Scale Formation and Degradation of Diffusion Coatings Deposited on 9% Cr Steel in Molten Solar Salt

Abstract
:1. Introduction
2. Materials and Methods
2.1. Coating Manufacturing
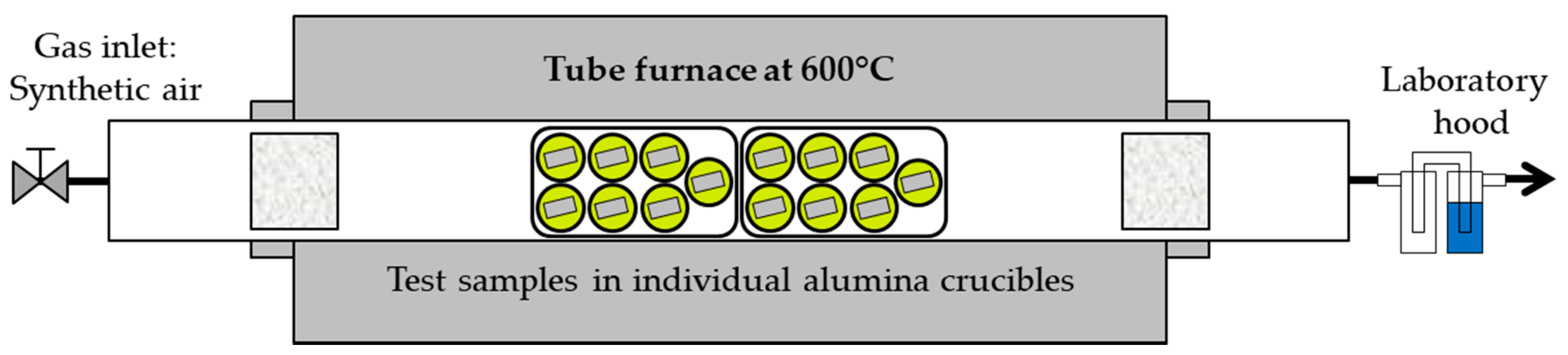
2.2. Exposure in Molten Solar Salt
2.3. Post-Exposure Characterization
3. Results and Discussion
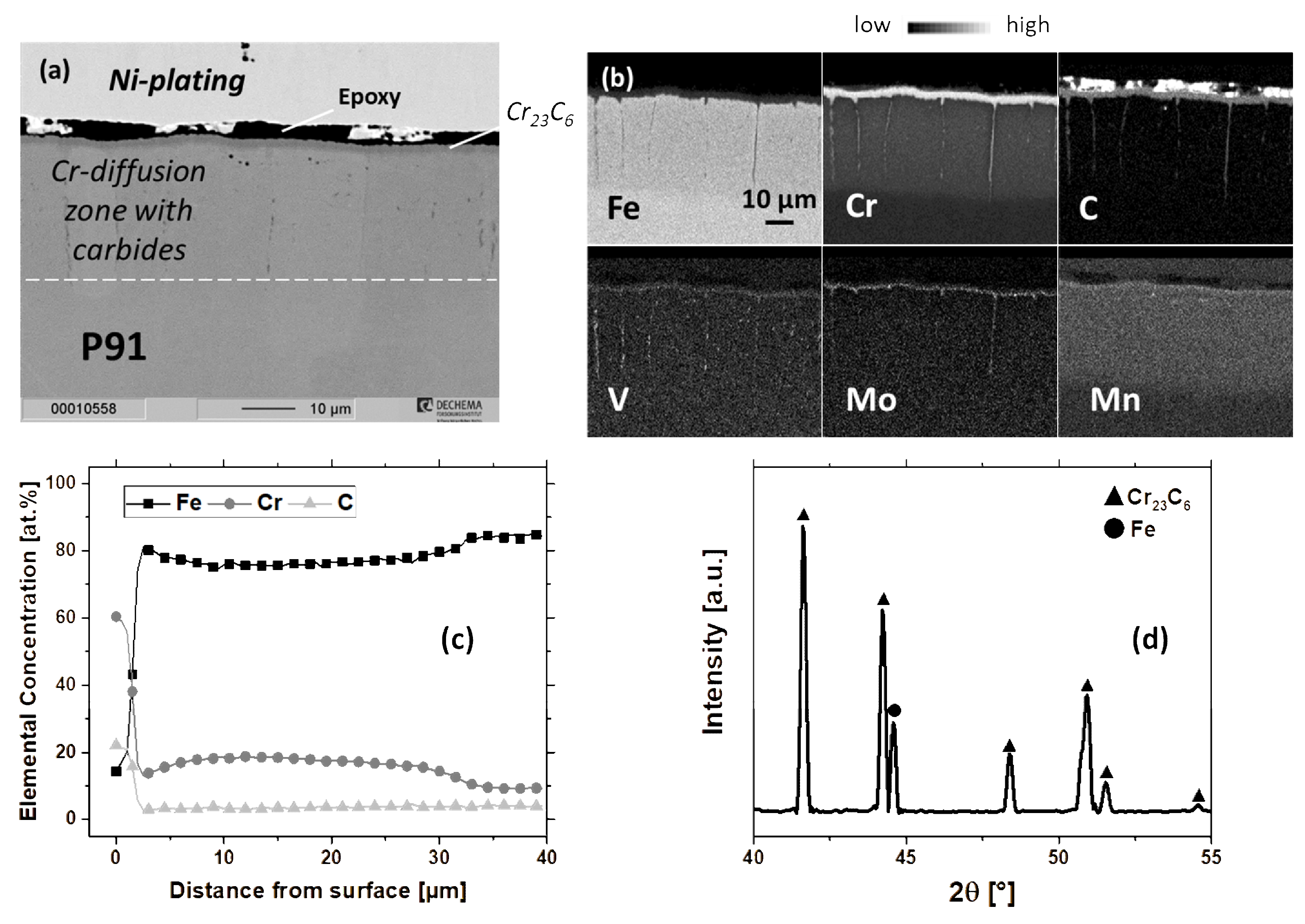
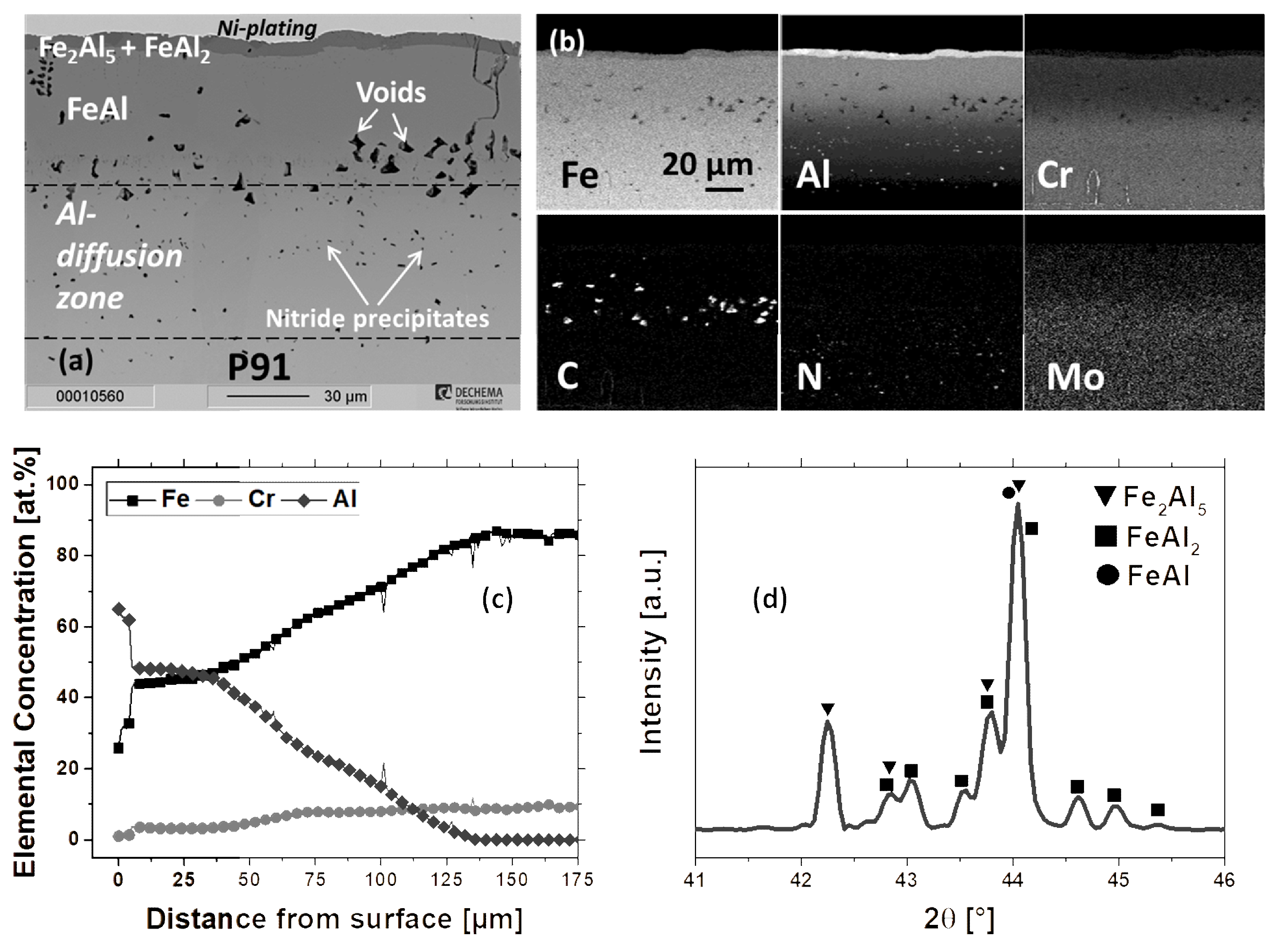
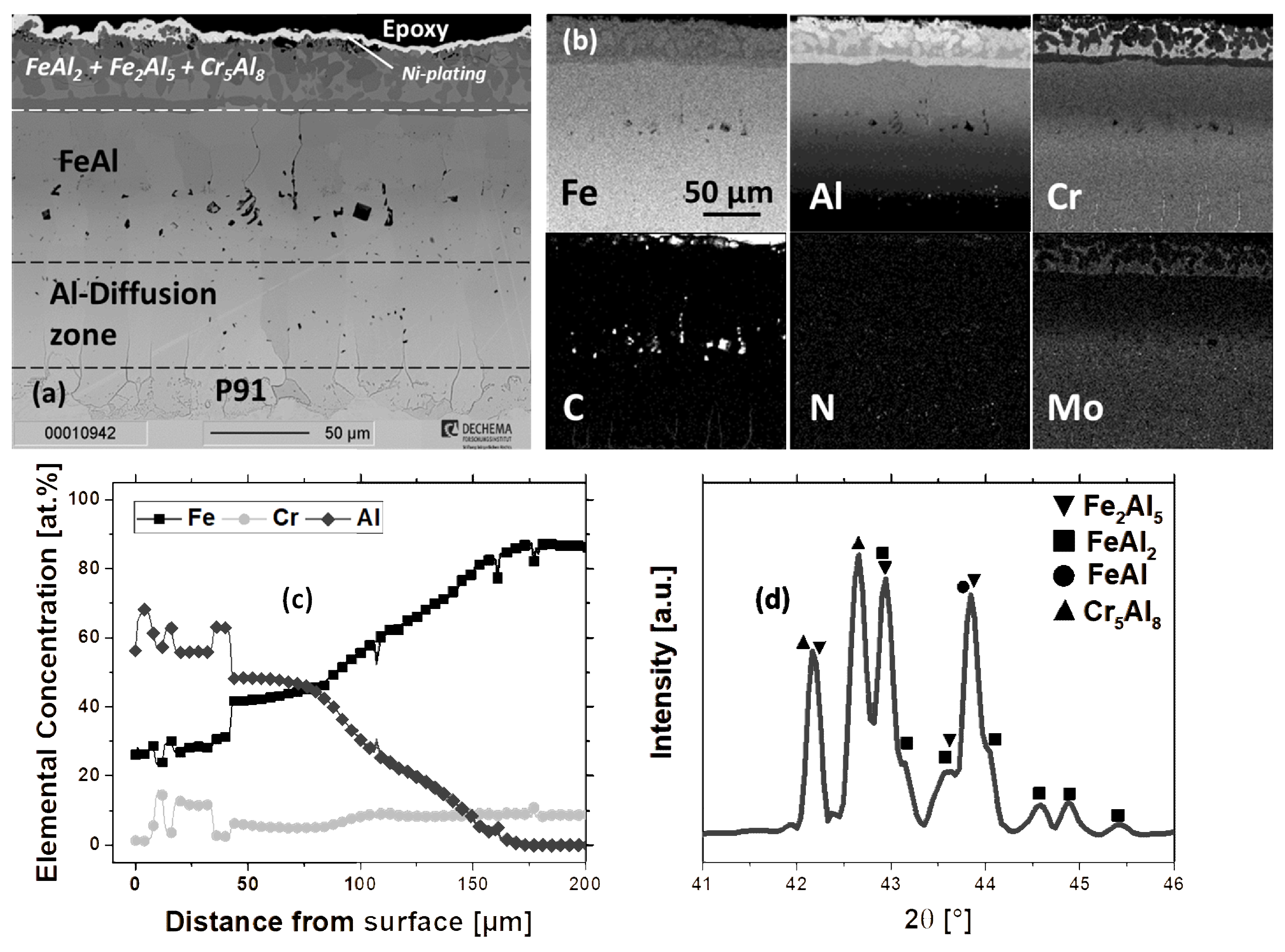
3.1. Microstructure of as-deposited Coatings
3.1.1. Cr-Diffusion Coating
3.1.2. Al-Diffusion Coating
3.1.3. Cr/Al-Diffusion Coating
3.2. Exposure in Molten Solar Salt
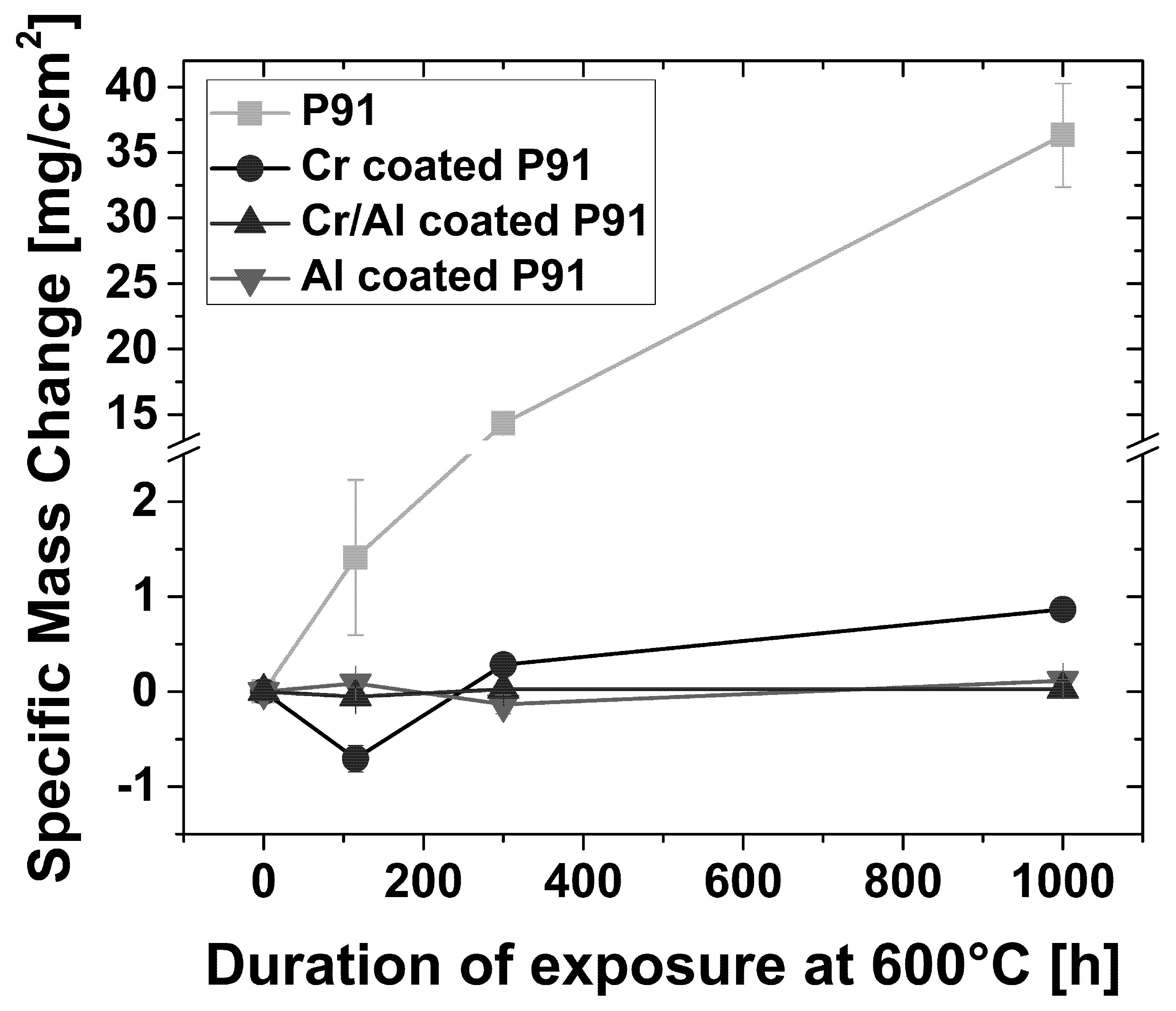
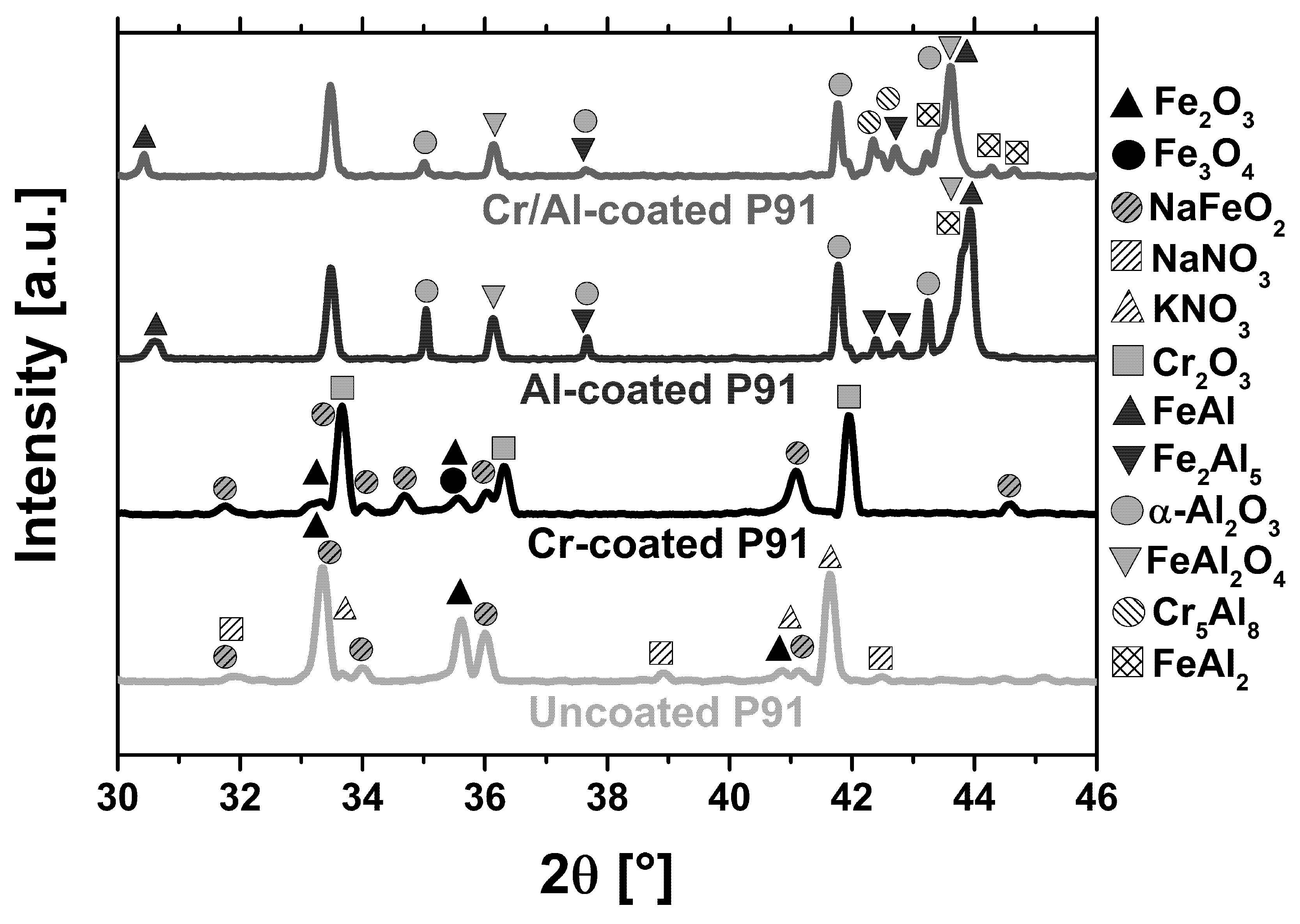
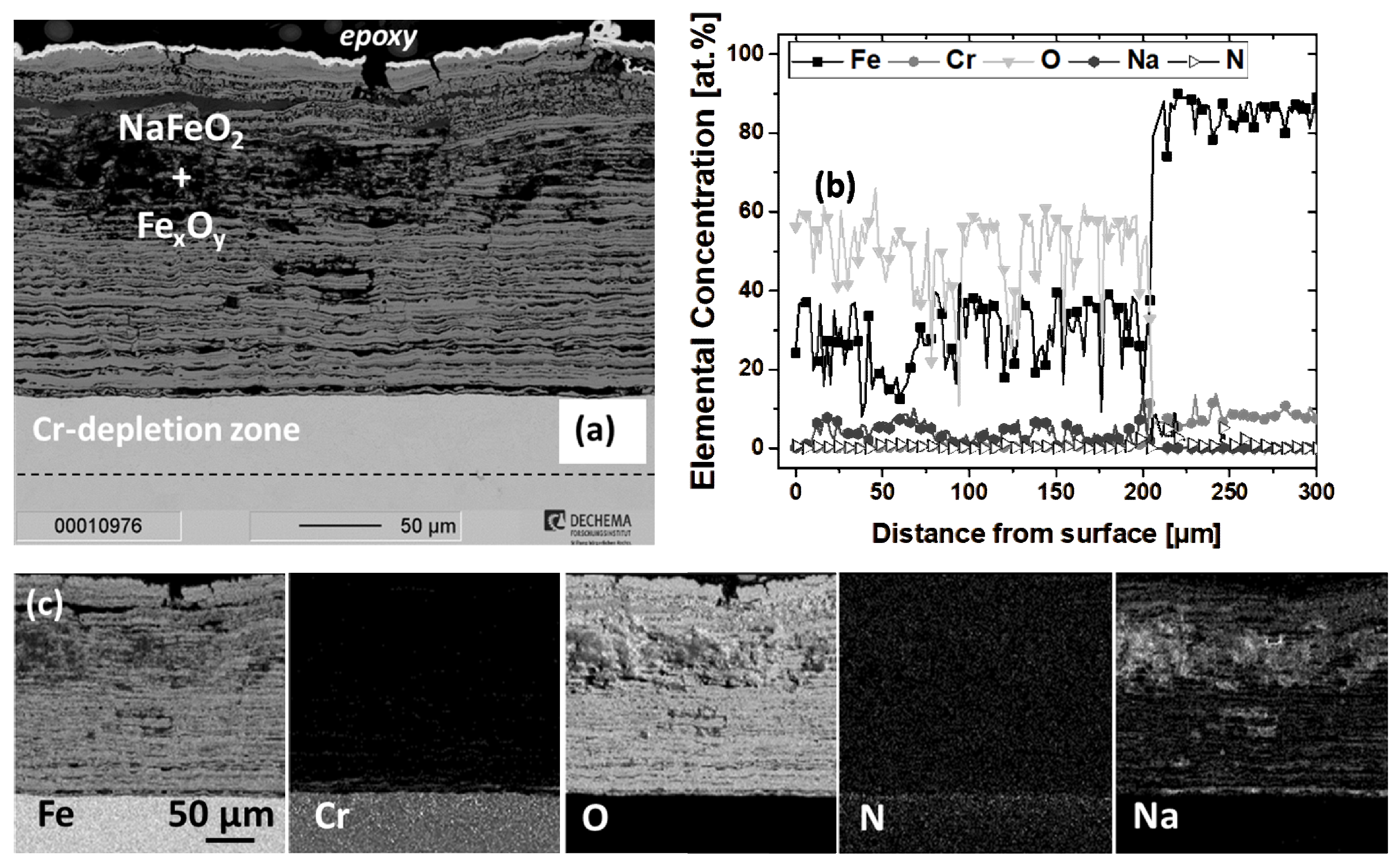
3.2.1. Scaling Behavior of Uncoated P91
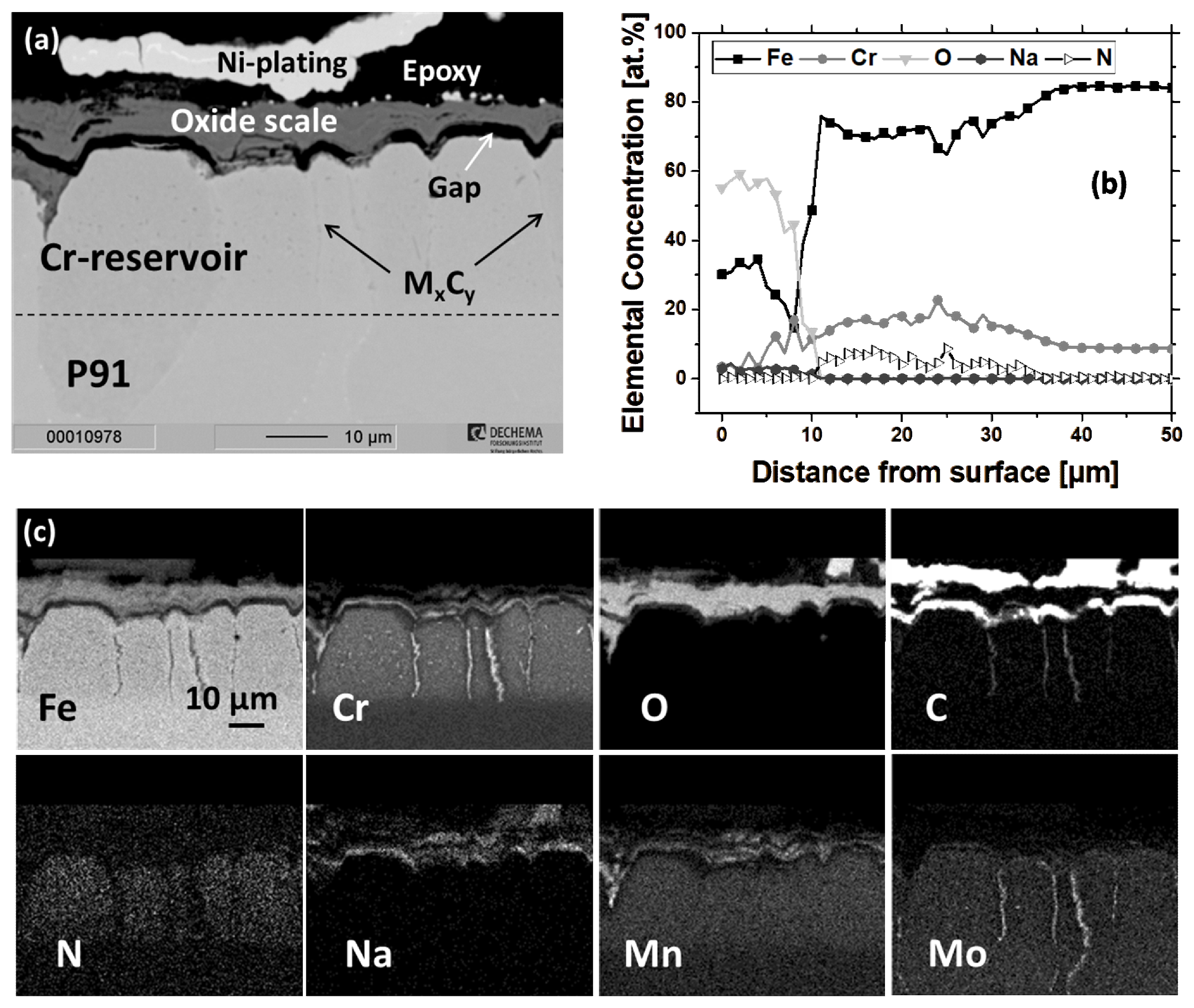
3.2.2. Scaling Behavior of Chromized P91
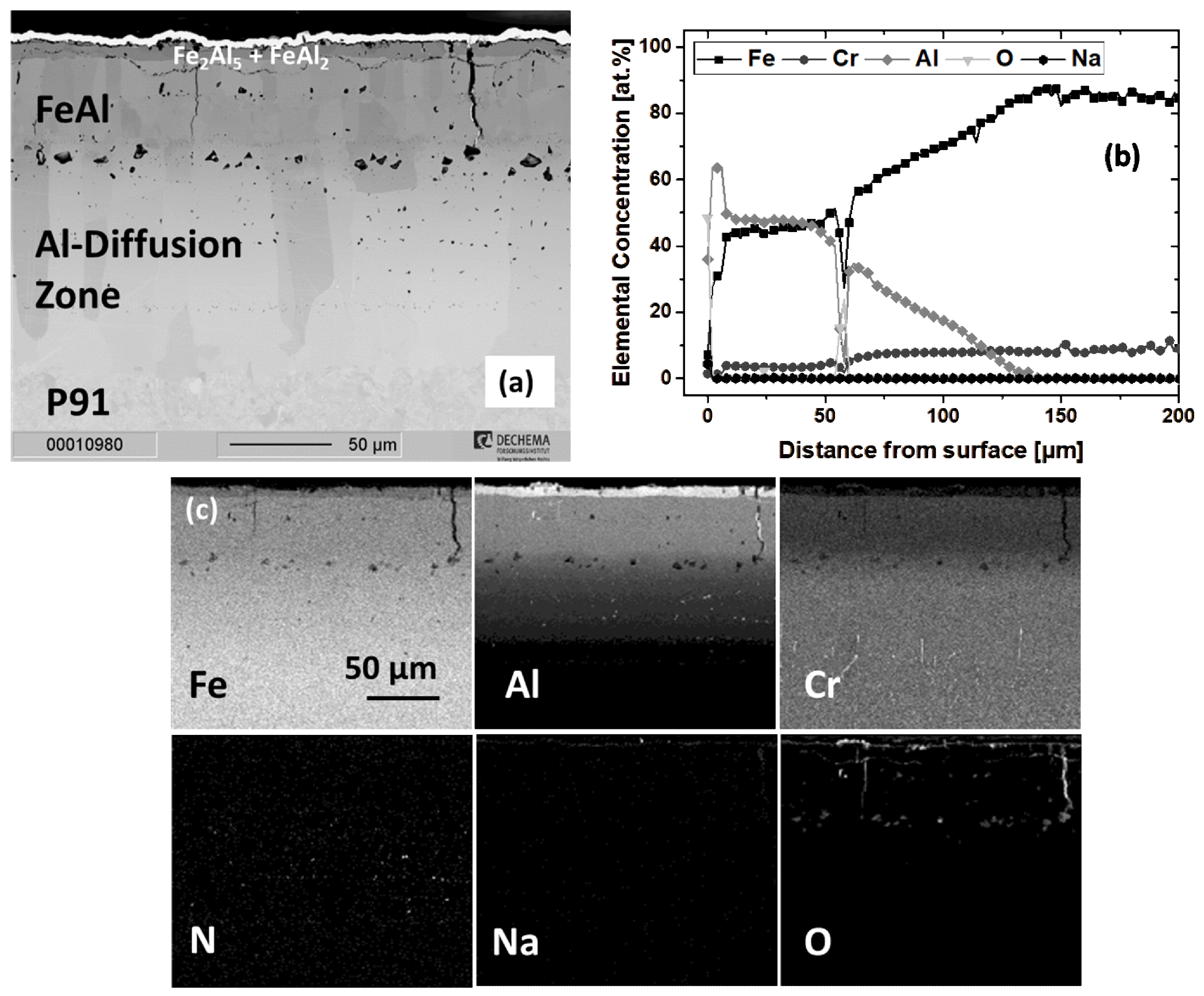
3.2.3. Scaling Behavior of Al-Coated P91
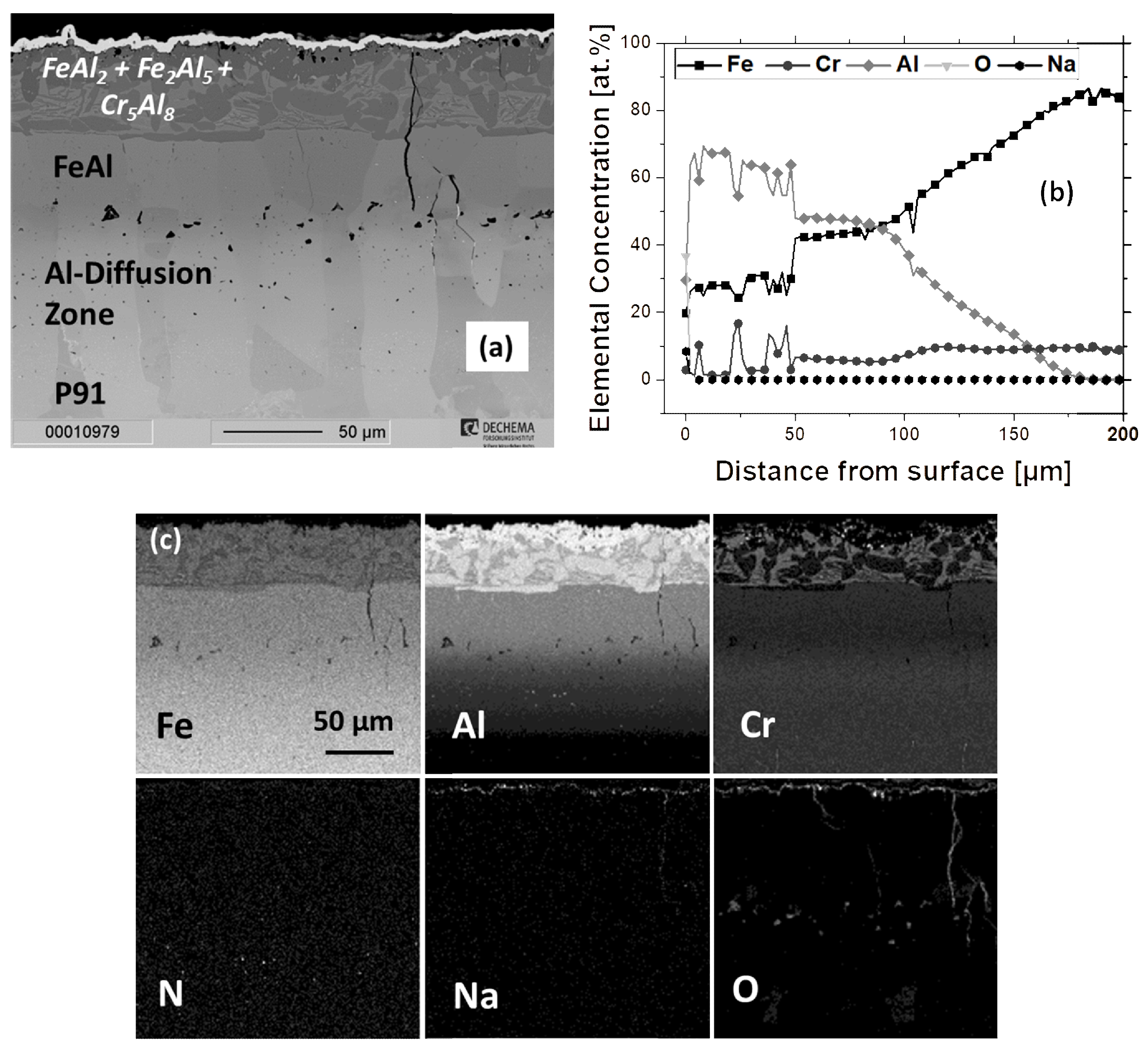
3.2.4. Scaling Behavior of Cr/Al-Coated P91
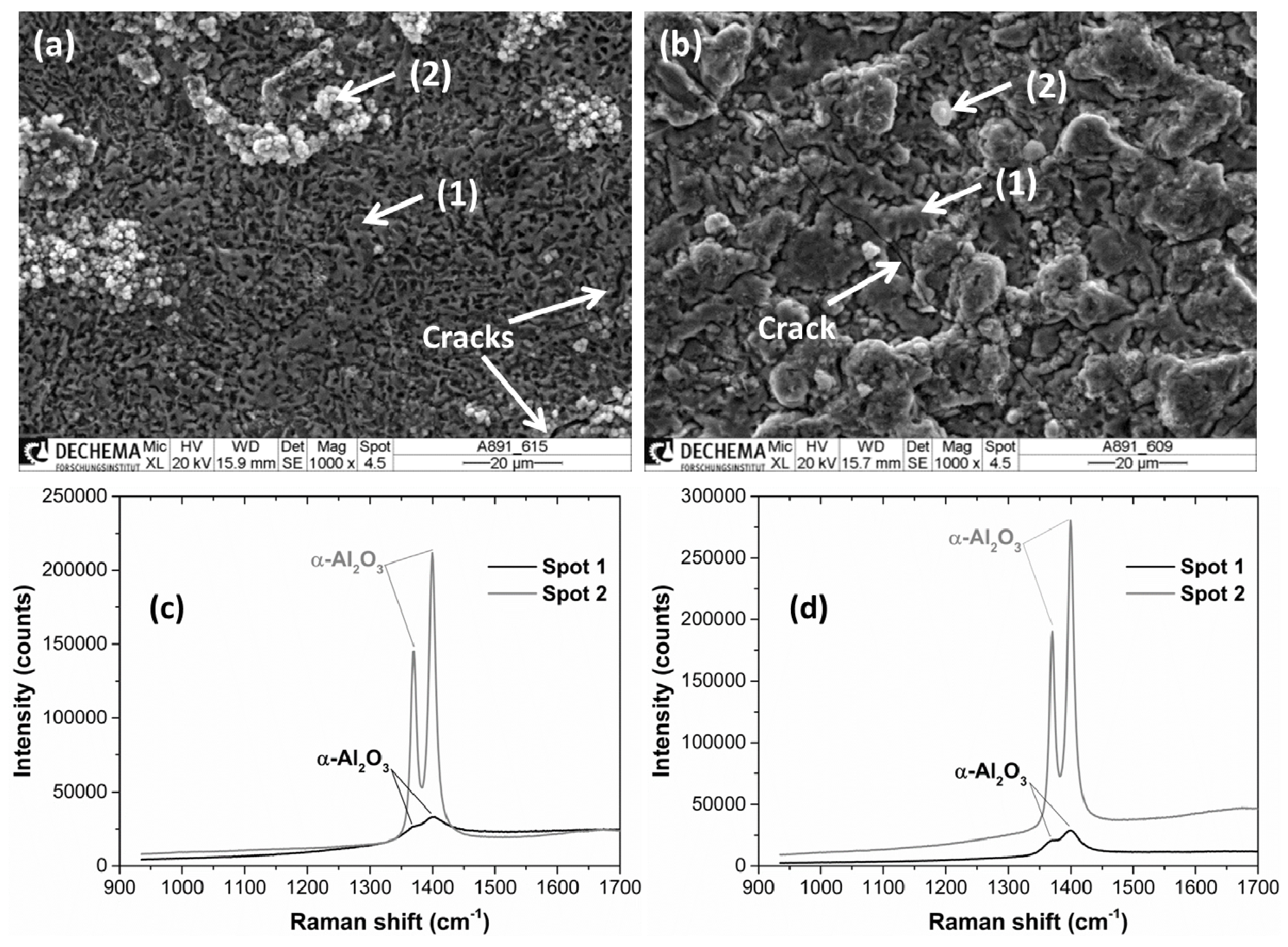
3.2.5. Formation of Corundum on Al- and Cr/Al-Coated P91
4. Conclusions
5. Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bonk, A.; Sau, S.; Uranga, N.; Hernaiz, M.; Bauer, T. Advanced heat transfer fluids for direct molten salt line-focusing CSP plants. Prog. Energy Combust. Sci. 2018, 67, 69–87. [Google Scholar] [CrossRef]
- Soleimani-Dorcheh, A.; Durham, R.N.; Galetz, M.C. Corrosion behavior of stainless and low-chromium steels and IN625 in molten nitrate salts at 600 C. Sol. Energy Mater. Sol. Cells 2016, 144, 109–116. [Google Scholar] [CrossRef]
- Barlev, D.; Vidu, R.; Stroeve, P. Innovation in concentrated solar power. Sol. Energy Mater. Sol. Cells 2011, 95, 2703–2725. [Google Scholar] [CrossRef]
- Vignarooban, K.; Xu, X.; Arvay, A.; Hsu, K.; Kannan, A.M. Heat transfer fluids for concentrating solar power systems—A review. Appl. Energy 2015, 146, 383–396. [Google Scholar] [CrossRef]
- Boretti, A.; Casteletto, S.; Al-Zubaidy, S. Concentrating solar power tower technology: Present status and outlook. Nonlinear Eng. 2019, 8, 10–31. [Google Scholar] [CrossRef]
- Agüero, A.; Audigié, P.; Rodríguez, S.; Encinas-Sánchez, V.; de Miguel, M.T.; Pérez, F.J. Protective coatings for high temperature molten salt heat storage systems in solar concentration power plants. In AIP Conference Proceedings; AIP Publishing: Melville, NY, USA, 2018; Volume 2033. [Google Scholar]
- Bonk, A.; Braun, M.; Hanke, A.; Forstner, J.; Rückle, D.; Kaesche, S.; Sötz, V.A.; Bauer, T. Influence of different atmospheres on molten salt chemistry and its effect on steel corrosion. In AIP Conference Proceedings; AIP Publishing: Melville, NY, USA, 2018; Volume 2033. [Google Scholar]
- Bradshaw, R.W.; Carling, R.W. A review of the chemical and physical properties of molten alkali nitrate salts and their effect on materials used for solar central receivers. Proc. Electrochem. Soc. 1987, 1987, 959–969. [Google Scholar] [CrossRef]
- Kruizenga, A.; Gill, D. Corrosion of iron stainless steels in molten nitrate salt. Energy Procedia 2014, 49, 878–887. [Google Scholar] [CrossRef]
- Spiegel, M.; Mentz, J. High temperature corrosion beneath nitrate melts. Mater. Corros. 2014, 65, 276–281. [Google Scholar] [CrossRef]
- Summers, K.L.; Chidambaram, D. Corrosion behavior of structural materials for potential use in nitrate salts based solar thermal power plants. J. Electrochem. Soc. 2017, 164, 5357–5363. [Google Scholar] [CrossRef]
- Soleimani-Dorcheh, A.; Galetz, M.C. Slurry aluminizing: A solution for molten nitrate salt corrosion in concentrated solar power plants. Sol. Energy Mater. Sol. Cells 2016, 146, 8–15. [Google Scholar] [CrossRef]
- Fernandez, A.G.; Lasanta, M.I.; Perez, F.J. Molten salt corrosion of stainless steels and low-Cr steel in CSP plants. Oxid. Met. 2012, 78, 329–348. [Google Scholar] [CrossRef]
- McConohy, G.; Kruizenga, A. Molten nitrate salts at 600 and 680 C: Thermophysical property changes and corrosion of high-temperature nickel alloys. Sol. Energy 2014, 103, 242–252. [Google Scholar] [CrossRef]
- Rapp, R.A.; Goto, K.S. The hot corrosion of metals by molten salts. In Proceedings of the Second International Symposium on Molten Salts; Selman, R., Braunstein, J., Eds.; Electrochemical Society: Pennington, NJ, USA, 1981; pp. 159–177. [Google Scholar]
- Tortorelli, P.F.; Bishop, P.S.; DiStefano, J.R. Selection of Corrosion-Resistant Materials for Use in Molten Nitrate Salts ORNL/TM-11162; Oak Ridge National Lab.: Oak Ridge, TN, USA, 1989.
- Bradshaw, R.W.; Clift, W.M. Effect of Chloride Content of Molten Nitrate Salt on Corrosion of A516 Carbon Steel; Sandia Report, SAND2010-7594; Sandia National Laboratories: Albuquerque, NM, USA; Livermore, CA, USA, 2010.
- Dorcheh, A.S.; Durham, R.N.; Galetz, M.C. High temperature corrosion in molten solar salt: The role of chloride impurities. Mater. Corros. 2017, 68, 943–951. [Google Scholar] [CrossRef]
- Encinas-Sánchez, V.; de Miguel, M.T.; Lasanta, M.I.; García-Martín, G.; Pérez, F.J. Electrochemical impedance spectroscopy (EIS): An efficient technique for monitoring corrosion processes in molten salt environments in CSP applications. Sol. Energy Mater. Sol. Cells 2019, 191, 157–163. [Google Scholar] [CrossRef]
- Bradshaw, R.W.; Goods, S.H. Corrosion Resistance of Stainless Steels During Thermal Cycling in Alkali Nitrate Molten Salts; Sandia Report, SAND2001-8518; Sandia National Laboratories: Albuquerque, NM, USA; Livermore, CA, USA, 2001.
- Goods, S.H.; Bradshaw, R.W. Corrosion of stainless steels and carbon steel by molten mixtures of commercial nitrate salts. J. Mater. Eng. Perform. 2004, 13, 78–87. [Google Scholar] [CrossRef]
- Slusser, J.W.; Titcomb, J.B.; Heffelfinger, M.T.; Dunbobbin, B.R. Corrosion in molten nitrate-nitrite salts. JOM 1985, 37, 24–27. [Google Scholar] [CrossRef]
- Fähsing, D. Ferritisch Martensitische Stähle: Verbesserung der Oxidationsbeständigkeit in Wasserdampfhaltigen Hochtemperaturatmosphären. Ph.D. Thesis, RWTH Aachen, Shaker Verlag, Herzogenrath, Germany, 2015. [Google Scholar]
- Audigié, P.; Bizien, N.; Baráibar, I.; Rodriguez, S.; Pastor, A.; Hernández, M.; Agüero, A. Aluminide slurry coatings for protection of ferritic steel in molten nitrate corrosion for concentrated solar power technology. In AIP Conference Proceedings; AIP Publishing: Melville, NY, USA, 2017; Volume 1850, p. 070002. [Google Scholar]
- Audigié, P.; Encinas-Sánchez, V.; Juez-Lorenzo, M.; Rodríguez, S.; Gutiérrez, M.; Pérez, F.J.; Agüero, A. High temperature molten salt corrosion behavior of aluminide and nickel-aluminide coatings for heat storage in concentrated solar power plants. Surf. Coat. Technol. 2018, 349, 1148–1157. [Google Scholar] [CrossRef]
- Fähsing, D.; Oskay, C.; Meißner, T.M.; Galetz, M.C. Corrosion testing of diffusion-coated steel in molten salt for concentrated solar power tower systems. Surf. Coat. Technol. 2018, 354, 46–55. [Google Scholar] [CrossRef]
- Material Data Sheet P91/T91; Thyssen Krupp Materials International: Essen, Germany, 2011.
- Goward, G.W. Protective Coatings for High Temperature Alloys: State of Technology. In Proceedings of the Symposium on Properties of High-Temperature Alloys with Emphasis on Environmental Effects, New York, NY, USA, 17 October 1976. [Google Scholar]
- Duret, C.; Pichoir, R. Protective Coatings for High Temperature Materials: Chemical Vapour Deposition and Pack Cementation Processes. In Coatings for High Temperature Applications; Lang, E., Ed.; Applied Science Publishers: London, UK, 1983; pp. 33–78. [Google Scholar]
- Mevrel, R.; Duret, C.; Pichoir, R. Pack cementation processes. Mater. Sci. Technol. 1986, 2, 201–206. [Google Scholar] [CrossRef]
- Rohr, V.; Donchev, A.; Schütze, M.; Milewska, A.; Pérez, F.J. Diffusion coatings for high temperature corrosion protection of 9–12% Cr steels. Corros. Eng. Sci. Technol. 2005, 40, 226–232. [Google Scholar] [CrossRef]
- ISO 17245:2015 Corrosion of Metals and Alloys—Test Method for High Temperature Corrosion Testing of Metallic Materials by Immersing in Molten Salt or Other Liquids under Static Conditions; ISO: Geneva, Switzerland, 2015.
- Schmidt, D.; Galetz, M.C.; Schütze, M. Ferritic–martensitic steels: Improvement of the oxidation behavior in steam environments via diffusion coatings. Surf. Coat. Technol. 2013, 237, 23–29. [Google Scholar] [CrossRef]
- Bianco, R.; Rapp, R.A. Pack cementation diffusion coatings. In Metallurgical and Ceramic Protective Coatings; Stern, K.H., Ed.; Chapman & Hall: London, UK, 1996; pp. 236–260. [Google Scholar]
- Meier, G.H.; Cheng, C.; Perkins, R.A.; Bakker, W. Diffusion chromizing of ferrous alloys. Surf. Coat. Technol. 1989, 39, 53–64. [Google Scholar] [CrossRef]
- Agüero, A.; Muelas, R.; Pastor, A.; Osgerby, S. Long exposure steam oxidation testing and mechanical properties of slurry aluminide coatings for steam turbine components. Surf. Coat. Technol. 2005, 200, 1219–1224. [Google Scholar] [CrossRef]
- Bates, B.L.; Zhang, Y.; Dryepondt, S.; Pint, B.A. Creep behavior of pack cementation aluminide coatings on grade 91 ferritic–martensitic alloy. Surf. Coat. Technol. 2014, 240, 32–39. [Google Scholar] [CrossRef]
- Rohr, V.; Schütze, M.; Fortuna, E.; Tsipas, D.N.; Milewska, A.; Pérez, F.J. Development of novel diffusion coatings for 9–12% Cr ferritic-martensitic steels. Mater. Corros. 2005, 56, 874–881. [Google Scholar] [CrossRef]
- Zhang, Y.; Pint, B.A.; Cooley, K.M.; Haynes, J.A. Formation of aluminide coatings on Fe-based alloys by chemical vapor deposition. Surf. Coat. Technol. 2008, 202, 3839–3849. [Google Scholar] [CrossRef]
- Naji, A.; Galetz, M.C.; Schütze, M. Improvements in the thermodynamic and kinetic considerations on the coating design for diffusion coatings formed via pack cementation. Mater. Corros. 2015, 66, 863–868. [Google Scholar] [CrossRef]
- Touloukian, Y.S.; Kirby, R.K.; Taylor, R.E.; Desai, P.D. Thermal Expansion of Metallic Elements and Alloy; Plenum Publishing Corporation: New York, NY, USA, 1970. [Google Scholar]
- Fähsing, D.; Rudolphi, M.; Konrad, L.; Galetz, M.C. Fireside Corrosion of Chromium-and Aluminum-Coated Ferritic–Martensitic Steels. Oxid. Met. 2017, 88, 155–164. [Google Scholar] [CrossRef]
- Zhang, Y.; Pint, B.A.; Cooley, K.M.; Haynes, J.A. Effect of nitrogen on the formation and oxidation behavior of iron aluminide coatings. Surf. Coat. Technol. 2005, 200, 1231–1235. [Google Scholar] [CrossRef]
- Palm, M. The Al–Cr–Fe system–Phases and phase equilibria in the Al-rich corner. J. Alloy Compd. 1997, 252, 192–200. [Google Scholar] [CrossRef]
- Pavlyuchkov, D.; Przepiórzynski, B.; Kowalski, W.; Velikanova, T.Y.; Grushko, B. Al–Cr–Fe phase diagram. Isothermal Sections in the region above 50 at% Al. Calphad 2014, 45, 194–203. [Google Scholar] [CrossRef]
- Das, D.K.; Singh, V.; Joshi, S.V. Evolution of aluminide coating microstructure on nickel-base cast superalloy CM-247 in a single-step high-activity aluminizing process. Metall. Mater. Trans. A 1998, 29, 2173–2188. [Google Scholar] [CrossRef]
- Fitzpatrick, M.E.; Fry, A.T.; Holdway, P.; Kandil, F.A.; Shackleton, J.; Suominen, L. Determination of Residual Stresses by X-ray Diffraction Measurement Good Practice Guide 52; National Physical Laboratory: Teddington, Middlesex, UK, 2005. [Google Scholar]
- Kamminga, J.D.; de Keijser, T.H.; Mittemeijer, E.J.; Delhez, R. New methods for diffraction stress measurement: A critical evaluation of new and existing methods. J. Appl. Cryst. 2000, 33, 1059–1066. [Google Scholar] [CrossRef]
- Boulesteix, C.; Kolarik, V.; Pedraza, F. Steam oxidation of aluminide coatings under high pressure and for long exposures. Corros. Sci. 2018, 144, 328–338. [Google Scholar] [CrossRef] [Green Version]
- Grabke, H.J. Oxidation of NiAl and FeAl. Intermetallics 1999, 7, 1153–1158. [Google Scholar] [CrossRef]
- Stott, F.H.; Wood, G.C.; Stringer, J. The influence of alloying elements on the development and maintenance of protective scales. Oxid. Met. 1995, 44, 113–145. [Google Scholar] [CrossRef]
- Brady, M.P.; Yamamoto, Y.; Santella, M.L.; Walker, L.R. Composition, microstructure, and water vapor effects on internal/external oxidation of alumina-forming austenitic stainless steels. Oxid. Met. 2009, 72, 311–333. [Google Scholar] [CrossRef]
- Heinonen, M.H.; Kokko, K.; Punkkinen, M.P.J.; Nurmi, E.; Kollár, J.; Vitos, L. Initial oxidation of Fe–Al and Fe–Cr–Al alloys: Cr as an alumina booster. Oxid. Met. 2011, 76, 331–346. [Google Scholar] [CrossRef]
- Renusch, D.; Grimsditch, M.; Koshelev, I.; Veal, B.W. Strain Determination in Thermally-Grown Alumina Scales Using Fluorescence Spectroscopy. Oxid. Met. 1997, 48, 471–795. [Google Scholar] [CrossRef]
- Lambotte, G.; Chartrand, P. Thermodynamic modeling of the (Al2O3 + Na2O), (Al2O3 + Na2O + SiO2), and (Al2O3 + Na2O + AlF3 + NaF) systems. J. Chem. Thermodyn. 2013, 57, 306–334. [Google Scholar] [CrossRef]
- Fernandez, A.G.; Rey, A.; Lasanta, I.; Mato, S.; Brady, M.P.; Perez, F.J. Corrosion of alumina-forming austenitic steel in molten nitrate salts by gravimetric analysis and impedance spectroscopy. Mater. Corros. 2014, 65, 267–275. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alloy | Fe | C | Cr | Ni | Mn | Si | P | S | Mo | V | Al | Nb | N |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P91 | bal. | 0.08– 0.12 | 8– 9.5 | ≤0.4 | 0.3– 0.6 | 0.2– 0.5 | ≤0.02 | ≤0.01 | 0.85– 1.05 | 0.18– 0.25 | ≤0.04 | 0.06– 0.1 | 0.03– 0.07 |
| Diffusion Element | Powder Composition | Process Parameters | ||||
|---|---|---|---|---|---|---|
| Master Alloy | Activator | Inert Filler | Temperature | Time | Atmosphere | |
| Cr | Cr | MnCl2 | Al2O3 | 1050 °C | 2 h | Ar + 5% H2 |
| Al | Al | NH4Cl | Al2O3 | 1000 °C | 1 h | Ar + 5% H2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oskay, C.; Meißner, T.M.; Dobler, C.; Grégoire, B.; Galetz, M.C. Scale Formation and Degradation of Diffusion Coatings Deposited on 9% Cr Steel in Molten Solar Salt. Coatings 2019, 9, 687. https://doi.org/10.3390/coatings9100687
Oskay C, Meißner TM, Dobler C, Grégoire B, Galetz MC. Scale Formation and Degradation of Diffusion Coatings Deposited on 9% Cr Steel in Molten Solar Salt. Coatings. 2019; 9(10):687. https://doi.org/10.3390/coatings9100687
Chicago/Turabian StyleOskay, Ceyhun, Tobias M. Meißner, Carmen Dobler, Benjamin Grégoire, and Mathias C. Galetz. 2019. "Scale Formation and Degradation of Diffusion Coatings Deposited on 9% Cr Steel in Molten Solar Salt" Coatings 9, no. 10: 687. https://doi.org/10.3390/coatings9100687