensitive to NRASG12V oncogene levels in an acute myeloid leukemia cell line. Mol. Cancer Res. 2022, OF1–OF13. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Kurata, M.; Onishi, I.; Sugita, K.; Matsumura, M.; Ishibashi, S.; Ikeda, M.; Yamamoto, K.; Kitagawa, M. CRISPR screening identifies M1AP as a new MYC regulator with a promoter-reporter system. PeerJ 2020, 8, e9046. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kambayashi, H.; Yamamoto, A.; Onishi, I.; Sugita, K.; Matsumura, M.; Ishibashi, S.; Ikeda, M.; Yamamoto, K.; Kitagawa, M.; et al. Efficient identification of the MYC regulator with the use of the CRISPR library and context-matched database screenings. Int. J. Mol. Sci. 2022, 23, 7723. [Google Scholar] [CrossRef] [PubMed]
- Evron, T.; Caspi, M.; Kazelnik, M.; Shor-Nareznoy, Y.; Armoza-Eilat, S.; Kariv, R.; Manber, Z.; Elkon, R.; Sklan, E.H.; Rosin-Arbesfeld, R. A CRISPR knockout screen reveals new regulators of canonical Wnt signaling. Oncogenesis 2021, 10, 63. [Google Scholar] [CrossRef] [PubMed]
- Degirmenci, U.; Wang, M.; Hu, J. Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells 2020, 9, 198. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Nguyen, M.Q.; Wilski, N.A.; Purwin, T.J.; Vernon, M.; Tiago, M.; Aplin, A.E. A genome-wide screen identifies PDPK1 as a target to enhance the efficacy of MEK1/2 inhibitors in NRAS mutant melanoma. Cancer Res. 2022, 82, 2625–2639. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Luo, D.; Yu, J.; Zhang, M.; Zheng, X.; Xu, G.; Wang, J.; Wang, H.; Xu, Y.; Jiang, K.; et al. Genome-wide CRISPR-cas9 knockout screening identifies GRB7 as a driver for MEK inhibitor resistance in KRAS mutant colon cancer. Oncogene 2022, 41, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Wolff, E.C.; Folk, J.E. Hypusine: Its post-translational formation in eukaryotic initiation factor 5A and its potential role in cellular regulation. Biofactors 1993, 4, 95–104. [Google Scholar]
- Hanauske-Abel, H.M.; Park, M.H.; Hanauske, A.R.; Popowicz, A.M.; Lalande, M.; Folk, J.E. Inhibition of the G1-S transition of the cell cycle by inhibitors of deoxyhypusine hydroxylation. BBA Mol. Cell Res. 1994, 1221, 115–124. [Google Scholar] [CrossRef]
- Epis, M.R.; Giles, K.M.; Kalinowski, F.C.; Barker, A.; Cohen, R.J.; Leedman, P.J. Regulation of expression of deoxyhypusine hydroxylase (DOHH), the enzyme that catalyzes the activation of eIF5A, by miR-331-3p and miR-642-5p in prostate cancer cells. J. Biol. Chem. 2012, 287, 35251–35259. [Google Scholar] [CrossRef] [Green Version]
- Yadav, D.; Ghosh, K.; Basu, S.; Roeder, R.G.; Biswas, D. Multivalent role of human TFIID in recruiting elongation components at the promoter-proximal region for transcriptional control. Cell Rep. 2019, 26, 1303–1317.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamtchueng, C.; Stebenne, M.E.; Delannoy, A.; Wilhelm, E.; Leger, H.; Benecke, A.G.; Bell, B. Alternative splicing of TAF6: Downstream transcriptome impacts and upstream RNA splice control elements. PLoS ONE 2014, 9, e102399. [Google Scholar] [CrossRef]
- Wei, W.; Nahta, R.; Huper, G.; Marks, J.R. TAFII70 Isoform-specific growth suppression correlates with its ability to complex with the GADD45a protein. Mol. Cancer Res. 2004, 2, 442–452. [Google Scholar] [CrossRef]
- Delannoy, A.; Wilhelm, E.; Eilebrecht, S.; Alvarado-Cuevas, E.M.; Benecke, A.G.; Bell, B. BIM and NOXA are mitochondrial effectors of TAF6delta-driven apoptosis. Cell Death Dis. 2018, 9, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, E.; Pellay, F.X.; Benecke, A.; Bell, B. TAF6delta controls apoptosis and gene expression in the absence of p53. PLoS ONE 2008, 3, e2721. [Google Scholar] [CrossRef] [Green Version]
- Schwerk, C.; Schulze-Osthoff, K. Regulation of apoptosis by alternative Pre-mRNA splicing. Mol. Cell 2005, 19, 1–13. [Google Scholar] [CrossRef]
- Singh, R.; Bassett, E.; Chakravarti, A.; Parthun, M.R. Replication-dependent histone isoforms: A new source of complexity in chromatin structure and function. Nucleic Acids Res. 2018, 46, 8665–8678. [Google Scholar] [CrossRef]
- Su, C.H.; Tzeng, T.Y.; Cheng, C.; Hsu, M.T. An H2A histone isotype regulates estrogen receptor target genes by mediating enhancer-promoter-3’-UTR interactions in breast cancer cells. Nucleic Acids Res. 2014, 42, 3073–3088. [Google Scholar] [CrossRef]
- Monteiro, F.L.; Vitorino, R.; Wang, J.; Cardoso, H.; Laranjeira, H.; Simoes, J.; Caldas, M.; Henrique, R.; Amado, F.; Williams, C.; et al. The histone H2A isoform Hist2h2ac is a novel regulator of proliferation and epithelial-mesenchymal transition in mammary epithelial and in breast cancer cells. Cancer Lett. 2017, 396, 42–52. [Google Scholar] [CrossRef]
- Shah, S.; Verma, T.; Rashid, M.; Gadewal, N.; Gupta, S. Histone H2A isoforms: Potential implications in epigenome plasticity and diseases in eukaryotes. J. Biosci. 2020, 45, 4. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clone | Gene Name | Sequence of gRNA |
|---|---|---|
| SAM-1 | CAT | GGCCAAGATTGGAAGCCCAA |
| CCL19 | CCCTGTTGCCCCCCTCTTTC | |
| DOHH | TTTTATTCCTCACCTACTTA | |
| SLC38A4 | AAAAGACGCAATTTACCAAC | |
| SAM-2 | ASCL5 | TAGGGCGATGGCTCTTGTTA |
| CAPN1 | CGGAAGGGCACCGCGGGGAA | |
| DHRS3 | TTTTTTTCTGGAGACGGGGT | |
| KCNJ11 | TCCGGATCTCTCCCACTAAC | |
| MXD1 | TTGCGAATCCTGTCACCAGT | |
| TAF6 | GTTTCCCTGCCTCCGTTTTG | |
| SAM-3 | ERAP1 | TCGGTCCCCAACTTGAGCAC |
| FAM107A | TGAAGTTCCAATGACATTCA | |
| HIST1H2AC | TTGTCTTCCAATTAACTAAG | |
| KRT32 | ATTTGGCTAAAGCAGGAGTC | |
| SEC14L3 | TCTGTCCCCAAGCCAAGCAG | |
| ZNF219 | ACTCCTTCCCTGGTATGTCC | |
| SAM-4 | MOK | AAGGCTATCGTCCACGTAGT |
| RIPK2 | TGGGACGGGCGGCTGGGAAG | |
| ZAN | GGACTGCAAACGGCTGGACG | |
| SAM-5 | UBLCP1 | TGTTCCGAATGAAGCTTAAA |
| ZNF77 | CCGCCCCTGCCTGTCCTGAT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tatsumi, A.; Hirakochi, H.; Inoue, S.; Tanaka, Y.; Furuno, H.; Ikeda, M.; Ishibashi, S.; Taguchi, T.; Yamamoto, K.; Onishi, I.; et al. Identification of NRAS Downstream Genes with CRISPR Activation Screening. Biology 2022, 11, 1551. https://doi.org/10.3390/biology11111551
Tatsumi A, Hirakochi H, Inoue S, Tanaka Y, Furuno H, Ikeda M, Ishibashi S, Taguchi T, Yamamoto K, Onishi I, et al. Identification of NRAS Downstream Genes with CRISPR Activation Screening. Biology. 2022; 11(11):1551. https://doi.org/10.3390/biology11111551
Chicago/Turabian StyleTatsumi, Akiya, Haruka Hirakochi, Satomi Inoue, Yosuke Tanaka, Hidehiro Furuno, Masumi Ikeda, Sachiko Ishibashi, Towako Taguchi, Kouhei Yamamoto, Iichiroh Onishi, and et al. 2022. "Identification of NRAS Downstream Genes with CRISPR Activation Screening" Biology 11, no. 11: 1551. https://doi.org/10.3390/biology11111551