
Autophagy a Close Relative of AML Biology

 ,
, {kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. The Role of Autophagy: From Normal Hematopoiesis to Leukemia
2.1. Autophagy in Normal Hematopoietic Stem Cell Maintenance and Function
2.2. Autophagy in Leukemia Initiation and Development
2.3. Contribution of Autophagy to AML Metabolism during Leukemic Development
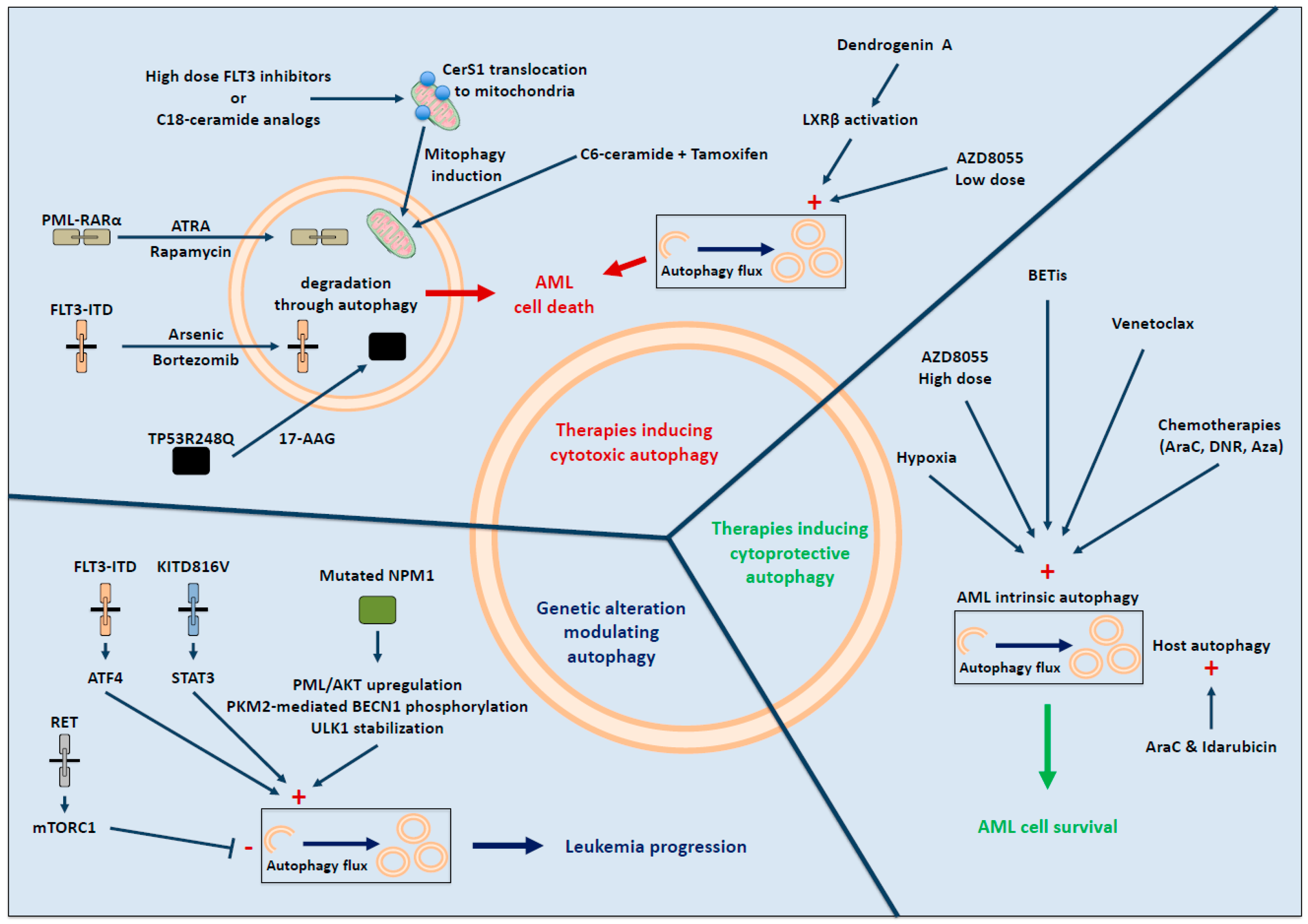
3. Recurrent Genetic AML Alterations Associated with Autophagy
4. Autophagic Response upon Therapy in AML
4.1. Therapy-Induced Cytotoxic Autophagy
4.2. Autophagy and Therapy Resistance in AML
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Döhner, H.; Campbell, P.J. Genomic Classification in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 375, 900–901. [Google Scholar] [CrossRef]
- Auberger, P.; Puissant, A. Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood 2017, 129, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Xu, A.; Huang, Y.; Cao, J.; Zhu, H.; Yang, B.; Shao, X.; He, Q.; Ying, M. The role of autophagy in targeted therapy for acute myeloid leukemia. Autophagy 2020, 1–15. [Google Scholar] [CrossRef]
- Boya, P.; Reggiori, F.; Codogno, P. Emerging regulation and functions of autophagy. Nat. Cell Biol. 2013, 15, 713–720. [Google Scholar] [CrossRef]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Stuani, L.; Sabatier, M.; Saland, E.; Cognet, G.; Poupin, N.; Bosc, C.; Castelli, F.A.; Gales, L.; Turtoi, E.; Montersino, C.; et al. Mitochondrial metabolism supports resistance to IDH mutant inhibitors in acute myeloid leukemia. J. Exp. Med. 2021, 218, e20200924. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Chen, G.; Li, W.; Kepp, O.; Zhu, Y.; Chen, Q. Mitophagy, Mitochondrial Homeostasis, and Cell Fate. Front. Cell Dev. Biol. 2020, 8, 467. [Google Scholar] [CrossRef]
- Bernardini, J.P.; Lazarou, M.; Dewson, G. Parkin and mitophagy in cancer. Oncogene 2017, 36, 1315–1327. [Google Scholar] [CrossRef]
- Montava-Garriga, L.; Ganley, I.G. Outstanding Questions in Mitophagy: What We Do and Do Not Know. J. Mol. Biol. 2020, 432, 206–230. [Google Scholar] [CrossRef]
- Liu, F.; Lee, J.Y.; Wei, H.; Tanabe, O.; Engel, J.D.; Morrison, S.J.; Guan, J.-L. FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 2010, 116, 4806–4814. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, M.; Soilleux, E.J.; Djordjevic, G.; Tripp, R.; Lutteropp, M.; Sadighi-Akha, E.; Stranks, A.J.; Glanville, J.; Knight, S.; Jacobsen, S.-E.W.; et al. The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 2011, 208, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Puerto, M.C.; Folkerts, H.; Wierenga, A.T.J.; Schepers, K.; Schuringa, J.J.; Coffer, P.J.; Vellenga, E. Autophagy Proteins ATG5 and ATG7 Are Essential for the Maintenance of Human CD34(+) Hematopoietic Stem-Progenitor Cells. Stem Cells 2016, 34, 1651–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katajisto, P.; Döhla, J.; Chaffer, C.L.; Pentinmikko, N.; Marjanovic, N.; Iqbal, S.; Zoncu, R.; Chen, W.; Weinberg, R.A.; Sabatini, D.M. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 2015, 348, 340–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takubo, K.; Nagamatsu, G.; Kobayashi, C.I.; Nakamura-Ishizu, A.; Kobayashi, H.; Ikeda, E.; Goda, N.; Rahimi, Y.; Johnson, R.S.; Soga, T.; et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 2013, 12, 49–61. [Google Scholar] [CrossRef] [Green Version]
- Vannini, N.; Girotra, M.; Naveiras, O.; Nikitin, G.; Campos, V.; Giger, S.; Roch, A.; Auwerx, J.; Lutolf, M.P. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat. Commun. 2016, 7, 13125. [Google Scholar] [CrossRef]
- Ito, K.; Turcotte, R.; Cui, J.; Zimmerman, S.E.; Pinho, S.; Mizoguchi, T.; Arai, F.; Runnels, J.M.; Alt, C.; Teruya-Feldstein, J.; et al. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science 2016, 354, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Cai, J.; Zhang, S.; Yuan, N.; Fang, Y.; Wang, Z.; Li, X.; Cao, D.; Xu, F.; Lin, W.; et al. Autophagy Sustains Hematopoiesis Through Targeting Notch. Stem Cells Dev. 2015, 24, 2660–2673. [Google Scholar] [CrossRef]
- Park, S.M.; Ou, J.; Chamberlain, L.; Simone, T.M.; Yang, H.; Virbasius, C.-M.; Ali, A.M.; Zhu, L.J.; Mukherjee, S.; Raza, A.; et al. U2AF35(S34F) Promotes Transformation by Directing Aberrant ATG7 Pre-mRNA 3’ End Formation. Mol. Cell 2016, 62, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Visconte, V.; Przychodzen, B.; Han, Y.; Nawrocki, S.T.; Thota, S.; Kelly, K.R.; Patel, B.J.; Hirsch, C.; Advani, A.S.; Carraway, H.E.; et al. Complete mutational spectrum of the autophagy interactome: A novel class of tumor suppressor genes in myeloid neoplasms. Leukemia 2017, 31, 505–510. [Google Scholar] [CrossRef]
- Watson, A.S.; Riffelmacher, T.; Stranks, A.; Williams, O.; De Boer, J.; Cain, K.; MacFarlane, M.; McGouran, J.; Kessler, B.; Khandwala, S.; et al. Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 2015, 1, 15008. [Google Scholar] [CrossRef]
- Bosc, C.; Broin, N.; Fanjul, M.; Saland, E.; Farge, T.; Courdy, C.; Batut, A.; Masoud, R.; Larrue, C.; Skuli, S.; et al. Autophagy regulates fatty acid availability for oxidative phosphorylation through mitochondria-endoplasmic reticulum contact sites. Nat. Commun. 2020, 11, 4056. [Google Scholar] [CrossRef]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER-mitochondria contact sites. Nature 2013, 495, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Minhajuddin, M.; Adane, B.; Khan, N.; Stevens, B.M.; Mack, S.C.; Lai, S.; Rich, J.N.; Inguva, A.; Shannon, K.M.; et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell 2018, 23, 86–100.e6. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Huang, Z.; Wang, Z.; Yin, C.; Zhou, L.; Zhang, L.; Huang, K.; Zhou, H.; Jiang, X.; Li, J.; et al. Identification of novel molecular markers for prognosis estimation of acute myeloid leukemia: Over-expression of PDCD7, FIS1 and Ang2 may indicate poor prognosis in pretreatment patients with acute myeloid leukemia. PLoS ONE 2014, 9, e84150. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Shaid, S.; Vakhrusheva, O.; Koschade, S.E.; Klann, K.; Thölken, M.; Baker, F.; Zhang, J.; Oellerich, T.; Sürün, D.; et al. Loss of the selective autophagy receptor p62 impairs murine myeloid leukemia progression and mitophagy. Blood 2019, 133, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, Y.; Yin, J.; Wang, C.; Yang, M.; Gu, J.; He, M.; Xu, H.; Fu, W.; Zhang, W.; et al. A mitophagy inhibitor targeting p62 attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Lett. 2021, 510, 24–36. [Google Scholar] [CrossRef]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [Green Version]
- Heydt, Q.; Larrue, C.; Saland, E.; Bertoli, S.; Sarry, J.-E.; Besson, A.; Manenti, S.; Joffre, C.; Mansat-De Mas, V. Oncogenic FLT3-ITD supports autophagy via ATF4 in acute myeloid leukemia. Oncogene 2018, 37, 787–797. [Google Scholar] [CrossRef] [Green Version]
- Rudat, S.; Pfaus, A.; Cheng, Y.Y.; Holtmann, J.; Ellegast, J.M.; Bühler, C.; Marcantonio, D.D.; Martinez, E.; Göllner, S.; Wickenhauser, C.; et al. RET-mediated autophagy suppression as targetable co-dependence in acute myeloid leukemia. Leukemia 2018, 32, 2189–2202. [Google Scholar] [CrossRef]
- Paschka, P.; Marcucci, G.; Ruppert, A.S.; Mrózek, K.; Chen, H.; Kittles, R.A.; Vukosavljevic, T.; Perrotti, D.; Vardiman, J.W.; Carroll, A.J.; et al. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): A Cancer and Leukemia Group B Study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 3904–3911. [Google Scholar] [CrossRef]
- Larrue, C.; Heydt, Q.; Saland, E.; Boutzen, H.; Kaoma, T.; Sarry, J.-E.; Joffre, C.; Récher, C. Oncogenic KIT mutations induce STAT3-dependent autophagy to support cell proliferation in acute myeloid leukemia. Oncogenesis 2019, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; La Starza, R.; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N. Engl. J. Med. 2005, 352, 254–266. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Yang, Z.; Tang, Y.; Tao, Y.; Zhan, Q.; Lei, L.; Jing, Y.; Jiang, X.; Jin, H.; et al. Glycolytic Enzyme PKM2 Mediates Autophagic Activation to Promote Cell Survival in NPM1-Mutated Leukemia. Int. J. Biol. Sci. 2019, 15, 882–894. [Google Scholar] [CrossRef] [Green Version]
- Zou, Q.; Tan, S.; Yang, Z.; Zhan, Q.; Jin, H.; Xian, J.; Zhang, S.; Yang, L.; Wang, L.; Zhang, L. NPM1 Mutant Mediated PML Delocalization and Stabilization Enhances Autophagy and Cell Survival in Leukemic Cells. Theranostics 2017, 7, 2289–2304. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Tao, Y.; Wang, L.; Yang, L.; Jing, Y.; Jiang, X.; Lei, L.; Yang, Z.; Wang, X.; Peng, M.; et al. NPM1 mutant maintains ULK1 protein stability via TRAF6-dependent ubiquitination to promote autophagic cell survival in leukemia. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21192. [Google Scholar]
- Folkerts, H.; Hilgendorf, S.; Wierenga, A.T.J.; Jaques, J.; Mulder, A.B.; Coffer, P.J.; Schuringa, J.J.; Vellenga, E. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Dis. 2017, 8, e2927. [Google Scholar] [CrossRef] [Green Version]
- Fan, Y.; Peng, X.; Li, B.; Zhao, G. Development of Autophagy Signature-Based Prognostic Nomogram for Refined Glioma Survival Prognostication. BioMed Res. Int. 2020, 2020, 1872962. [Google Scholar] [CrossRef]
- Dai, Y.-J.; Hu, F.; He, S.-Y.; Wang, Y.-Y. Epigenetic landscape analysis of lncRNAs in acute myeloid leukemia with DNMT3A mutations. Ann. Transl. Med. 2020, 8, 318. [Google Scholar] [CrossRef]
- Isakson, P.; Bjørås, M.; Bøe, S.O.; Simonsen, A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 2010, 116, 2324–2331. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, L.; Kang, R.; Yang, M.; Liu, L.; Zhao, Y.; Yu, Y.; Xie, M.; Yin, X.; Livesey, K.M.; et al. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARα oncoprotein. Autophagy 2011, 7, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Schläfli, A.M.; Isakson, P.; Garattini, E.; Simonsen, A.; Tschan, M.P. The autophagy scaffold protein ALFY is critical for the granulocytic differentiation of AML cells. Sci. Rep. 2017, 7, 12980. [Google Scholar] [CrossRef] [Green Version]
- Larrue, C.; Saland, E.; Boutzen, H.; Vergez, F.; David, M.; Joffre, C.; Hospital, M.-A.; Tamburini, J.; Delabesse, E.; Manenti, S.; et al. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood 2016, 127, 882–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.-J.; Wang, L.-N.; Zhang, Z.-H.; Liang, C.; Li, Y.; Luo, J.-S.; Peng, C.-J.; Zhang, X.-L.; Ke, Z.-Y.; Huang, L.-B.; et al. Arsenic trioxide induces autophagic degradation of the FLT3-ITD mutated protein in FLT3-ITD acute myeloid leukemia cells. J. Cancer 2020, 11, 3476–3482. [Google Scholar] [CrossRef] [Green Version]
- Allende-Vega, N.; Villalba, M. Metabolic stress controls mutant p53 R248Q stability in acute myeloid leukemia cells. Sci. Rep. 2019, 9, 5637. [Google Scholar] [CrossRef]
- Willems, L.; Chapuis, N.; Puissant, A.; Maciel, T.T.; Green, A.S.; Jacque, N.; Vignon, C.; Park, S.; Guichard, S.; Herault, O.; et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia 2012, 26, 1195–1202. [Google Scholar] [CrossRef] [Green Version]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Maciel, T.T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef] [Green Version]
- Dany, M.; Gencer, S.; Nganga, R.; Thomas, R.J.; Oleinik, N.; Baron, K.D.; Szulc, Z.M.; Ruvolo, P.; Kornblau, S.; Andreeff, M.; et al. Targeting FLT3-ITD signaling mediates ceramide-dependent mitophagy and attenuates drug resistance in AML. Blood 2016, 128, 1944–1958. [Google Scholar] [CrossRef] [Green Version]
- Morad, S.A.F.; MacDougall, M.R.; Abdelmageed, N.; Kao, L.-P.; Feith, D.J.; Tan, S.-F.; Kester, M.; Loughran, T.P.; Wang, H.-G.; Cabot, M.C. Pivotal role of mitophagy in response of acute myelogenous leukemia to a ceramide-tamoxifen-containing drug regimen. Exp. Cell Res. 2019, 381, 256–264. [Google Scholar] [CrossRef]
- Segala, G.; David, M.; de Medina, P.; Poirot, M.C.; Serhan, N.; Vergez, F.; Mougel, A.; Saland, E.; Carayon, K.; Leignadier, J.; et al. Dendrogenin A drives LXR to trigger lethal autophagy in cancers. Nat. Commun. 2017, 8, 1903. [Google Scholar] [CrossRef] [PubMed]
- Serhan, N.; Mouchel, P.-L.; de Medina, P.; Segala, G.; Mougel, A.; Saland, E.; Rives, A.; Lamaziere, A.; Despres, G.; Sarry, J.-E.; et al. Dendrogenin A synergizes with Cytarabine to Kill Acute Myeloid Leukemia Cells In Vitro and In Vivo. Cancers 2020, 12, 1725. [Google Scholar] [CrossRef]
- Mouchel, P.-L.; Serhan, N.; Betous, R.; Farge, T.; Saland, E.; De Medina, P.; Hoffmann, J.-S.; Sarry, J.-E.; Poirot, M.; Silvente-Poirot, S.; et al. Dendrogenin A Enhances Anti-Leukemic Effect of Anthracycline in Acute Myeloid Leukemia. Cancers 2020, 12, 2933. [Google Scholar] [CrossRef]
- Bosnjak, M.; Ristic, B.; Arsikin, K.; Mircic, A.; Suzin-Zivkovic, V.; Perovic, V.; Bogdanovic, A.; Paunovic, V.; Markovic, I.; Bumbasirevic, V.; et al. Inhibition of mTOR-dependent autophagy sensitizes leukemic cells to cytarabine-induced apoptotic death. PLoS ONE 2014, 9, e94374. [Google Scholar] [CrossRef] [PubMed]
- Putyrski, M.; Vakhrusheva, O.; Bonn, F.; Guntur, S.; Vorobyov, A.; Brandts, C.; Dikic, I.; Ernst, A. Disrupting the LC3 Interaction Region (LIR) Binding of Selective Autophagy Receptors Sensitizes AML Cell Lines to Cytarabine. Front. Cell Dev. Biol. 2020, 8, 208. [Google Scholar] [CrossRef]
- Sumitomo, Y.; Koya, J.; Nakazaki, K.; Kataoka, K.; Tsuruta-Kishino, T.; Morita, K.; Sato, T.; Kurokawa, M. Cytoprotective autophagy maintains leukemia-initiating cells in murine myeloid leukemia. Blood 2016, 128, 1614–1624. [Google Scholar] [CrossRef] [Green Version]
- Piya, S.; Kornblau, S.M.; Ruvolo, V.R.; Mu, H.; Ruvolo, P.P.; McQueen, T.; Davis, R.E.; Hail, N.; Kantarjian, H.; Andreeff, M.; et al. Atg7 suppression enhances chemotherapeutic agent sensitivity and overcomes stroma-mediated chemoresistance in acute myeloid leukemia. Blood 2016, 128, 1260–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsunaga, T.; Takemoto, N.; Sato, T.; Takimoto, R.; Tanaka, I.; Fujimi, A.; Akiyama, T.; Kuroda, H.; Kawano, Y.; Kobune, M.; et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat. Med. 2003, 9, 1158–1165. [Google Scholar] [CrossRef]
- Drolle, H.; Wagner, M.; Vasold, J.; Kütt, A.; Deniffel, C.; Sotlar, K.; Sironi, S.; Herold, T.; Rieger, C.; Fiegl, M. Hypoxia regulates proliferation of acute myeloid leukemia and sensitivity against chemotherapy. Leuk. Res. 2015, 39, 779–785. [Google Scholar] [CrossRef]
- Dykstra, K.M.; Fay, H.R.S.; Massey, A.C.; Yang, N.; Johnson, M.; Portwood, S.; Guzman, M.L.; Wang, E.S. Inhibiting autophagy targets human leukemic stem cells and hypoxic AML blasts by disrupting mitochondrial homeostasis. Blood Adv. 2021, 5, 2087–2100. [Google Scholar] [CrossRef]
- Qiu, L.; Zhou, G.; Cao, S. Targeted inhibition of ULK1 enhances daunorubicin sensitivity in acute myeloid leukemia. Life Sci. 2020, 243, 117234. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Han, Y.; Visconte, V.; Przychodzen, B.; Espitia, C.M.; Phillips, J.; Anwer, F.; Advani, A.; Carraway, H.E.; Kelly, K.R.; et al. The novel autophagy inhibitor ROC-325 augments the antileukemic activity of azacitidine. Leukemia 2019, 33, 2971–2974. [Google Scholar] [CrossRef]
- Torgersen, M.L.; Engedal, N.; Bøe, S.-O.; Hokland, P.; Simonsen, A. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t(8;21) AML cells. Blood 2013, 122, 2467–2476. [Google Scholar] [CrossRef] [Green Version]
- Peterse, E.F.P.; Niessen, B.; Addie, R.D.; de Jong, Y.; Cleven, A.H.G.; Kruisselbrink, A.B.; van den Akker, B.E.W.M.; Molenaar, R.J.; Cleton-Jansen, A.-M.; Bovée, J.V.M.G. Targeting glutaminolysis in chondrosarcoma in context of the IDH1/2 mutation. Br. J. Cancer 2018, 118, 1074–1083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, M.R.; Liu, Y.; Neltner, J.; Pu, H.; Morris, A.; Sunkara, M.; Pittman, T.; Kyprianou, N.; Horbinski, C. Autophagy and Oxidative Stress in Gliomas with IDH1 Mutations. Acta Neuropathol. (Berl.) 2014, 127, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Folkerts, H.; Wierenga, A.T.; van den Heuvel, F.A.; Woldhuis, R.R.; Kluit, D.S.; Jaques, J.; Schuringa, J.J.; Vellenga, E. Elevated VMP1 expression in acute myeloid leukemia amplifies autophagy and is protective against venetoclax-induced apoptosis. Cell Death Dis. 2019, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.E.; Eom, J.-I.; Jeung, H.-K.; Cheong, J.-W.; Lee, J.Y.; Kim, J.S.; Min, Y.H. Targeting AMPK-ULK1-mediated autophagy for combating BET inhibitor resistance in acute myeloid leukemia stem cells. Autophagy 2017, 13, 761–762. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy Levy, J.M.; Towers, C.G.; Thorburn, A. Targeting Autophagy in Cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joffre, C.; Ducau, C.; Poillet-Perez, L.; Courdy, C.; Mansat-De Mas, V. Autophagy a Close Relative of AML Biology. Biology 2021, 10, 552. https://doi.org/10.3390/biology10060552
Joffre C, Ducau C, Poillet-Perez L, Courdy C, Mansat-De Mas V. Autophagy a Close Relative of AML Biology. Biology. 2021; 10(6):552. https://doi.org/10.3390/biology10060552
Chicago/Turabian StyleJoffre, Carine, Charlotte Ducau, Laura Poillet-Perez, Charly Courdy, and Véronique Mansat-De Mas. 2021. "Autophagy a Close Relative of AML Biology" Biology 10, no. 6: 552. https://doi.org/10.3390/biology10060552