
Soil Bacterial Community Shifts Are Driven by Soil Nutrient Availability along a Teak Plantation Chronosequence in Tropical Forests in China

 ,
,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Region
2.2. Experimental Design and Soil Sample Collection
2.3. Soil DNA Isolation and PCR Amplification
2.4. Illumina Miseq Sequencing
2.5. Processing of Sequencing Data
2.6. Soil Physicochemical Parameters and Enzyme Activity Measurements
2.7. Statistical Analysis
3. Results
3.1. Soil Physicochemical Properties
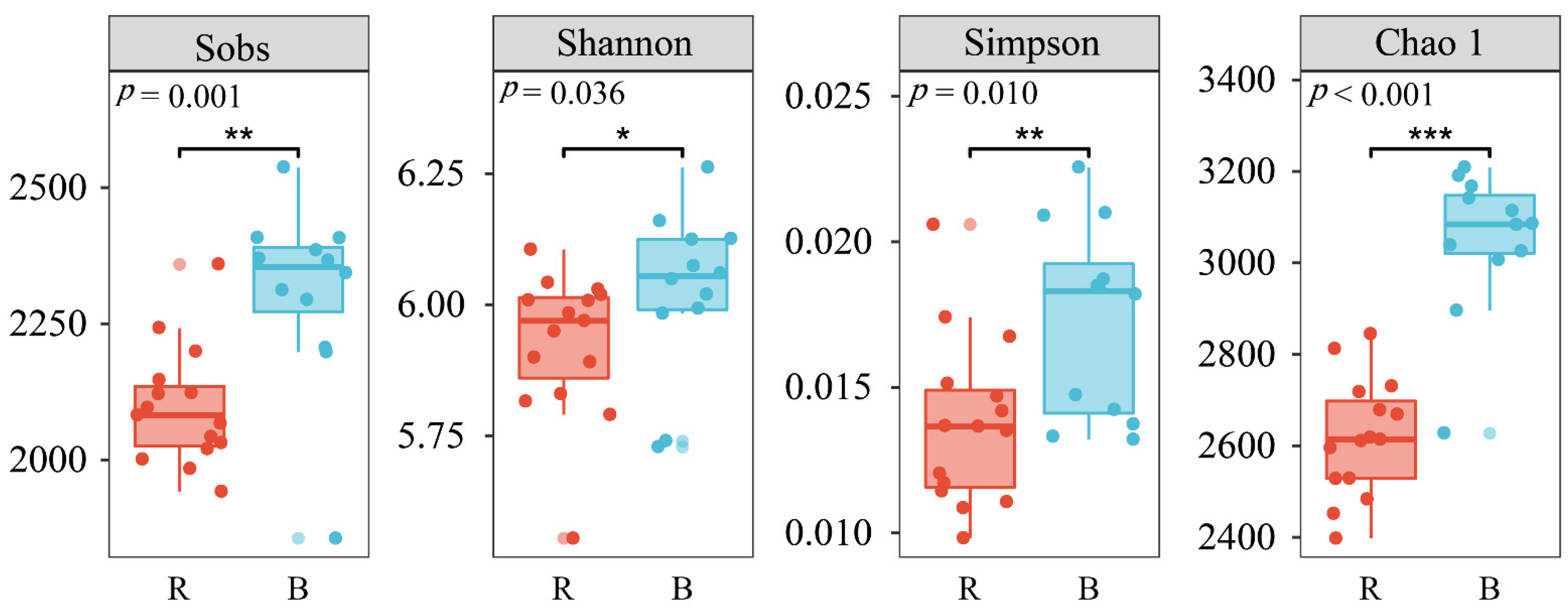
3.2. Changes in Soil Bacterial Alpha Diversity
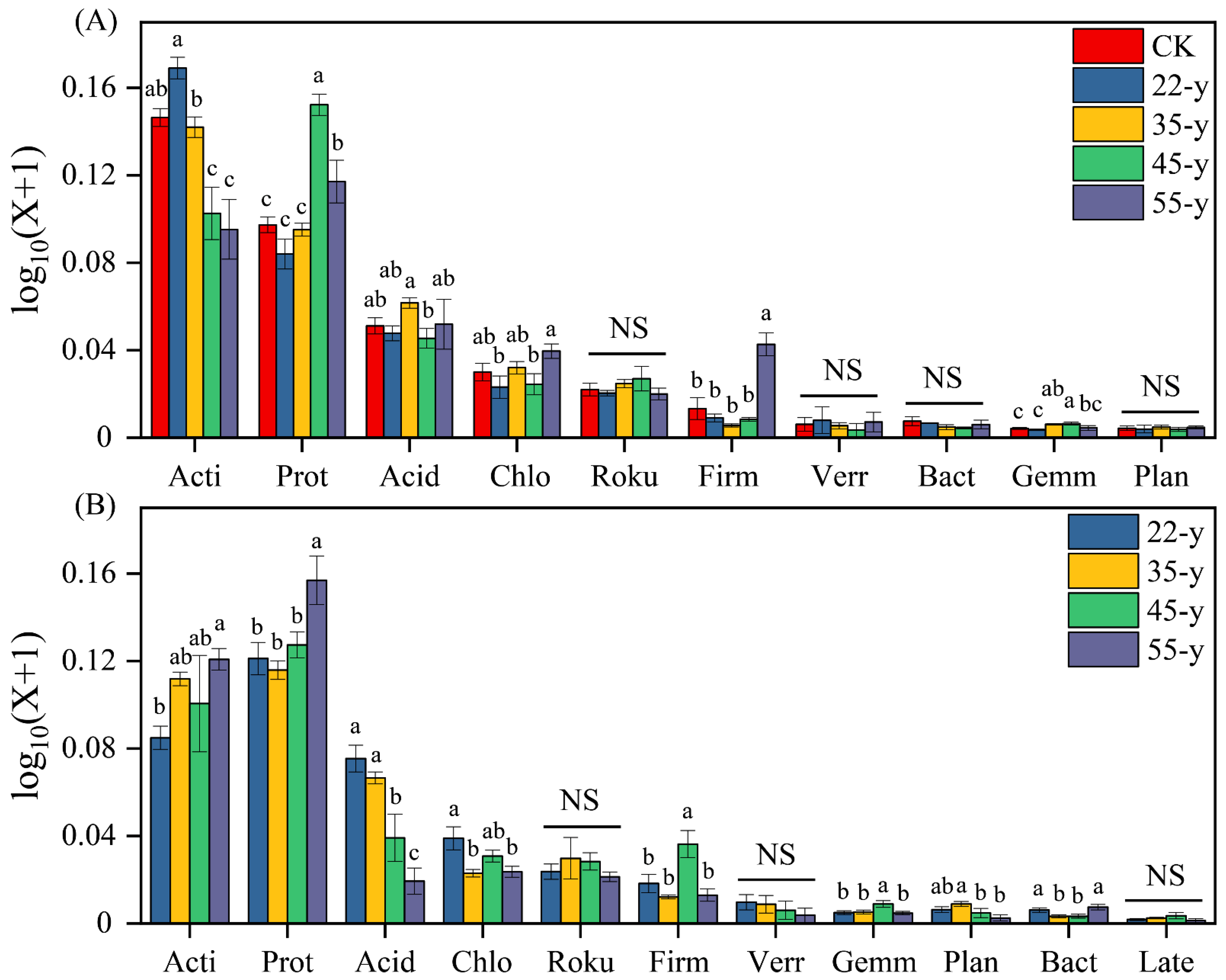
3.3. Soil Bacterial Community Composition and Structure with Stand Development
3.4. Soil Bacterial Co-Occurrence Networks
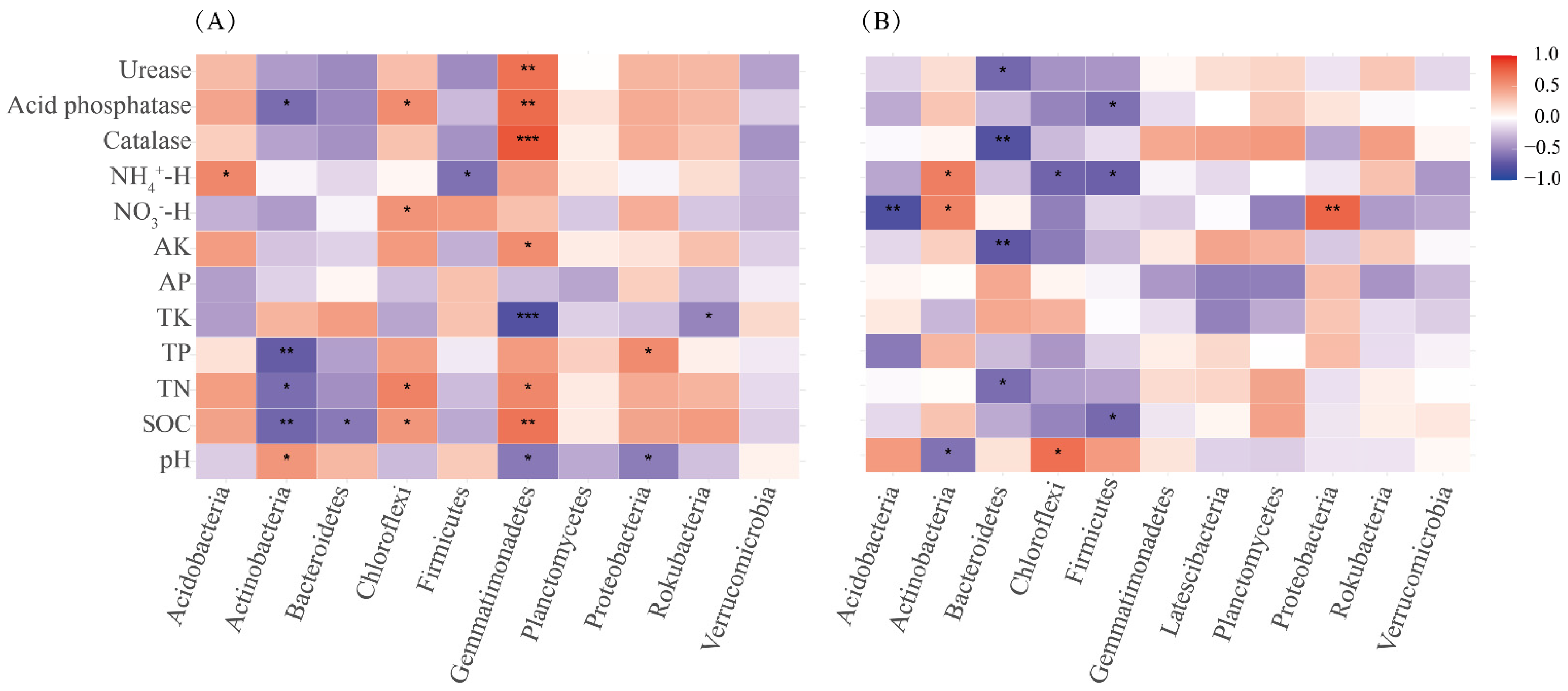
3.5. Relationships between Soil Properties and Soil Bacterial Community
4. Discussion
4.1. Responses of Soil Properties to the Chronosequence
4.2. Shifts in Bacterial Communities between Successional Series
4.3. Correlation between Bacterial Community and Edaphic Factors
4.4. Co-Occurrence Network of the Bacterial Communities
4.5. Implications of Bacterial Community Variation in Teak Plantations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 22-y | 22-year-old stand |
| 35-y | 35-year-old stand |
| 45-y | 45-year-old stand |
| 55-y | 55-year-old stand |
| CK | control |
| DBH | diameter at breast height |
| OTUs | Operational taxonomic units |
| RDP | Ribosomal Database Project |
| SOC | Soil organic carbon |
| TN | Total nitrogen |
| TP | Total phosphorus |
| TK | Total potassium |
| AP | Available phosphorus |
| AK | Available potassium |
| -H | Ammonium-N |
| -H | Nitrate-N |
| HSD | Honestly significant difference |
| NMDS | Non-metric multidimensional scaling |
| ANOSIM | Analysis of similarities |
| PERMANOVA | Permutation multivariate analysis of variance |
| RDA | Redundancy analysis |
| APL | Average path length |
| MD | Modularity |
| ACC | Average clustering coefficient |
| BC | Betweenness centrality |
References
- Russell, A.E.; Parton, W.J., Jr. Modeling the Effects of Global Change on Ecosystem Processes in a Tropical Rainforest. Forests 2020, 11, 213. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.B.; Li, T.; Cui, H.L.; Li, L.F.; Allen Samuel, C.; Chen, L.; Li, Y.P.; Zhao, Z.W. Shifts in the arbuscular mycorrhizal fungal community composition of Betula alnoides along young, middle-aged plantation and adjacent natural forest. IForest 2020, 13, 447. [Google Scholar] [CrossRef]
- Qiang, W.; He, L.L.; Zhang, Y.; Liu, B.; Liu, Y.; Liu, Q.H.; Pang, X.Y. Aboveground vegetation and soil physicochemical properties jointly drive the shift of soil microbial community during subalpine secondary succession in southwest China. Catena 2021, 202, 105251. [Google Scholar] [CrossRef]
- Wardle, D.A.; Bardgett, R.D.; Klironomos, J.N.; Setälä, H.; van der Putten, W.; Wall, D.H. Ecological linkages between aboveground and belowground biota. Science 2004, 304, 1629–1633. [Google Scholar] [CrossRef]
- Xu, M.P.; Lu, X.Q.; Xu, Y.D.; Zhong, Z.K.; Zhang, W.; Ren, C.J.; Han, X.H.; Yang, G.H.; Feng, Y.Z. Dynamics of bacterial community in litter and soil along a chronosequence of Robinia pseudoacacia plantations. Sci. Total Environ. 2020, 703, 135613. [Google Scholar] [CrossRef]
- Wang, G.Z.; Liu, Y.G.; Cui, M.; Zhou, Z.Y.; Zhang, Q.; Li, Y.J.; Ha, W.X.; Pang, D.B.; Luo, J.F.; Zhou, J.X. Effects of secondary succession on soil fungal and bacterial compositions and diversities in a karst area. Plant Soil 2021, 465, 1–12. [Google Scholar] [CrossRef]
- Zeng, Q.C.; An, S.S.; Liu, Y. Soil bacterial community response to vegetation succession after fencing in the grassland of China. Sci. Total Environ. 2017, 609, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Ren, H.D.; Li, S.; Leng, X.H.; Yao, X.H. Soil bacterial community structure and co-occurrence pattern during vegetation restoration in karst rocky desertification area. Front. Microbiol. 2017, 8, 2377. [Google Scholar] [CrossRef] [Green Version]
- Shang, R.G.; Li, S.F.; Huang, X.B.; Liu, W.D.; Lang, X.D.; Su, J.R. Effects of Soil Properties and Plant Diversity on Soil Microbial Community Composition and Diversity during Secondary Succession. Forests 2021, 12, 805. [Google Scholar] [CrossRef]
- Hu, Y.G.; Zhang, Z.S.; Huang, L.; Qi, Q.; Liu, L.C.; Zhao, Y.; Wang, Z.R.; Zhou, H.K.; Lv, X.Y.; Mao, Z.C.; et al. Shifts in soil microbial community functional gene structure across a 61-year desert revegetation chronosequence. Geoderma 2019, 347, 126–134. [Google Scholar] [CrossRef]
- Adesemoye, A.O.; Kloepper, J.W. Plant–microbes interactions in enhanced fertilizer-use efficiency. Appl. Microbiol. Biotechnol. 2009, 85, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Bender, S.F.; Conen, F.; Van der Heijden, M.G.A. Mycorrhizal effects on nutrient cycling, nutrient leaching and N2O production in experimental grassland. Soil Biol. Biochem. 2015, 80, 283–292. [Google Scholar] [CrossRef]
- Li, J.J.; Li, L.J.; Arif, M.; Ding, D.D.; Hu, X.; Zheng, J.; Yuan, Z.X.; Li, C.X. Artificial plantation responses to periodic submergence in massive dam and reservoir riparian zones: Changes in soil properties and bacterial community characteristics. Biology 2021, 10, 819. [Google Scholar] [CrossRef]
- Na, X.F.; Xu, T.T.; Li, M.; Zhou, Z.N.; Ma, S.L.; Wang, J.; He, J.; Jiao, B.Z.; Ma, F. Variations of bacterial community diversity within the rhizosphere of three phylogenetically related perennial shrub plant species across environmental gradients. Front. Microbiol. 2018, 9, 709. [Google Scholar] [CrossRef] [Green Version]
- Rolfe, S.A.; Griffiths, J.; Ton, J. Crying out for help with root exudates: Adaptive mechanisms by which stressed plants assemble health-promoting soil microbiomes. Curr. Opin. Microbiol. 2019, 49, 73–82. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Pieterse, C.M.; Bakker, P.A. The rhizosphere microbiome and plant health. Trends Plant Sci. 2012, 17, 478–486. [Google Scholar] [CrossRef]
- Landesman, W.J.; Nelson, D.M.; Fitzpatrick, M.C. Soil properties and tree species drive ß-diversity of soil bacterial communities. Soil Biol. Biochem. 2014, 76, 201–209. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, G.B.; Xue, S.; Wang, G.L. Soil bacterial community dynamics reflect changes in plant community and soil properties during the secondary succession of abandoned farmland in the Loess Plateau. Soil Biol. Biochem. 2016, 97, 40–49. [Google Scholar] [CrossRef]
- Liu, G.Y.; Chen, L.L.; Shi, X.R.; Yuan, Z.Y.; Yuan, L.Y.; Lock, T.R.; Kallenbach, R.L. Changes in rhizosphere bacterial and fungal community composition with vegetation restoration in planted forests. Land Degrad. Dev. 2019, 30, 1147–1157. [Google Scholar] [CrossRef]
- Huang, X.M.; Liu, S.R.; Wang, H.; Hu, Z.D.; Li, Z.G.; You, Y.M. Changes of soil microbial biomass carbon and community composition through mixing nitrogen-fixing species with Eucalyptus urophylla in subtropical China. Soil Biol. Biochem. 2014, 73, 42–48. [Google Scholar] [CrossRef]
- Deng, J.J.; Yin, Y.; Zhu, W.X.; Zhou, Y.B. Variations in soil bacterial community diversity and structures among different revegetation types in the Baishilazi Nature Reserve. Front. Microbiol. 2018, 9, 2874. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.J.; Liu, Y.J.; Luo, J.J.; Qin, M.S.; Johnson, N.C.; Öpik, M.; Vasar, M.; Chai, Y.X.; Zhou, X.L.; Mao, L. Dynamics of arbuscular mycorrhizal fungal community structure and functioning along a nitrogen enrichment gradient in an alpine meadow ecosystem. New Phytol. 2018, 220, 1222–1235. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.P.; Wang, J.Y.; Zhu, Y.F.; Han, X.H.; Ren, C.J.; Yang, G.H. Plant Biomass and Soil Nutrients Mainly Explain the Variation of Soil Microbial Communities During Secondary Succession on the Loess Plateau. Microb. Ecol. 2021, 81, 1–13. [Google Scholar] [CrossRef]
- Ma, H.B.; Zou, W.T.; Yang, J.C.; Hogan, J.A.; Xu, H.; Chen, J. Dominant tree species shape soil microbial community via regulating assembly processes in planted subtropical forests. Forests 2019, 10, 978. [Google Scholar] [CrossRef] [Green Version]
- Dai, Z.M.; Lv, X.F.; Ma, B.; Chen, N.; Chang, S.X.; Lin, J.H.; Wang, X.H.; Su, W.Q.; Liu, H.T.; Huang, Y.L.; et al. Concurrent and rapid recovery of bacteria and protist communities in Canadian boreal forest ecosystems following wildfire. Soil Biol. Biochem. 2021, 163, 108452. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, R.B.; Sun, T.T.; Liang, Y.T.; Jiang, Y.J.; Sun, B. Organic amendments shift the phosphorus-correlated microbial co-occurrence pattern in the peanut rhizosphere network during long-term fertilization regimes. Appl. Soil Ecol. 2018, 124, 229–239. [Google Scholar] [CrossRef]
- Li, S.; Wu, F.Z. Diversity and co-occurrence patterns of soil bacterial and fungal communities in seven intercropping systems. Front. Microbiol. 2018, 9, 1521. [Google Scholar] [CrossRef]
- Zhang, L.; Zhong, M.M.; Li, X.C.; Lu, W.X.; Li, J. River bacterial community structure and co-occurrence patterns under the influence of different domestic sewage types. J. Environ. Manag. 2020, 266, 110590. [Google Scholar] [CrossRef]
- Zhu, H.Z.; Zhang, Z.F.; Zhou, N.; Jiang, C.Y.; Wang, B.J.; Cai, L.; Liu, S.J. Diversity, distribution and co-occurrence patterns of bacterial communities in a karst cave system. Front. Microbiol. 2019, 10, 1726. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Li, H.Z.; Wang, Y.L.; Wu, B.W.; Wu, M.S.; Chen, L.; Li, X.C.; Zhang, Y.; Wang, X.P.; Shi, M.M. Leaf and root endospheres harbor lower fungal diversity and less complex fungal co-occurrence patterns than rhizosphere. Front. Microbiol. 2019, 10, 1015. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, W.D.; Lee, Y.K.; Xie, J.J.; Zhang, H.F. Spatial heterogeneity and co-occurrence of mucosal and luminal microbiome across swine intestinal tract. Front. Microbiol. 2018, 9, 48. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.R.; Liu, Q.; Li, X.R.; Wang, M.; Liu, X.S.; Yang, J.P.; Xu, J.S.; Jiang, Y. Spatial patterns and co-occurrence networks of microbial communities related to environmental heterogeneity in deep-sea surface sediments around Yap Trench, Western Pacific Ocean. Sci. Total Environ. 2021, 759, 143799. [Google Scholar] [CrossRef]
- Barberán, A.; Bates, S.T.; Casamayor, E.O.; Fierer, N. Using network analysis to explore co-occurrence patterns in soil microbial communities. ISME J. 2012, 6, 343–351. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Liang, K.N.; Zhou, Z.Z.; Wang, X.Y.; Huang, G.H. UPLC-ESI-MS/MS-based widely targeted metabolomics analysis of wood metabolites in teak (Tectona grandis). Molecules 2020, 25, 2189. [Google Scholar] [CrossRef]
- Yu, Z.; Liang, K.N.; Wang, X.B.; Huang, G.H.; Lin, M.P.; Zhou, Z.Z.; Chen, Y.L. Alterations in arbuscular mycorrhizal community along a chronosequence of teak (Tectona grandis) plantations in tropical forests of China. Front. Microbiol. 2021, 12, 737068. [Google Scholar] [CrossRef]
- Ren, C.J.; Zhou, Z.H.; Guo, Y.X.; Yang, G.H.; Zhao, F.Z.; Wei, G.H.; Han, X.H.; Feng, L.; Feng, Y.Z.; Ren, G.X. Contrasting patterns of microbial community and enzyme activity between rhizosphere and bulk soil along an elevation gradient. Catena 2021, 196, 104921. [Google Scholar] [CrossRef]
- Ollion, J.; Cochennec, J.; Loll, F.; Escudé, C.; Boudier, T. TANGO: A generic tool for high-throughput 3D image analysis for studying nuclear organization. Bioinformatics 2013, 29, 1840–1841. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Lu, R. Methods for Soil and Agriculture Chemistry Analysis; Chinese Agricultural Science and Technology Press: Beijing, China, 1999. [Google Scholar]
- Liu, Z.Q.; Li, D.F.; Zhang, J.E.; Saleem, M.; Zhang, Y.; Ma, R.; He, Y.N.; Yang, J.Y.; Xiang, H.M.; Wei, H. Effect of simulated acid rain on soil CO2, CH4 and N2O emissions and microbial communities in an agricultural soil. Geoderma 2020, 366, 114222. [Google Scholar] [CrossRef]
- Zhen, Z.; Wang, S.B.; Luo, S.W.; Ren, L.; Liang, Y.Q.; Yang, R.C.; Li, Y.; Zhang, Y.Q.; Deng, S.Q.; Zou, L.N. Significant impacts of both total amount and availability of heavy metals on the functions and assembly of soil microbial communities in different land use patterns. Front. Microbiol. 2019, 10, 2293. [Google Scholar] [CrossRef]
- Ji, L.; Yang, Y.C.; Yang, N.; Khan, A.; Yang, L.X. Seasonal variation of diversity and co-occurrence patterns of arbuscular mycorrhizal fungal communities in mixed broadleaf-conifer forests. Appl. Soil Ecol. 2021, 158, 103782. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, W.W.; Wang, G.; Yang, S.Q.; Pereira, P. Effects of long-term afforestation and natural grassland recovery on soil properties and quality in Loess Plateau (China). Sci. Total Environ. 2021, 770, 144833. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Luo, Z.Q.; Zhang, C.H.; Qu, X.J.; Chen, M.; Song, T.; Yuan, J. Seasonal Variation in the Rhizosphere and Non-Rhizosphere Microbial Community Structures and Functions of Camellia yuhsienensis Hu. Microorganisms 2020, 8, 1385. [Google Scholar] [CrossRef]
- Srivastava, D.S.; Cadotte, M.W.; MacDonald, A.A.M.; Marushia, R.G.; Mirotchnick, N. Phylogenetic diversity and the functioning of ecosystems. Ecol. Lett. 2012, 15, 637–648. [Google Scholar] [CrossRef]
- Nannipieri, P.; Ascher, J.; Ceccherini, M.T.; Landi, L.; Pietramellara, G.; Renella, G. Microbial diversity and soil functions. Eur. J. Soil Sci. 2003, 54, 655–670. [Google Scholar] [CrossRef]
- Huang, F.Y.; Liu, Z.H.; Mou, H.Y.; Zhang, P.; Jia, Z.K. Effects of different long-term farmland mulching practices on the loessial soil fungal community in a semiarid region of China. Appl. Soil Ecol. 2019, 137, 111–119. [Google Scholar] [CrossRef]
- Bi, B.Y.; Zhang, H.; Yuan, Y.; Wu, Z.H.; Wang, Y.; Han, F.P. Dynamic changes of soil microbial community in Pinus sylvestris var. mongolica plantations in the Mu Us Sandy Land. J. Environ. Manag. 2021, 287, 112306. [Google Scholar]
- Xu, M.P.; Jian, J.N.; Wang, J.Y.; Zhang, Z.J.; Yang, G.H.; Han, X.H.; Ren, C.J. Response of root nutrient resorption strategies to rhizosphere soil microbial nutrient utilization along Robinia pseudoacacia plantation chronosequence. For. Ecol. Manag. 2021, 489, 119053. [Google Scholar] [CrossRef]
- Ezeokoli, O.T.; Mashigo, S.K.; Maboeta, M.S.; Bezuidenhout, C.C.; Khasa, D.P.; Adeleke, R.A. Arbuscular mycorrhizal fungal community differentiation along a post-coal mining reclamation chronosequence in South Africa: A potential indicator of ecosystem recovery. Appl. Soil Ecol. 2020, 147, 103429. [Google Scholar] [CrossRef]
- Hart, M.M.; Aleklett, K.; Chagnon, P.L.; Egan, C.; Ghignone, S.; Helgason, T.; Lekberg, Y.; Öpik, M.; Pickles, B.J.; Waller, L. Navigating the labyrinth: A guide to sequence-based, community ecology of arbuscular mycorrhizal fungi. New Phytol. 2015, 207, 235–247. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.T.; Li, T.; Li, Y.Z.; Zhao, D.Q.; Han, J.; Liu, Y.; Liao, Y.C. Relationship between the microbial community and catabolic diversity in response to conservation tillage. Soil Tillage Res. 2020, 196, 104431. [Google Scholar] [CrossRef]
- Bulgarelli, D.; Garrido-Oter, R.; Münch, P.C.; Weiman, A.; Dröge, J.; Pan, Y.; McHardy, A.C.; Schulze-Lefert, P.S. Structure and function of the bacterial root microbiota in wild and domesticated barley. Cell Host Microbe. 2015, 17, 392–403. [Google Scholar] [CrossRef] [Green Version]
- Li, H.Y.; Qiu, Y.Z.; Yao, T.; Han, D.R.; Gao, Y.M.; Zhang, J.G.; Ma, Y.C.; Zhang, H.R.; Yang, X.L. Nutrients available in the soil regulate the changes of soil microbial community alongside degradation of alpine meadows in the northeast of the Qinghai-Tibet Plateau. Sci. Total Environ. 2021, 792, 148363. [Google Scholar] [CrossRef]
- Cao, H.Y.; Du, Y.J.; Gao, G.L.; Rao, L.Y.; Ding, G.D.; Zhang, Y. Afforestation with Pinus sylvestris var. mongolica remodelled soil bacterial community and potential metabolic function in the Horqin Desert. Glob. Ecol. Conserv. 2021, 29, e01716. [Google Scholar]
- Lozano, Y.M.; Hortal, S.; Armas, C.; Pugnaire, F.I. Interactions among soil, plants, and microorganisms drive secondary succession in a dry environment. Soil Biol. Biochem. 2014, 78, 298–306. [Google Scholar] [CrossRef]
- Maestre, F.T.; Delgado-Baquerizo, M.; Jeffries, T.C.; Eldridge, D.J.; Ochoa, V.; Gozalo, B.; Quero, J.L.; Garcia-Gomez, M.; Gallardo, A.; Ulrich, W. Increasing aridity reduces soil microbial diversity and abundance in global drylands. Proc. Natl. Acad. Sci. USA 2015, 112, 15684–15689. [Google Scholar] [CrossRef] [Green Version]
- Dang, P.; Yu, X.; Le, H.; Liu, J.L.; Shen, Z.; Zhao, Z. Effects of stand age and soil properties on soil bacterial and fungal community composition in Chinese pine plantations on the Loess Plateau. PLoS ONE 2017, 12, e0186501. [Google Scholar] [CrossRef] [Green Version]
- Jangid, K.; Whitman, W.B.; Condron, L.M.; Turner, B.L.; Williams, M.A. Soil bacterial community succession during long-term ecosystem development. Mol. Ecol. 2013, 22, 3415–3424. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.F.; Ma, M.C.; Jiang, X.; Guan, D.W.; Wei, D.; Cao, F.M.; Kang, Y.W.; Chu, C.B.; Wu, S.H.; Li, J. Influence of 37 Years of Nitrogen and Phosphorus Fertilization on Composition of Rhizosphere Arbuscular Mycorrhizal Fungi Communities in Black Soil of Northeast China. Front. Microbiol. 2020, 11, 2206. [Google Scholar] [CrossRef]
- Vasar, M.; Davison, J.; Sepp, S.-K.; Öpik, M.; Moora, M.; Koorem, K.; Meng, Y.; Oja, J.; Akhmetzhanova, A.A.; Al-Quraishy, S. Arbuscular Mycorrhizal Fungal Communities in the Soils of Desert Habitats. Microorganisms 2021, 9, 229. [Google Scholar] [CrossRef]
- Jiang, Y.L.; Lei, Y.B.; Yang, Y.; Korpelainen, H.; Niinemets, Ü.; Li, C.Y. Divergent assemblage patterns and driving forces for bacterial and fungal communities along a glacier forefield chronosequence. Soil Biol. Biochem. 2018, 118, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Neina, D. The role of soil pH in plant nutrition and soil remediation. Appl. Environ. Soil Sci. 2019, 2019, 5794869. [Google Scholar] [CrossRef]
- Xu, H.F.; Du, H.; Zeng, F.P.; Song, T.Q.; Peng, W.X. Diminished rhizosphere and bulk soil microbial abundance and diversity across succession stages in Karst area, southwest China. Appl. Soil Ecol. 2021, 158, 103799. [Google Scholar] [CrossRef]
- Zörb, C.; Senbayram, M.; Peiter, E. Potassium in agriculture–status and perspectives. J. Plant Physiol. 2014, 171, 656–669. [Google Scholar] [CrossRef]
- Sheng, M.; Chen, X.D.; Zhang, X.L.; Hamel, C.; Cui, X.W.; Chen, J.; Chen, H.; Tang, M. Changes in arbuscular mycorrhizal fungal attributes along a chronosequence of black locust (Robinia pseudoacacia) plantations can be attributed to the plantation-induced variation in soil properties. Sci. Total Environ. 2017, 599, 273–283. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, G.B.; Xue, S.; Wang, G.L. Changes in rhizospheric microbial community structure and function during the natural recovery of abandoned cropland on the Loess Plateau, China. Ecol. Eng. 2015, 96, 3374–3385. [Google Scholar] [CrossRef]
- Liu, J.; Jia, X.Y.; Yan, W.M.; Zhong, Y.Q.W.; Shangguan, Z.P. Changes in soil microbial community structure during long-term secondary succession. Land Degrad. Dev. 2020, 31, 1151–1166. [Google Scholar] [CrossRef]
- Eiler, A.; Heinrich, F.; Bertilsson, S. Coherent dynamics and association networks among lake bacterioplankton taxa. ISME J. 2012, 6, 330–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, M.; Eguíluz, V.M.; Duarte, C.M.; Voolstra, C.R. Rare symbionts may contribute to the resilience of coral–algal assemblages. ISME J. 2018, 12, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.G.; Zhang, J.; Liu, Y.; Shi, P.; Wei, G.H. Co-occurrence patterns of soybean rhizosphere microbiome at a continental scale. Soil Biol. Biochem. 2018, 118, 178–186. [Google Scholar] [CrossRef]
- Krishna, M.; Gupta, S.; Delgado–Baquerizo, M.; Morriën, E.; Garkoti, S.; Chaturvedi, R.; Ahmad, S. Successional trajectory in soil bacterial communities are shaped by plant-driven changes in soilduring secondary succession. Sci. Rep. 2020, 10, 9864. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zheng, M.M.; Song, W.F.; Chen, R.F.; Zhao, X.Q.; Wen, S.L.; Zheng, Z.S.; Shen, R.F. Biogeographic patterns and co-occurrence networks of diazotrophic and arbuscular mycorrhizal fungal communities in the acidic soil ecosystem of southern China. Appl. Soil Ecol. 2021, 158, 103798. [Google Scholar] [CrossRef]
- Fan, K.K.; Weisenhorn, P.; Gilbert, J.A.; Chu, H.Y. Wheat rhizosphere harbors a less complex and more stable microbial co-occurrence pattern than bulk soil. Soil Biol. Biochem. 2018, 125, 251–260. [Google Scholar] [CrossRef]
- Wagg, C.; Schlaeppi, K.; Banerjee, S.; Kuramae, E.E.; van der Heijden, M.G.A. Fungal-bacterial diversity and microbiome complexity predict ecosystem functioning. Nat. Commun. 2019, 10, 4841. [Google Scholar] [CrossRef]
- Wu, L.W.; Yang, Y.F.; Chen, S.; Zhao, M.X.; Zhu, Z.W.; Yang, S.H.; Qu, Y.Y.; Ma, Q.; He, Z.L.; Zhou, J.Z. Long-term successional dynamics of microbial association networks in anaerobic digestion processes. Water Res. 2016, 104, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Wang, Y.; Zheng, W.; Hou, F.B.; Hu, Y.X.; Guo, S.L. Converting croplands to orchards changes soil microbial community composition and co-occurrence patterns. Land Degrad. Dev. 2021, 32, 2509–2519. [Google Scholar] [CrossRef]
- Zheng, W.; Zhao, Z.Y.; Gong, Q.L.; Zhai, B.N.; Li, Z.Y. Responses of fungal–bacterial community and network to organic inputs vary among different spatial habitats in soil. Soil Biol. Biochem. 2018, 125, 54–63. [Google Scholar] [CrossRef]
- Chen, J.; Li, J.J.; Shen, W.J.; Xu, H.; Li, Y.D.; Luo, T.S. The Structure and Species Co-Occurrence Networks of Soil Denitrifying Bacterial Communities Differ Between A Coniferous and A Broadleaved Forests. Microorganisms 2019, 7, 361. [Google Scholar] [CrossRef] [Green Version]
- Ren, G.L.; Wang, J.L.; Qu, L.; Li, W.; Hu, M.; Bian, L.H.; Zhang, Y.T.; Le, J.J.; Dou, X.M.; Chen, X.H. Compositions and Co-occurrence Patterns of Bacterial Communities Associated with Polymer-and ASP-Flooded Petroleum Reservoir Blocks. Front. Microbiol. 2020, 11, 3011. [Google Scholar] [CrossRef]
- Laurent, P.; Raaijmakers, J.; Philippe, L.; Van Der Putten, W.H. Going back to the roots: The microbial ecology of the rhizosphere. Nat. Rev. Microbiol. 2013, 11, 789–799. [Google Scholar]
- Piao, Z.; Yang, L.Z.; Zhao, L.P.; Yin, S.X. Actinobacterial community structure in soils receiving long-term organic and inorganic amendments. Appl. Environ. Microbiol. 2008, 74, 526–530. [Google Scholar] [CrossRef] [Green Version]
- Blothe, M.; Akob, D.M.; Kostka, J.E.; Goschel, K.; Drake, H.L.; Kusel, K. pH gradient-induced heterogeneity of Fe (III)-reducing microorganisms in coal mining-associated lake sediments. Appl. Environ. Microbiol. 2008, 74, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Pankratov, T.A.; Ivanova, A.O.; Dedysh, S.N.; Liesack, W. Bacterial populations and environmental factors controlling cellulose degradation in an acidic Sphagnum peat. Environ. Microbiol. 2011, 13, 1800–1814. [Google Scholar] [CrossRef]
- Yao, M.J.; Rui, J.P.; Li, J.B.; Wang, J.M.; Cao, W.D.; Li, X.Z. Soil bacterial community shifts driven by restoration time and steppe types in the degraded steppe of Inner Mongolia. Catena 2018, 165, 228–236. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stand Age | Longitude (N) | Latitude (E) | Altitude (m) | Mean DBH (m) | Mean Height (m) | Canopy Closure (%) |
|---|---|---|---|---|---|---|
| CK | 18°42′16.14″ | 108°47′46.78″ | 118 | - | - | - |
| 22-y | 18°42′20.93″ | 108°48′57.56″ | 161 | 0.20 | 15.44 | 0.65 |
| 35-y | 18°41′52.93″ | 108°47′2.09″ | 106 | 0.33 | 23.66 | 0.70 |
| 45-y | 18°41′58.85″ | 108°47′5.47″ | 83 | 0.31 | 20.58 | 0.67 |
| 55-y | 18°42′11.48″ | 108°49′28.27″ | 154 | 0.38 | 24.00 | 0.75 |
| Alpha Diversity | Soil | CK | 22-y | 35-y | 45-y | 55-y | Age |
|---|---|---|---|---|---|---|---|
| Sobs | R | 2057.00 ± 60.48 | 2037.00 ± 32.47 | 2087.00 ± 56.36 | 2063.00 ± 34.70 | 2241.33 ± 68.13 | ns |
| B | - | 2378.67 ± 19.03 | 2369.67 ± 95.57 | 2293.33 ± 50.52 | 2185.67 ± 168.03 | ns | |
| Shannon | R | 5.85 ± 0.05 | 6.02 ± 0.01 | 5.99 ± 0.06 | 5.96 ± 0.04 | 6.00 ± 0.09 | ns |
| B | - | 6.18 ± 0.04 | 5.93 ± 0.10 | 5.94 ± 0.10 | 5.87 ± 0.16 | ns | |
| Simpson | R | 0.02 ± 0.00 | 0.01 ± 0.00 | 0.01 ± 0.00 | 0.02 ± 0.00 | 0.02 ± 0.00 | ns |
| B | - | 0.01 ± 0.00 | 0.02 ± 0.00 | 0.02 ± 0.00 | 0.02 ± 0.00 | ns | |
| Chao 1 | R | 2562.04 ± 84.45 | 2561.24 ± 54.80 | 2582.40 ± 53.85 | 2595.71 ± 67.34 | 2791.59 ± 38.01 | ns |
| B | - | 3070.74 ± 49.04 | 3081.95 ± 94.98 | 3073.89 ± 25.80 | 2967.665 ± 172.92 | ns | |
| Coverage (%) | R | 98.25 | 97.98 | 98.36 | 98.13 | 98.06 | |
| B | 97.84 | 97.94 | 97.68 | 97.70 |
| Treatments | Nodes | Edges | Average Path Length (APL) | Modularity (MD) | Average Clustering Coefficient (ACC) | Average Degree |
|---|---|---|---|---|---|---|
| 22-y | 79 | 144 | 3.981 | 1.683 | 0.554 | 3.646 |
| 35-y | 91 | 237 | 4.134 | 0.965 | 0.591 | 5.852 |
| 45-y | 86 | 221 | 3.511 | 1.828 | 0.581 | 5.140 |
| 55-y | 81 | 172 | 5.960 | 0.674 | 0.574 | 4.247 |
| Variables | Bulk | Rhizosphere | ||
|---|---|---|---|---|
| R2 | p | R2 | p | |
| pH | 0.327 | 0.019 * | 0.264 | 0.007 ** |
| SOC | 0.287 | 0.032 * | −0.040 | 0.611 |
| TN | 0.058 | 0.292 | 0.029 | 0.347 |
| TP | −0.116 | 0.756 | 0.213 | 0.024 * |
| TK | 0.351 | 0.009 ** | 0.335 | 0.001 ** |
| AP | 0.343 | 0.012 * | 0.174 | 0.038 * |
| AK | 0.472 | 0.001 ** | 0.195 | 0.035 * |
| -H | 0.116 | 0.229 | 0.180 | 0.045 * |
| -H | 0.090 | 0.260 | −0.029 | 0.570 |
| Catalase | 0.344 | 0.011 * | 0.015 | 0.361 |
| Acid phosphatase | 0.225 | 0.079 | 0.023 | 0.354 |
| Urease | 0.011 | 0.434 | 0.585 | 0.001 ** |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, Z.; Liang, K.; Huang, G.; Wang, X.; Lin, M.; Chen, Y.; Zhou, Z. Soil Bacterial Community Shifts Are Driven by Soil Nutrient Availability along a Teak Plantation Chronosequence in Tropical Forests in China. Biology 2021, 10, 1329. https://doi.org/10.3390/biology10121329
Yu Z, Liang K, Huang G, Wang X, Lin M, Chen Y, Zhou Z. Soil Bacterial Community Shifts Are Driven by Soil Nutrient Availability along a Teak Plantation Chronosequence in Tropical Forests in China. Biology. 2021; 10(12):1329. https://doi.org/10.3390/biology10121329
Chicago/Turabian StyleYu, Zhi, Kunnan Liang, Guihua Huang, Xianbang Wang, Mingping Lin, Yinglong Chen, and Zaizhi Zhou. 2021. "Soil Bacterial Community Shifts Are Driven by Soil Nutrient Availability along a Teak Plantation Chronosequence in Tropical Forests in China" Biology 10, no. 12: 1329. https://doi.org/10.3390/biology10121329