
Phosphoethanolamine Transferases as Drug Discovery Targets for Therapeutic Treatment of Multi-Drug Resistant Pathogenic Gram-Negative Bacteria

Abstract
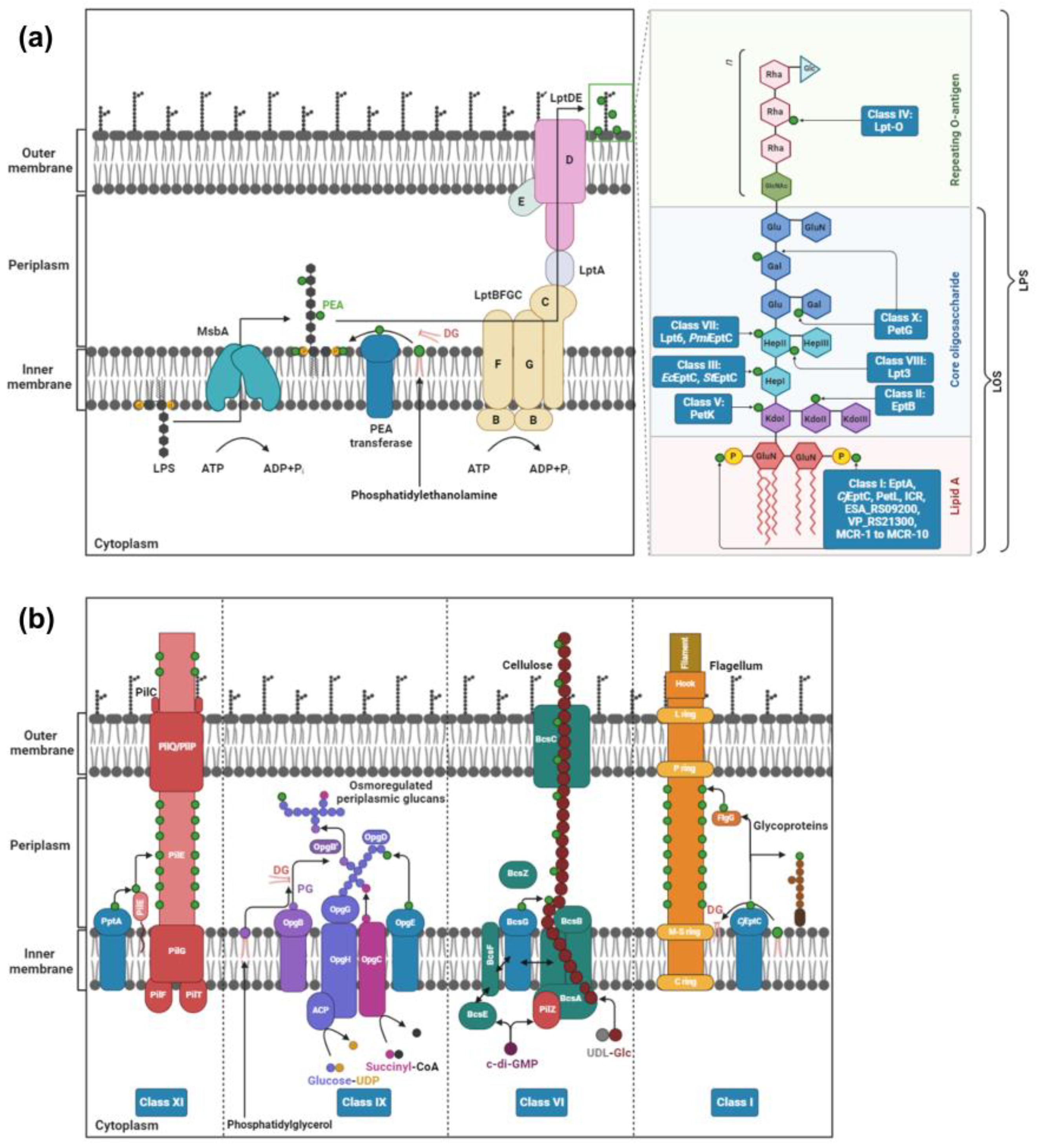
:1. Introduction
2. PEA Transferases
2.1. Class I: Lipid A PEA Transferases
2.2. Class II: PEA Transferases That Modify KdoII of LPS
2.3. Class III: PEA Transferases That Modify HepI of LPS
2.4. Class IV: PEA Transferases That Modify O-Antigen
2.5. Class V: PEA Transferases That Modify KdoI
2.6. Class VI: PEA Transferases That Modify Cellulose
2.7. Class VII: PEA Transferases Modifying O-6 Position of HepII of the LPS/LOS
2.8. Class VIII: PEA Transferases That Modify the O-3 Position of HepII of LOS
2.9. Class IX: PEA Transferases That Modify Osmoregulated Periplasmic Glucans
2.10. Class X: PEA Transferases That Modify Gal Residues of LPS
2.11. Class XI: PEA Transferases That Modify the Pilin Subunit PilE
3. Regulation of PEA Transferase Expression in Pathogenic Gram-Negative Bacteria

4. Structure of PEA Transferases
4.1. Overall Structure

4.2. Disulfide Bonds
4.3. Active Site Architecture
4.4. Model of Enzyme Activity
4.5. Co-Crystals with Receiver Substrates
5. Progress in Drug Development of PEA Transferase Inhibitors
5.1. Inhibitors Based on Analogues
5.2. Targeting Zinc Co-Ordination in the Active Site of EptA
5.3. Miscellaneous Compounds with No Known Mode of Action

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Structure | Target | Organism | Working Concentration | Activity | Target Verification | Reference |
|---|---|---|---|---|---|---|---|
| Ethanolamine |  | MCR-1 | E. coli | 10 mM | Complete reversal of PxB resistance | The crystal structure of ethanolamine binding to MCR-1. | [118] |
| Compounds 6p |  | MCR-1 | E. coli | 25 μM | Complete reversal of colistin resistance | Inhibit the reaction of PEA transfer catalyzed by MCR-1 in an enzymatic assay. | [134] |
| Compounds 6q |  | MCR-1 | E. coli | 25 μM | Complete reversal of colistin resistance | Inhibit the reaction of PEA transfer catalyzed by MCR-1 in an enzymatic assay. | [134] |
| Auranofin |  | MCR-1 and MCR variants | E. coli and other G-ve species * | 7.37 μM | Complete/partial reversal colistin resistance | Inhibit the reaction of PEA transfer catalyzed by MCR-1 in an enzymatic assay. Displace Zn2+ by Au+ ion in a zinc release assay (PAR assay) and X-ray crystallography. | [135] |
| Silver nitrate |  | MCR-1 and MCR variants | E. coli and other G-ve species ** | 2.96 μM | Complete/partial reversal of colistin resistance | Inhibit the reaction of PEA transfer catalyzed by MCR-1 in an enzymatic assay. Displace Zn2+ by Ag+ ion in a zinc release assay (PAR assay) and X-ray crystallography. | [136] |
| EDTA/Baicalin # |  | MCR-1 | Salmonella isolates | EDTA: 213.86 μM/Baicalin: 2800.41 μM | Complete reversal of colistin resistance | Directly inhibit mcr-1 expression. | [140] |
| PBT2/zinc ## |  | EptA | N. gonorrhoeae | PBT2: 0.5 μM/zinc: 2.5 μM | All tested strains became sensitive to tetracycline, colistin, PxB, LL-37, and PG-1. | Reduce PEA decoration of lipid A by MS analysis. | [138,139] |
| Compound 137 |  | EptA | N. gonorrhoeae | 100 μM | Reduced PxB MIC by 4-fold for FA1090 and 2-fold for WHO reference strains. | Reduce PEA decoration of lipid A by MS analysis. | [149] |
| Compound 2 |  | MCR-1 | A. baumannii, E. coli, K. pneumoniae | 30 μM | Reversed colistin resistance mediated by MCR-1 and chromosome-encoded enzymes. | Reduce PEA decoration of lipid A by MS analysis. | [150] |
| IMD-0354 |  | Lipid A modifying enzymes | A. baumannii, E. coli, K. pneumoniae, P. aeruginosa | 5 μM | Reversed colistin resistance mediated by MCR-1 and chromosome-encoded enzymes. | Reduce or completely abrogate PEA and Ara4N modification of lipid A by MS analysis. | [151] |
5.4. Rational Drug Development Programs
6. Concluding Remarks and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Antimicrobial Resistance Global Report on Surveillance: 2014 Summary; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- Lomazzi, M.; Moore, M.; Johnson, A.; Balasegaram, M.; Borisch, B. Antimicrobial resistance–moving forward? BMC Public Health 2019, 19, 858. [Google Scholar] [CrossRef] [PubMed]
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef]
- O’Neill, J. Review on Antimicrobial Resistance Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations; Review on Antimicrobial Resistance: London, UK, 2014. [Google Scholar]
- Tagliabue, A.; Rappuoli, R. Changing Priorities in Vaccinology: Antibiotic Resistance Moving to the Top. Front. Immunol. 2018, 9, 1068. [Google Scholar] [CrossRef]
- WHO. Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections, Including Tuberculosis; (WHO/EMP/IAU/2017.12); WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Kapoor, G.; Saigal, S.; Elongavan, A. Action and resistance mechanisms of antibiotics: A guide for clinicians. J. Anaesthesiol. Clin. Pharmacol. 2017, 33, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.S.; Dwibedy, S.K.; Padhy, I. Polymyxins, the last-resort antibiotics: Mode of action, resistance emergence, and potential solutions. J. Biosci. 2021, 46, 85. [Google Scholar] [CrossRef] [PubMed]
- Ayoub Moubareck, C. Polymyxins and Bacterial Membranes: A Review of Antibacterial Activity and Mechanisms of Resistance. Membranes 2020, 10, 181. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, J.H.; Harper, M.; Boyce, J.D. Mechanisms of Polymyxin Resistance. Adv. Exp. Med. Biol. 2019, 1145, 55–71. [Google Scholar] [CrossRef]
- Llewellyn, A.C.; Zhao, J.; Song, F.; Parvathareddy, J.; Xu, Q.; Napier, B.A.; Laroui, H.; Merlin, D.; Bina, J.E.; Cotter, P.A.; et al. NaxD is a deacetylase required for lipid A modification and Francisella pathogenesis. Mol. Microbiol. 2012, 86, 611–627. [Google Scholar] [CrossRef]
- Cox, A.D.; Wright, J.C.; Li, J.; Hood, D.W.; Moxon, E.R.; Richards, J.C. Phosphorylation of the lipid A region of meningococcal lipopolysaccharide: Identification of a family of transferases that add phosphoethanolamine to lipopolysaccharide. J. Bacteriol. 2003, 185, 3270–3277. [Google Scholar] [CrossRef]
- Anandan, A.; Evans, G.L.; Condic-Jurkic, K.; O’Mara, M.L.; John, C.M.; Phillips, N.J.; Jarvis, G.A.; Wills, S.S.; Stubbs, K.A.; Moraes, I.; et al. Structure of a lipid A phosphoethanolamine transferase suggests how conformational changes govern substrate binding. Proc. Natl. Acad. Sci. USA 2017, 114, 2218–2223. [Google Scholar] [CrossRef]
- Huang, J.; Li, C.; Song, J.; Velkov, T.; Wang, L.; Zhu, Y.; Li, J. Regulating polymyxin resistance in Gram-negative bacteria: Roles of two-component systems PhoPQ and PmrAB. Futur. Microbiol. 2020, 15, 445–459. [Google Scholar] [CrossRef]
- Wei, W.; Srinivas, S.; Lin, J.; Tang, Z.; Wang, S.; Ullah, S.; Kota, V.G.; Feng, Y. Defining ICR-Mo, an intrinsic colistin resistance determinant from Moraxella osloensis. PLoS Genet. 2018, 14, e1007389. [Google Scholar] [CrossRef] [PubMed]
- Gaballa, A.; Wiedmann, M.; Carroll, L.M. More than mcr: Canonical plasmid- and transposon-encoded mobilized colistin resistance genes represent a subset of phosphoethanolamine transferases. Front. Cell. Infect. Microbiol. 2023, 13, 1060519. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhu, Y.; Han, M.L.; Li, M.; Song, J.; Velkov, T.; Li, C.; Li, J. Comparative analysis of phosphoethanolamine transferases involved in polymyxin resistance across 10 clinically relevant Gram-negative bacteria. Int. J. Antimicrob. Agents 2018, 51, 586–593. [Google Scholar] [CrossRef]
- Harper, M.; Wright, A.; St Michael, F.; Li, J.; Deveson Lucas, D.; Ford, M.; Adler, B.; Cox, A.D.; Boyce, J.D. Characterization of Two Novel Lipopolysaccharide Phosphoethanolamine Transferases in Pasteurella multocida and Their Role in Resistance to Cathelicidin-2. Infect. Immun. 2017, 85, e00557-17. [Google Scholar] [CrossRef]
- Klein, G.; Muller-Loennies, S.; Lindner, B.; Kobylak, N.; Brade, H.; Raina, S. Molecular and structural basis of inner core lipopolysaccharide alterations in Escherichia coli: Incorporation of glucuronic acid and phosphoethanolamine in the heptose region. J. Biol. Chem. 2013, 288, 8111–8127. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.; Alarcon, M.; Huerta, J.; Navarro, B.; Aguayo, D. Phosphoethanolamine addition to the Heptose I of the Lipopolysaccharide modifies the inner core structure and has an impact on the binding of Polymyxin B to the Escherichia coli outer membrane. Arch. Biochem. Biophys. 2017, 620, 28–34. [Google Scholar] [CrossRef]
- Zhao, Y.; Meng, Q.; Lai, Y.; Wang, L.; Zhou, D.; Dou, C.; Gu, Y.; Nie, C.; Wei, Y.; Cheng, W. Structural and mechanistic insights into polymyxin resistance mediated by EptC originating from Escherichia coli. FEBS J. 2019, 286, 750–764. [Google Scholar] [CrossRef]
- Tamayo, R.; Choudhury, B.; Septer, A.; Merighi, M.; Carlson, R.; Gunn, J.S. Identification of cptA, a PmrA-regulated locus required for phosphoethanolamine modification of the Salmonella enterica serovar Typhimurium lipopolysaccharide core. J. Bacteriol. 2005, 187, 3391–3399. [Google Scholar] [CrossRef]
- Chen, H.D.; Groisman, E.A. The biology of the PmrA/PmrB two-component system: The major regulator of lipopolysaccharide modifications. Annu. Rev. Microbiol. 2013, 67, 83–112. [Google Scholar] [CrossRef]
- Sun, Q.; Knirel, Y.A.; Lan, R.; Wang, J.; Senchenkova, S.N.; Jin, D.; Shashkov, A.S.; Xia, S.; Perepelov, A.V.; Chen, Q.; et al. A novel plasmid-encoded serotype conversion mechanism through addition of phosphoethanolamine to the O-antigen of Shigella flexneri. PLoS ONE 2012, 7, e46095. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.C.; Burnett, A.J.N.; Hiscock, L.; Maly, K.E.; Weadge, J.T. The Escherichia coli cellulose synthase subunit G (BcsG) is a Zn2+-dependent phosphoethanolamine transferase. J. Biol. Chem. 2020, 295, 6225–6235. [Google Scholar] [CrossRef] [PubMed]
- Thongsomboon, W.; Serra, D.O.; Possling, A.; Hadjineophytou, C.; Hengge, R.; Cegelski, L. Phosphoethanolamine cellulose: A naturally produced chemically modified cellulose. Science 2018, 359, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Vella, P.; Schnell, R.; Polyakova, A.; Bourenkov, G.; Li, F.; Cimdins, A.; Schneider, T.R.; Lindqvist, Y.; Galperin, M.Y.; et al. Structural and Functional Characterization of the BcsG Subunit of the Cellulose Synthase in Salmonella Typhimurium. J. Mol. Biol. 2018, 430, 3170–3189. [Google Scholar] [CrossRef]
- Bontemps-Gallo, S.; Cogez, V.; Robbe-Masselot, C.; Quintard, K.; Dondeyne, J.; Madec, E.; Lacroix, J.M. Biosynthesis of osmoregulated periplasmic glucans in Escherichia coli: The phosphoethanolamine transferase is encoded by opgE. BioMed Res. Int. 2013, 2013, 371429. [Google Scholar] [CrossRef]
- Warren, M.J.; Jennings, M.P. Identification and characterization of pptA: A gene involved in the phase-variable expression of phosphorylcholine on pili of Neisseria meningitidis. Infect. Immun. 2003, 71, 6892–6898. [Google Scholar] [CrossRef]
- Naessan, C.L.; Egge-Jacobsen, W.; Heiniger, R.W.; Wolfgang, M.C.; Aas, F.E.; Rohr, A.; Winther-Larsen, H.C.; Koomey, M. Genetic and functional analyses of PptA, a phospho-form transferase targeting type IV pili in Neisseria gonorrhoeae. J. Bacteriol. 2008, 190, 387–400. [Google Scholar] [CrossRef]
- Aas, F.E.; Egge-Jacobsen, W.; Winther-Larsen, H.C.; Lovold, C.; Hitchen, P.G.; Dell, A.; Koomey, M. Neisseria gonorrhoeae type IV pili undergo multisite, hierarchical modifications with phosphoethanolamine and phosphocholine requiring an enzyme structurally related to lipopolysaccharide phosphoethanolamine transferases. J. Biol. Chem. 2006, 281, 27712–27723. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Samantha, A.; Vrielink, A. Lipid A Phosphoethanolamine Transferase: Regulation, Structure and Immune Response. J. Mol. Biol. 2020, 432, 5184–5196. [Google Scholar] [CrossRef]
- Lee, H.; Hsu, F.F.; Turk, J.; Groisman, E.A. The PmrA-regulated pmrC gene mediates phosphoethanolamine modification of lipid A and polymyxin resistance in Salmonella enterica. J. Bacteriol. 2004, 186, 4124–4133. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D.; Nickel, G.C.; Bajaksouzian, S.; Lavender, H.; Murthy, A.R.; Jacobs, M.R.; Bonomo, R.A. Resistance to colistin in Acinetobacter baumannii associated with mutations in the PmrAB two-component system. Antimicrob. Agents Chemother. 2009, 53, 3628–3634. [Google Scholar] [CrossRef]
- Arroyo, L.A.; Herrera, C.M.; Fernandez, L.; Hankins, J.V.; Trent, M.S.; Hancock, R.E. The pmrCAB operon mediates polymyxin resistance in Acinetobacter baumannii ATCC 17978 and clinical isolates through phosphoethanolamine modification of lipid A. Antimicrob. Agents Chemother. 2011, 55, 3743–3751. [Google Scholar] [CrossRef]
- Cheng, H.Y.; Chen, Y.F.; Peng, H.L. Molecular characterization of the PhoPQ-PmrD-PmrAB mediated pathway regulating polymyxin B resistance in Klebsiella pneumoniae CG43. J. Biomed. Sci. 2010, 17, 60. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.X.; Karbarz, M.J.; Wang, X.; Raetz, C.R.; McGrath, S.C.; Cotter, R.J.; Trent, M.S. Periplasmic cleavage and modification of the 1-phosphate group of Helicobacter pylori lipid A. J. Biol. Chem. 2004, 279, 55780–55791. [Google Scholar] [CrossRef]
- Tran, A.X.; Whittimore, J.D.; Wyrick, P.B.; McGrath, S.C.; Cotter, R.J.; Trent, M.S. The lipid A 1-phosphatase of Helicobacter pylori is required for resistance to the antimicrobial peptide polymyxin. J. Bacteriol. 2006, 188, 4531–4541. [Google Scholar] [CrossRef]
- Nowicki, E.M.; O’Brien, J.P.; Brodbelt, J.S.; Trent, M.S. Extracellular zinc induces phosphoethanolamine addition to Pseudomonas aeruginosa lipid A via the ColRS two-component system. Mol. Microbiol. 2015, 97, 166–178. [Google Scholar] [CrossRef]
- Herrera, C.M.; Henderson, J.C.; Crofts, A.A.; Trent, M.S. Novel coordination of lipopolysaccharide modifications in Vibrio cholerae promotes CAMP resistance. Mol. Microbiol. 2017, 106, 582–596. [Google Scholar] [CrossRef]
- Henderson, J.C.; Fage, C.D.; Cannon, J.R.; Brodbelt, J.S.; Keatinge-Clay, A.T.; Trent, M.S. Antimicrobial peptide resistance of Vibrio cholerae results from an LPS modification pathway related to nonribosomal peptide synthetases. ACS Chem. Biol. 2014, 9, 2382–2392. [Google Scholar] [CrossRef]
- Hobbs, M.M.; Anderson, J.E.; Balthazar, J.T.; Kandler, J.L.; Carlson, R.W.; Ganguly, J.; Begum, A.A.; Duncan, J.A.; Lin, J.T.; Sparling, P.F.; et al. Lipid A’s structure mediates Neisseria gonorrhoeae fitness during experimental infection of mice and men. mBio 2013, 4, e00892-13. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; John, C.M.; Jarvis, G.A. Phosphoryl moieties of lipid A from Neisseria meningitidis and N. gonorrhoeae lipooligosaccharides play an important role in activation of both MyD88- and TRIF-dependent TLR4-MD-2 signaling pathways. J. Immunol. 2010, 185, 6974–6984. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Carlson, R.W.; Muszynski, A.; Choudhury, B.; Kim, K.S.; Stephens, D.S.; Watanabe, H. Modification of lipooligosaccharide with phosphoethanolamine by LptA in Neisseria meningitidis enhances meningococcal adhesion to human endothelial and epithelial cells. Infect. Immun. 2008, 76, 5777–5789. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, Y.L.; Ambrose, K.D.; Zughaier, S.; Zhou, X.; Miller, Y.K.; Shafer, W.M.; Stephens, D.S. Cationic antimicrobial peptide resistance in Neisseria meningitidis. J. Bacteriol. 2005, 187, 5387–5396. [Google Scholar] [CrossRef]
- Lewis, L.A.; Shafer, W.M.; Dutta Ray, T.; Ram, S.; Rice, P.A. Phosphoethanolamine residues on the lipid A moiety of Neisseria gonorrhoeae lipooligosaccharide modulate binding of complement inhibitors and resistance to complement killing. Infect. Immun. 2013, 81, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Packiam, M.; Yedery, R.D.; Begum, A.A.; Carlson, R.W.; Ganguly, J.; Sempowski, G.D.; Ventevogel, M.S.; Shafer, W.M.; Jerse, A.E. Phosphoethanolamine decoration of Neisseria gonorrhoeae lipid A plays a dual immunostimulatory and protective role during experimental genital tract infection. Infect. Immun. 2014, 82, 2170–2179. [Google Scholar] [CrossRef]
- Wanty, C.; Anandan, A.; Piek, S.; Walshe, J.; Ganguly, J.; Carlson, R.W.; Stubbs, K.A.; Kahler, C.M.; Vrielink, A. The structure of the Neisserial lipooligosaccharide phosphoethanolamine transferase A (LptA) required for resistance to polymyxin. J. Mol. Biol. 2013, 425, 3389–3402. [Google Scholar] [CrossRef]
- John, C.M.; Liu, M.; Phillips, N.J.; Yang, Z.; Funk, C.R.; Zimmerman, L.I.; Griffiss, J.M.; Stein, D.C.; Jarvis, G.A. Lack of lipid A pyrophosphorylation and functional lptA reduces inflammation by Neisseria commensals. Infect. Immun. 2012, 80, 4014–4026. [Google Scholar] [CrossRef]
- Handing, J.W.; Criss, A.K. The lipooligosaccharide-modifying enzyme LptA enhances gonococcal defence against human neutrophils. Cell. Microbiol. 2015, 17, 910–921. [Google Scholar] [CrossRef]
- Zughaier, S.M.; Kandler, J.L.; Balthazar, J.T.; Shafer, W.M. Phosphoethanolamine Modification of Neisseria gonorrhoeae Lipid A Reduces Autophagy Flux in Macrophages. PLoS ONE 2015, 10, e0144347. [Google Scholar] [CrossRef]
- Trombley, M.P.; Post, D.M.; Rinker, S.D.; Reinders, L.M.; Fortney, K.R.; Zwickl, B.W.; Janowicz, D.M.; Baye, F.M.; Katz, B.P.; Spinola, S.M.; et al. Phosphoethanolamine Transferase LptA in Haemophilus ducreyi Modifies Lipid A and Contributes to Human Defensin Resistance In Vitro. PLoS ONE 2015, 10, e0124373. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Wang, X.; Guo, W. A phosphoethanolamine transferase specific for the 4′-phosphate residue of Cronobacter sakazakii lipid A. J. Appl. Microbiol. 2016, 121, 1444–1456. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Qiao, J.; Zhou, Q.; Huang, D.; Li, H.; Wang, J.; Wang, X. Identification of a phosphoethanolamine transferase for lipid A modification in Vibrio parahaemolyticus. Food Control 2021, 125, 108033. [Google Scholar] [CrossRef]
- St Michael, F.; Harper, M.; Parnas, H.; John, M.; Stupak, J.; Vinogradov, E.; Adler, B.; Boyce, J.D.; Cox, A.D. Structural and genetic basis for the serological differentiation of Pasteurella multocida Heddleston serotypes 2 and 5. J. Bacteriol. 2009, 191, 6950–6959. [Google Scholar] [CrossRef] [PubMed]
- Stogios, P.J.; Cox, G.; Zubyk, H.L.; Evdokimova, E.; Wawrzak, Z.; Wright, G.D.; Savchenko, A. Substrate Recognition by a Colistin Resistance Enzyme from Moraxella catarrhalis. ACS Chem. Biol. 2018, 13, 1322–1332. [Google Scholar] [CrossRef]
- Yin, W.; Li, H.; Shen, Y.; Liu, Z.; Wang, S.; Shen, Z.; Zhang, R.; Walsh, T.R.; Shen, J.; Wang, Y. Novel Plasmid-Mediated Colistin Resistance Gene mcr-3 in Escherichia coli. mBio 2017, 8, e00543-17. [Google Scholar] [CrossRef]
- Carattoli, A.; Villa, L.; Feudi, C.; Curcio, L.; Orsini, S.; Luppi, A.; Pezzotti, G.; Magistrali, C.F. Novel plasmid-mediated colistin resistance mcr-4 gene in Salmonella and Escherichia coli, Italy 2013, Spain and Belgium, 2015 to 2016. Eurosurveillance 2017, 22, 30589. [Google Scholar] [CrossRef]
- Borowiak, M.; Fischer, J.; Hammerl, J.A.; Hendriksen, R.S.; Szabo, I.; Malorny, B. Identification of a novel transposon-associated phosphoethanolamine transferase gene, mcr-5, conferring colistin resistance in d-tartrate fermenting Salmonella enterica subsp. enterica serovar Paratyphi B. J. Antimicrob. Chemother. 2017, 72, 3317–3324. [Google Scholar] [CrossRef]
- AbuOun, M.; Stubberfield, E.J.; Duggett, N.A.; Kirchner, M.; Dormer, L.; Nunez-Garcia, J.; Randall, L.P.; Lemma, F.; Crook, D.W.; Teale, C.; et al. mcr-1 and mcr-2 variant genes identified in Moraxella species isolated from pigs in Great Britain from 2014 to 2015. J. Antimicrob. Chemother. 2017, 72, 2745–2749. [Google Scholar] [CrossRef]
- Yang, Y.Q.; Li, Y.X.; Lei, C.W.; Zhang, A.Y.; Wang, H.N. Novel plasmid-mediated colistin resistance gene mcr-7.1 in Klebsiella pneumoniae. J. Antimicrob. Chemother. 2018, 73, 1791–1795. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Zhou, Y.; Wang, Z.; Wang, Y.; Zhang, S.; Shen, Z. Emergence of Colistin Resistance Gene mcr-8 and Its Variant in Raoultella ornithinolytica. Front. Microbiol. 2019, 10, 228. [Google Scholar] [CrossRef]
- Wang, C.; Feng, Y.; Liu, L.; Wei, L.; Kang, M.; Zong, Z. Identification of novel mobile colistin resistance gene mcr-10. Emerg. Microbes. Infect. 2020, 9, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Carroll, L.M.; Gaballa, A.; Guldimann, C.; Sullivan, G.; Henderson, L.O.; Wiedmann, M. Identification of Novel Mobilized Colistin Resistance Gene mcr-9 in a Multidrug-Resistant, Colistin-Susceptible Salmonella enterica Serotype Typhimurium Isolate. mBio 2019, 10, e00853-19. [Google Scholar] [CrossRef] [PubMed]
- Snesrud, E.; Maybank, R.; Kwak, Y.I.; Jones, A.R.; Hinkle, M.K.; McGann, P. Chromosomally Encoded mcr-5 in Colistin-Nonsusceptible Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2018, 62, e00679-18. [Google Scholar] [CrossRef]
- Singh, S.; Pathak, A.; Kumar, A.; Rahman, M.; Singh, A.; Gonzalez-Zorn, B.; Prasad, K.N. Emergence of Chromosome-Borne Colistin Resistance Gene mcr-1 in Clinical Isolates of Klebsiella pneumoniae from India. Antimicrob. Agents Chemother. 2018, 62, e01885-17. [Google Scholar] [CrossRef] [PubMed]
- Martins-Sorenson, N.; Snesrud, E.; Xavier, D.E.; Cacci, L.C.; Iavarone, A.T.; McGann, P.; Riley, L.W.; Moreira, B.M. A novel plasmid-encoded mcr-4.3 gene in a colistin-resistant Acinetobacter baumannii clinical strain. J. Antimicrob. Chemother. 2020, 75, 60–64. [Google Scholar] [CrossRef]
- Kalova, A.; Gelbicova, T.; Overballe-Petersen, S.; Litrup, E.; Karpiskova, R. Characterisation of Colistin–Resistant Enterobacterales and Acinetobacter Strains Carrying mcr Genes from Asian Aquaculture Products. Antibiotics 2021, 10, 838. [Google Scholar] [CrossRef]
- Lythell, E.; Suardiaz, R.; Hinchliffe, P.; Hanpaibool, C.; Visitsatthawong, S.; Oliveira, A.S.F.; Lang, E.J.M.; Surawatanawong, P.; Lee, V.S.; Rungrotmongkol, T.; et al. Resistance to the “last resort” antibiotic colistin: A single-zinc mechanism for phosphointermediate formation in MCR enzymes. Chem. Commun. 2020, 56, 6874–6877. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Wang, Y.; Walsh, T.R.; Yi, L.X.; Zhang, R.; Spencer, J.; Doi, Y.; Tian, G.; Dong, B.; Huang, X.; et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: A microbiological and molecular biological study. Lancet Infect. Dis. 2016, 16, 161–168. [Google Scholar] [CrossRef]
- Xavier, B.B.; Lammens, C.; Ruhal, R.; Kumar-Singh, S.; Butaye, P.; Goossens, H.; Malhotra-Kumar, S. Identification of a novel plasmid-mediated colistin-resistance gene, mcr-2, in Escherichia coli, Belgium, June 2016. Eurosurveillance 2016, 21, 30280. [Google Scholar] [CrossRef]
- Poirel, L.; Kieffer, N.; Fernandez-Garayzabal, J.F.; Vela, A.I.; Larpin, Y.; Nordmann, P. MCR-2-mediated plasmid-borne polymyxin resistance most likely originates from Moraxella pluranimalium. J. Antimicrob. Chemother. 2017, 72, 2947–2949. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Zhou, Y.; Li, J.; Yin, W.; Wang, S.; Zhang, S.; Shen, J.; Shen, Z.; Wang, Y. Emergence of a novel mobile colistin resistance gene, mcr-8, in NDM-producing Klebsiella pneumoniae. Emerg. Microbes Infect. 2018, 7, 122. [Google Scholar] [CrossRef] [PubMed]
- Cullen, T.W.; Trent, M.S. A link between the assembly of flagella and lipooligosaccharide of the Gram-negative bacterium Campylobacter jejuni. Proc. Natl. Acad. Sci. USA 2010, 107, 5160–5165. [Google Scholar] [CrossRef] [PubMed]
- Scott, N.E.; Nothaft, H.; Edwards, A.V.; Labbate, M.; Djordjevic, S.P.; Larsen, M.R.; Szymanski, C.M.; Cordwell, S.J. Modification of the Campylobacter jejuni N-linked glycan by EptC protein-mediated addition of phosphoethanolamine. J. Biol. Chem. 2012, 287, 29384–29396. [Google Scholar] [CrossRef] [PubMed]
- Cullen, T.W.; O’Brien, J.P.; Hendrixson, D.R.; Giles, D.K.; Hobb, R.I.; Thompson, S.A.; Brodbelt, J.S.; Trent, M.S. EptC of Campylobacter jejuni mediates phenotypes involved in host interactions and virulence. Infect. Immun. 2013, 81, 430–440. [Google Scholar] [CrossRef]
- Lim, E.S.; Kim, J.S. Role of eptC in Biofilm Formation by Campylobacter jejuni NCTC11168 on Polystyrene and Glass Surfaces. J. Microbiol. Biotechnol. 2017, 27, 1609–1616. [Google Scholar] [CrossRef]
- Fage, C.D.; Brown, D.B.; Boll, J.M.; Keatinge-Clay, A.T.; Trent, M.S. Crystallographic study of the phosphoethanolamine transferase EptC required for polymyxin resistance and motility in Campylobacter jejuni. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 2730–2739. [Google Scholar] [CrossRef]
- Kanipes, M.I.; Lin, S.; Cotter, R.J.; Raetz, C.R. Ca2+-induced phosphoethanolamine transfer to the outer 3-deoxy-D-manno-octulosonic acid moiety of Escherichia coli lipopolysaccharide. A novel membrane enzyme dependent upon phosphatidylethanolamine. J. Biol. Chem. 2001, 276, 1156–1163. [Google Scholar] [CrossRef]
- Reynolds, C.M.; Kalb, S.R.; Cotter, R.J.; Raetz, C.R. A phosphoethanolamine transferase specific for the outer 3-deoxy-D-manno-octulosonic acid residue of Escherichia coli lipopolysaccharide. Identification of the eptB gene and Ca2+ hypersensitivity of an eptB deletion mutant. J. Biol. Chem. 2005, 280, 21202–21211. [Google Scholar] [CrossRef]
- Moon, K.; Six, D.A.; Lee, H.J.; Raetz, C.R.; Gottesman, S. Complex transcriptional and post-transcriptional regulation of an enzyme for lipopolysaccharide modification. Mol. Microbiol. 2013, 89, 52–64. [Google Scholar] [CrossRef]
- Klein, G.; Raina, S. Regulated Assembly of LPS, Its Structural Alterations and Cellular Response to LPS Defects. Int. J. Mol. Sci. 2019, 20, 356. [Google Scholar] [CrossRef]
- Dersch, P.; Khan, M.A.; Muhlen, S.; Gorke, B. Roles of Regulatory RNAs for Antibiotic Resistance in Bacteria and Their Potential Value as Novel Drug Targets. Front. Microbiol. 2017, 8, 803. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.F.; Rivera-Chavez, F.; Hiyoshi, H.; Baumler, A.J. 2420: Loss of eptB decreases systemic inflammation during Salmonella infection and allows for evasion of the host immune response. J. Clin. Transl. Sci. 2018, 1, 9. [Google Scholar] [CrossRef]
- Acuna, L.G.; Barros, M.J.; Penaloza, D.; Rodas, P.I.; Paredes-Sabja, D.; Fuentes, J.A.; Gil, F.; Calderon, I.L. A feed-forward loop between SroC and MgrR small RNAs modulates the expression of eptB and the susceptibility to polymyxin B in Salmonella Typhimurium. Microbiology 2016, 162, 1996–2004. [Google Scholar] [CrossRef]
- Knirel, Y.A.; Anisimov, A.P.; Kislichkina, A.A.; Kondakova, A.N.; Bystrova, O.V.; Vagaiskaya, A.S.; Shatalin, K.Y.; Shashkov, A.S.; Dentovskaya, S.V. Lipopolysaccharide of the Yersinia pseudotuberculosis Complex. Biomolecules 2021, 11, 1410. [Google Scholar] [CrossRef]
- Knirel, Y.A.; Lindner, B.; Vinogradov, E.; Shaikhutdinova, R.Z.; Senchenkova, S.N.; Kocharova, N.A.; Holst, O.; Pier, G.B.; Anisimov, A.P. Cold temperature-induced modifications to the composition and structure of the lipopolysaccharide of Yersinia pestis. Carbohydr. Res. 2005, 340, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Dentovskaya, S.V.; Anisimov, A.P.; Kondakova, A.N.; Lindner, B.; Bystrova, O.V.; Svetoch, T.E.; Shaikhutdinova, R.Z.; Ivanov, S.A.; Bakhteeva, I.V.; Titareva, G.M.; et al. Functional characterization and biological significance of Yersinia pestis lipopolysaccharide biosynthesis genes. Biochemistry 2011, 76, 808–822. [Google Scholar] [CrossRef]
- Trimble, M.J.; Mlynarcik, P.; Kolar, M.; Hancock, R.E. Polymyxin: Alternative Mechanisms of Action and Resistance. Cold Spring Harb. Perspect. Med. 2016, 6, a025288. [Google Scholar] [CrossRef]
- Wright, J.C.; Hood, D.W.; Randle, G.A.; Makepeace, K.; Cox, A.D.; Li, J.; Chalmers, R.; Richards, J.C.; Moxon, E.R. lpt6, a gene required for addition of phosphoethanolamine to inner-core lipopolysaccharide of Neisseria meningitidis and Haemophilus influenzae. J. Bacteriol. 2004, 186, 6970–6982. [Google Scholar] [CrossRef]
- Wenzel, C.Q.; St Michael, F.; Stupak, J.; Li, J.; Cox, A.D.; Richards, J.C. Functional characterization of Lpt3 and Lpt6, the inner-core lipooligosaccharide phosphoethanolamine transferases from Neisseria meningitidis. J. Bacteriol. 2010, 192, 208–216. [Google Scholar] [CrossRef]
- Lewis, L.A.; Choudhury, B.; Balthazar, J.T.; Martin, L.E.; Ram, S.; Rice, P.A.; Stephens, D.S.; Carlson, R.; Shafer, W.M. Phosphoethanolamine substitution of lipid A and resistance of Neisseria gonorrhoeae to cationic antimicrobial peptides and complement-mediated killing by normal human serum. Infect. Immun. 2009, 77, 1112–1120. [Google Scholar] [CrossRef]
- Mansson, M.; Hood, D.W.; Moxon, E.R.; Schweda, E.K. Structural characterization of a novel branching pattern in the lipopolysaccharide from nontypeable Haemophilus influenzae. Eur. J. Biochem. 2003, 270, 2979–2991. [Google Scholar] [CrossRef]
- Aquilini, E.; Merino, S.; Knirel, Y.A.; Regue, M.; Tomas, J.M. Functional identification of Proteus mirabilis eptC gene encoding a core lipopolysaccharide phosphoethanolamine transferase. Int. J. Mol. Sci. 2014, 15, 6689–6702. [Google Scholar] [CrossRef]
- Sun, Y.; Wen, S.; Zhao, L.; Xia, Q.; Pan, Y.; Liu, H.; Wei, C.; Chen, H.; Ge, J.; Wang, H. Association among biofilm formation, virulence gene expression, and antibiotic resistance in Proteus mirabilis isolates from diarrhetic animals in Northeast China. BMC Vet. Res. 2020, 16, 176. [Google Scholar] [CrossRef]
- Mackinnon, F.G.; Cox, A.D.; Plested, J.S.; Tang, C.M.; Makepeace, K.; Coull, P.A.; Wright, J.C.; Chalmers, R.; Hood, D.W.; Richards, J.C.; et al. Identification of a gene (lpt-3) required for the addition of phosphoethanolamine to the lipopolysaccharide inner core of Neisseria meningitidis and its role in mediating susceptibility to bactericidal killing and opsonophagocytosis. Mol. Microbiol. 2002, 43, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Plested, J.S.; Makepeace, K.; Jennings, M.P.; Gidney, M.A.; Lacelle, S.; Brisson, J.; Cox, A.D.; Martin, A.; Bird, A.G.; Tang, C.M.; et al. Conservation and accessibility of an inner core lipopolysaccharide epitope of Neisseria meningitidis. Infect. Immun. 1999, 67, 5417–5426. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, E.T.; Piekarowicz, A.; Swanson, K.V.; Griffiss, J.M.; Stein, D.C. Biochemical analysis of Lpt3, a protein responsible for phosphoethanolamine addition to lipooligosaccharide of pathogenic Neisseria. J. Bacteriol. 2006, 188, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Olaitan, A.O.; Morand, S.; Rolain, J.M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front. Microbiol. 2014, 5, 643. [Google Scholar] [CrossRef]
- Melaugh, W.; Phillips, N.J.; Campagnari, A.A.; Tullius, M.V.; Gibson, B.W. Structure of the major oligosaccharide from the lipooligosaccharide of Haemophilus ducreyi strain 35000 and evidence for additional glycoforms. Biochemistry 1994, 33, 13070–13078. [Google Scholar] [CrossRef]
- Kieffer, N.; Nordmann, P.; Poirel, L. Moraxella Species as Potential Sources of MCR-Like Polymyxin Resistance Determinants. Antimicrob. Agents Chemother. 2017, 61, e00129-17. [Google Scholar] [CrossRef]
- Gao, R.; Hu, Y.; Li, Z.; Sun, J.; Wang, Q.; Lin, J.; Ye, H.; Liu, F.; Srinivas, S.; Li, D.; et al. Dissemination and Mechanism for the MCR-1 Colistin Resistance. PLoS Pathog. 2016, 12, e1005957. [Google Scholar] [CrossRef]
- Mmatli, M.; Mbelle, N.M.; Osei Sekyere, J. Global epidemiology, genetic environment, risk factors and therapeutic prospects of mcr genes: A current and emerging update. Front. Cell. Infect. Microbiol. 2022, 12, 941358. [Google Scholar] [CrossRef] [PubMed]
- Lentz, S.A.M.; Dalmolin, T.V.; Barth, A.L.; Martins, A.F. mcr-1 Gene in Latin America: How is It Disseminated among Humans, Animals, and the Environment? Front. Public Health 2021, 9, 648940. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Li, M.; Spiller, O.B.; Andrey, D.O.; Hinchliffe, P.; Li, H.; MacLean, C.; Niumsup, P.; Powell, L.; Pritchard, M.; et al. Balancing mcr-1 expression and bacterial survival is a delicate equilibrium between essential cellular defence mechanisms. Nat. Commun. 2017, 8, 2054. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Ling, Z.; Dong, Y.; Qiao, L.; Shen, Y.; Liu, Z.; Wu, Y.; Li, W.; Zhang, R.; Walsh, T.R.; et al. Mobile Colistin Resistance Enzyme MCR-3 Facilitates Bacterial Evasion of Host Phagocytosis. Adv. Sci. 2021, 8, e2101336. [Google Scholar] [CrossRef] [PubMed]
- Dentovskaya, S.V.; Bakhteeva, I.V.; Titareva, G.M.; Shaikhutdinova, R.Z.; Kondakova, A.N.; Bystrova, O.V.; Lindner, B.; Knirel, Y.A.; Anisimov, A.P. Structural diversity and endotoxic activity of the lipopolysaccharide of Yersinia pestis. Biochemistry 2008, 73, 192–199. [Google Scholar] [CrossRef]
- Morgan, J.L.; McNamara, J.T.; Zimmer, J. Mechanism of activation of bacterial cellulose synthase by cyclic di-GMP. Nat. Struct. Mol. Biol. 2014, 21, 489–496. [Google Scholar] [CrossRef]
- Kahler, C.M.; Datta, A.; Tzeng, Y.L.; Carlson, R.W.; Stephens, D.S. Inner core assembly and structure of the lipooligosaccharide of Neisseria meningitidis: Capacity of strain NMB to express all known immunotype epitopes. Glycobiology 2005, 15, 409–419. [Google Scholar] [CrossRef]
- Ram, S.; Cox, A.D.; Wright, J.C.; Vogel, U.; Getzlaff, S.; Boden, R.; Li, J.; Plested, J.S.; Meri, S.; Gulati, S.; et al. Neisserial lipooligosaccharide is a target for complement component C4b. Inner core phosphoethanolamine residues define C4b linkage specificity. J. Biol. Chem. 2003, 278, 50853–50862. [Google Scholar] [CrossRef]
- Boda, S.K.; Fischer, N.G.; Ye, Z.; Aparicio, C. Dual Oral Tissue Adhesive Nanofiber Membranes for pH-Responsive Delivery of Antimicrobial Peptides. Biomacromolecules 2020, 21, 4945–4961. [Google Scholar] [CrossRef]
- Anonsen, J.H.; Egge-Jacobsen, W.; Aas, F.E.; Borud, B.; Koomey, M.; Vik, A. Novel protein substrates of the phospho-form modification system in Neisseria gonorrhoeae and their connection to O-linked protein glycosylation. Infect. Immun. 2012, 80, 22–30. [Google Scholar] [CrossRef]
- Kahler, C.M.; Nawrocki, K.L.; Anandan, A.; Vrielink, A.; Shafer, W.M. Structure-Function Relationships of the Neisserial EptA Enzyme Responsible for Phosphoethanolamine Decoration of Lipid A: Rationale for Drug Targeting. Front. Microbiol. 2018, 9, 1922. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, Y.L.; Datta, A.; Ambrose, K.; Lo, M.; Davies, J.K.; Carlson, R.W.; Stephens, D.S.; Kahler, C.M. The MisR/MisS two-component regulatory system influences inner core structure and immunotype of lipooligosaccharide in Neisseria meningitidis. J. Biol. Chem. 2004, 279, 35053–35062. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, Y.L.; Kahler, C.M.; Zhang, X.; Stephens, D.S. MisR/MisS two-component regulon in Neisseria meningitidis. Infect. Immun. 2008, 76, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.B.; Criss, A.K. Resistance of Neisseria gonorrhoeae to neutrophils. Front. Microbiol. 2011, 2, 77. [Google Scholar] [CrossRef]
- Wei, P.; Song, G.; Shi, M.; Zhou, Y.; Liu, Y.; Lei, J.; Chen, P.; Yin, L. Substrate analog interaction with MCR-1 offers insight into the rising threat of the plasmid-mediated transferable colistin resistance. FASEB J. 2018, 32, 1085–1098. [Google Scholar] [CrossRef]
- Son, S.J.; Huang, R.; Squire, C.J.; Leung, I.K.H. MCR-1: A promising target for structure-based design of inhibitors to tackle polymyxin resistance. Drug Discov. Today 2019, 24, 206–216. [Google Scholar] [CrossRef]
- Sun, J.; Xu, Y.; Gao, R.; Lin, J.; Wei, W.; Srinivas, S.; Li, D.; Yang, R.S.; Li, X.P.; Liao, X.P.; et al. Deciphering MCR-2 Colistin Resistance. mBio 2017, 8, e00625-17. [Google Scholar] [CrossRef]
- Ma, G.; Zhu, Y.; Yu, Z.; Ahmad, A.; Zhang, H. High resolution crystal structure of the catalytic domain of MCR-1. Sci. Rep. 2016, 6, 39540. [Google Scholar] [CrossRef]
- Coates, K.; Walsh, T.R.; Spencer, J.; Hinchliffe, P. 1.12 A resolution crystal structure of the catalytic domain of the plasmid-mediated colistin resistance determinant MCR-2. Acta Crystallogr. F Struct. Biol. Commun. 2017, 73, 443–449. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Piek, S.; Wang, Z.; Ganguly, J.; Lakey, A.M.; Bartley, S.N.; Mowlaboccus, S.; Anandan, A.; Stubbs, K.A.; Scanlon, M.J.; Vrielink, A.; et al. The role of oxidoreductases in determining the function of the Neisserial lipid A phosphoethanolamine transferase required for resistance to polymyxin. PLoS ONE 2014, 9, e106513. [Google Scholar] [CrossRef]
- Hinchliffe, P.; Yang, Q.E.; Portal, E.; Young, T.; Li, H.; Tooke, C.L.; Carvalho, M.J.; Paterson, N.G.; Brem, J.; Niumsup, P.R.; et al. Insights into the Mechanistic Basis of Plasmid-Mediated Colistin Resistance from Crystal Structures of the Catalytic Domain of MCR-1. Sci. Rep. 2017, 7, 39392. [Google Scholar] [CrossRef] [PubMed]
- Stojanoski, V.; Sankaran, B.; Prasad, B.V.; Poirel, L.; Nordmann, P.; Palzkill, T. Structure of the catalytic domain of the colistin resistance enzyme MCR-1. BMC Biol. 2016, 14, 81. [Google Scholar] [CrossRef]
- Hu, M.; Guo, J.; Cheng, Q.; Yang, Z.; Chan, E.W.C.; Chen, S.; Hao, Q. Crystal Structure of Escherichia coli originated MCR-1, a phosphoethanolamine transferase for Colistin Resistance. Sci. Rep. 2016, 6, 38793. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wei, W.; Lei, S.; Lin, J.; Srinivas, S.; Feng, Y. An Evolutionarily Conserved Mechanism for Intrinsic and Transferable Polymyxin Resistance. mBio 2018, 9, e02317-17. [Google Scholar] [CrossRef] [PubMed]
- Suardiaz, R.; Lythell, E.; Hinchliffe, P.; van der Kamp, M.; Spencer, J.; Fey, N.; Mulholland, A.J. Catalytic mechanism of the colistin resistance protein MCR-1. Org. Biomol. Chem. 2021, 19, 3813–3819. [Google Scholar] [CrossRef]
- Xu, Y.; Zhong, L.L.; Srinivas, S.; Sun, J.; Huang, M.; Paterson, D.L.; Lei, S.; Lin, J.; Li, X.; Tang, Z.; et al. Spread of MCR-3 Colistin Resistance in China: An Epidemiological, Genomic and Mechanistic Study. eBioMedicine 2018, 34, 139–157. [Google Scholar] [CrossRef]
- Zhang, H.; Hou, M.; Xu, Y.; Srinivas, S.; Huang, M.; Liu, L.; Feng, Y. Action and mechanism of the colistin resistance enzyme MCR-4. Commun. Biol. 2019, 2, 36. [Google Scholar] [CrossRef]
- Zhang, H.; Zong, Z.; Lei, S.; Srinivas, S.; Sun, J.; Feng, Y.; Huang, M.; Feng, Y. A Genomic, Evolutionary, and Mechanistic Study of MCR-5 Action Suggests Functional Unification across the MCR Family of Colistin Resistance. Adv. Sci. 2019, 6, 1900034. [Google Scholar] [CrossRef]
- Liu, Z.-X.; Han, Z.; Yu, X.-L.; Wen, G.; Zeng, C. Crystal Structure of the Catalytic Domain of MCR-1 (cMCR-1) in Complex with d-Xylose. Crystals 2018, 8, 172. [Google Scholar] [CrossRef]
- Lan, X.J.; Yan, H.T.; Lin, F.; Hou, S.; Li, C.C.; Wang, G.S.; Sun, W.; Xiao, J.H.; Li, S. Design, Synthesis and Biological Evaluation of 1-Phenyl-2-(phenylamino) Ethanone Derivatives as Novel MCR-1 Inhibitors. Molecules 2019, 24, 2719. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, Q.; Wang, R.; Wang, H.; Wong, Y.T.; Wang, M.; Hao, Q.; Yan, A.; Kao, R.Y.; Ho, P.L.; et al. Resensitizing carbapenem- and colistin-resistant bacteria to antibiotics using auranofin. Nat. Commun. 2020, 11, 5263. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, R.; Wang, M.; Liu, C.; Koohi-Moghadam, M.; Wang, H.; Ho, P.L.; Li, H.; Sun, H. Re-sensitization of mcr carrying multidrug resistant bacteria to colistin by silver. Proc. Natl. Acad. Sci. USA 2022, 119, e2119417119. [Google Scholar] [CrossRef] [PubMed]
- Bohlmann, L.; De Oliveira, D.M.P.; El-Deeb, I.M.; Brazel, E.B.; Harbison-Price, N.; Ong, C.Y.; Rivera-Hernandez, T.; Ferguson, S.A.; Cork, A.J.; Phan, M.D.; et al. Chemical Synergy between Ionophore PBT2 and Zinc Reverses Antibiotic Resistance. mBio 2018, 9, e02391-18. [Google Scholar] [CrossRef] [PubMed]
- Jen, F.E.; Everest-Dass, A.V.; El-Deeb, I.M.; Singh, S.; Haselhorst, T.; Walker, M.J.; von Itzstein, M.; Jennings, M.P. Neisseria gonorrhoeae Becomes Susceptible to Polymyxin B and Colistin in the Presence of PBT2. ACS Infect. Dis. 2020, 6, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Jen, F.E.; El-Deeb, I.M.; Zalucki, Y.M.; Edwards, J.L.; Walker, M.J.; von Itzstein, M.; Jennings, M.P. A drug candidate for Alzheimer’s and Huntington’s disease, PBT2, can be repurposed to render Neisseria gonorrhoeae susceptible to natural cationic antimicrobial peptides. J. Antimicrob. Chemother. 2021, 76, 2850–2853. [Google Scholar] [CrossRef]
- Cui, X.D.; Zhang, J.K.; Sun, Y.W.; Yan, F.B.; Zhao, J.F.; He, D.D.; Pan, Y.S.; Yuan, L.; Zhai, Y.J.; Hu, G.Z. Synergistic antibacterial activity of baicalin and EDTA in combination with colistin against colistin-resistant Salmonella. Poult. Sci. 2023, 102, 102346. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, S.; Wang, T.; Li, H.; Tang, S.; Wang, J.; Wang, Y.; Deng, X. Pterostilbene, a Potential MCR-1 Inhibitor That Enhances the Efficacy of Polymyxin B. Antimicrob. Agents Chemother. 2018, 62, e02146-17. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Sun, X.; Wen, Z.; Wang, D.; Peng, L. A Potential Inhibitor of MCR-1: An Attempt to Enhance the Efficacy of Polymyxin Against Multidrug-Resistant Bacteria. Curr. Microbiol. 2020, 77, 3256–3263. [Google Scholar] [CrossRef]
- Du, R.; Lv, Q.; Hu, W.; Hou, X.; Zhou, Y.; Deng, X.; Sun, L.; Li, L.; Deng, Y.; Wang, J. Phloretin potentiates polymyxin E activity against gram-negative bacteria. Life Sci. 2021, 287, 120085. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Guo, Y.; Liu, X.; Liu, S.; Niu, X.; Wang, Y.; Deng, X. Discovery of a potential MCR-1 inhibitor that reverses polymyxin activity against clinical mcr-1-positive Enterobacteriaceae. J. Infect. 2019, 78, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Li, L.; Zhan, B.; Shen, X.; Deng, X.; Tan, W.; Fang, T. Pogostone Enhances the Antibacterial Activity of Colistin against MCR-1-Positive Bacteria by Inhibiting the Biological Function of MCR-1. Molecules 2022, 27, 2819. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Q.; Du, R.; Ma, C.; Zhou, Y.; Shen, X.; Hou, X.; Xu, L.; Li, L.; Deng, X.; Wang, J. NMPA-approved traditional Chinese medicine-Pingwei Pill: New indication for colistin recovery against MCR-positive bacteria infection. Chin. Med. 2021, 16, 106. [Google Scholar] [CrossRef] [PubMed]
- Hanpaibool, C.; Ngamwongsatit, N.; Ounjai, P.; Yotphan, S.; Wolschann, P.; Mulholland, A.J.; Spencer, J.; Rungrotmongkol, T. Pyrazolones Potentiate Colistin Activity against MCR-1-Producing Resistant Bacteria: Computational and Microbiological Study. ACS Omega 2023, 8, 8366–8376. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Lv, X.; Wang, Y.; Zhou, Y.; Lu, N.; Deng, X.; Wang, J. Honokiol Restores Polymyxin Susceptibility to MCR-1-Positive Pathogens both In Vitro and In Vivo. Appl. Environ. Microbiol. 2020, 86, e02346-19. [Google Scholar] [CrossRef]
- Mullally, C.; Stubbs, K.A.; Thai, V.C.; Anandan, A.; Bartley, S.; Scanlon, M.J.; Jarvis, G.A.; John, C.M.; Lim, K.Y.L.; Sullivan, C.M.; et al. Novel small molecules that increase the susceptibility of Neisseria gonorrhoeae to cationic antimicrobial peptides by inhibiting lipid A phosphoethanolamine transferase. J. Antimicrob. Chemother. 2022, 77, 2441–2447. [Google Scholar] [CrossRef]
- Barker, W.T.; Martin, S.E.; Chandler, C.E.; Nguyen, T.V.; Harris, T.L.; Goodell, C.; Melander, R.J.; Doi, Y.; Ernst, R.K.; Melander, C. Small molecule adjuvants that suppress both chromosomal and mcr-1 encoded colistin-resistance and amplify colistin efficacy in polymyxin-susceptible bacteria. Bioorg. Med. Chem. 2017, 25, 5749–5753. [Google Scholar] [CrossRef]
- Barker, W.T.; Nemeth, A.M.; Brackett, S.M.; Basak, A.K.; Chandler, C.E.; Jania, L.A.; Zuercher, W.J.; Melander, R.J.; Koller, B.H.; Ernst, R.K.; et al. Repurposing Eukaryotic Kinase Inhibitors as Colistin Adjuvants in Gram-Negative Bacteria. ACS Infect. Dis. 2019, 5, 1764–1771. [Google Scholar] [CrossRef]
- Nemeth, A.M.; Basak, A.K.; Weig, A.W.; Marrujo, S.A.; Barker, W.T.; Jania, L.A.; Hendricks, T.A.; Sullivan, A.E.; O’Connor, P.M.; Melander, R.J.; et al. Structure-Function Studies on IMD-0354 Identifies Highly Active Colistin Adjuvants. ChemMedChem 2020, 15, 210–218. [Google Scholar] [CrossRef]






| Class | Enzyme | Organism | Encoded by | Sites of PEA Modification | Regulation | Function | Reference |
|---|---|---|---|---|---|---|---|
| I | EptA (LptA, PmrC, YjdB) | E. coli | Core genome | Lipid A headgroups | PmrA/B and PhoP/Q | Resistance to CAMPs | [17,23,33] |
| S. enterica Typhymurium | Core genome | Lipid A headgroups | PmrA/B and PhoP/Q | Resistance to CAMPs | [17,23,33,34] | ||
| A. baumannii | Core genome | Lipid A headgroups | PmrA/B | Resistance to CAMPs | [35,36] | ||
| S. flexneri | Core genome | Lipid A headgroups | PmrA/B and PhoP/Q | Resistance to CAMPs | [17] | ||
| K. pneumonia | Core genome | Lipid A headgroups | PmrA/B and PhoP/Q | Resistance to CAMPs | [17,37] | ||
| H. pylori | Core genome | Lipid A headgroups | Requires the removal of phosphate group by LpxEHP (Hp0021, Lipid A phosphatase) | Resistance to CAMPs | [38,39] | ||
| P. aeruginosa | Core genome | Lipid A headgroups | ColR/S | Resistance to CAMPs | [17,40] | ||
| V. cholera | Core genome | Lipid A headgroups | pH = 5.8 | Resistance to CAMPs | [41,42] | ||
| N. meningitidis | Genetic island | Lipid A headgroups | Phase variation of the open reading frame and MisR/S | Resistance to CAMPs Resistance to human serum Colonisation of mucosal surfaces | [12,17,43,44,45,46,47,48,49] | ||
| N. gonorrhoeae | Genetic island | Lipid A headgroups | Phase variation of the open reading frame and MisR/S | Resistance to CAMPs Resistance to human serum Evasion of phagocytosis by PNMs and macrophages Colonisation of mucosal surfaces Influence bacterial survival in mouse and human models | [12,17,43,44,45,46,47,48,50,51,52] | ||
| H. ducreyi | Core genome | Lipid A headgroups | Unknown | Resistance to CAMPs | [53] | ||
| ESA_RS09200 (ESA_02008) | C. sakazakii | Core genome | Lipid A headgroups | pH = 5.0 | Resistance to CAMPs Avoid TLR4/MD2 recognition Evasion of phagocytosis by macrophages | [54] | |
| VP_RS21300 | V. parahaemolyticus | Core genome | Lipid A headgroups | pH = 6.5 | Resistance to CAMPs Increase pathogenicity | [55] | |
| PetL | P. multocida | Core genome | Lipid A headgroups | Global regulator Fis and Hfq-dependent sRNA | Resistance to CAMPs | [18,56] | |
| ICR | Moraxella spp. (M. catarrhalis and M. osloensis) | Core genome | Lipid A headgroups | Unknown | Intrinsic resistance to CAMPs | [15,57] | |
| MCR-1 to MCR-10 | Enterobacteriaceae, Moraxella spp., P. aeruginosa and Acinetobacter spp. | Plasmid | Lipid A headgroups | Unknown | Resistance to CAMPs Mcr-3 increases pathogenicity and impairs phagocytosis. | [58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74] | |
| EptC | C. jejuni | Core genome | Lipid A headgroups HepI of LOS FlgG flagellar rod protein N-linked glycan (Glycoproteins) | Unknown | Resistance to CAMPs Mobility and flagella production Biofilm formation Recognition by a human TLR4/MD2 Colonization of chick ceca and BALB/cByJ mice | [75,76,77,78,79] | |
| II | EptB | E. coli | Core genome | KdoII of LPS | MgrR/RpoE (High Ca2+ concentration) ArcZ sRNA in an ArcA/B-dependent manner (Low oxygen concentration) | Resistance to CAMPs | [80,81,82,83,84] |
| S. enterica Typhymurium | Core genome | KdoII of LPS | MgrR | Resistance to CAMPs Protect bacteria from the binding and detoxifying by intelectin Enhance cytokine secretion in spleen of infected mouse | [85,86] | ||
| Y. pestis | Core genome | KdoII of LPS | Low temperatures (6 °C) | Resistance to CAMPs | [87,88,89,90] | ||
| III | EptC (CptA) | E. coli | Core genome | HepI of LPS | PhoB/R | Resistance to SDS and sublethal concentration of Zn2+ Not confer resistance to CAMPs | [19,20,21] |
| S. enterica Typhymurium | Core genome | HepI of LPS | PmrA/B | Modest resistance to CAMPs but not impairing the virulence in mouse model of infection. | [22,23] | ||
| IV | Lpt-O | S. flexneri | Plasmid | O-antigen of LPS | Unknown | Confer the MASF IV-1 positive phenotype in serotype Xv strains | [24] |
| V | PetK | P. multocida | Core genome | KdoI of LPS | Unknown | Resistance to CAMPs | [18] |
| VI | BcsG | E. coli | Core genome | Cellulose | Unknown | Cellulose production Maintain biofilm’s architecture and integrity | [25,26] |
| S. enterica Typhymurium | Core genome | Cellulose | Unknown | Cellulose production Maintain biofilm’s architecture and integrity | [27] | ||
| VII | Lpt6 | N. meningitidis | Genetic island | O-6 position of HepII of LOS | Unknown | Increase susceptibility to complement-mediated killing | [91,92] |
| N. gonorrhoeae | Genetic island | O-6 position of HepII of LOS | Unknown | Unknown | [93] | ||
| H. influenzae | Core genome | O-6 position of HepII of LPS | Unknown | Unknown | [91,94] | ||
| EptC | P. mirabilis | Core genome | O-6 position of HepII of LPS | Unknown | Resistance to CAMPs | [95,96] | |
| VIII | Lpt3 | N. meningitidis | Core genome | O-3 position of HepII of LOS | Unknown | Require for the binding of mAb B5 | [92,97,98] |
| N. gonorrhoeae | Core genome | O-3 position of HepII of LOS | Unknown | Unknown | [99] | ||
| P. multocida | Core genome | O-3 position of HepII of LPS | Unknown | Inhibit the binding of mAbs T1C6 and T6B2 | [56] | ||
| IX | OpgE | E. coli | Core genome | Osmoregulated periplasmic glycans | Unknown | Unknown | [28] |
| X | PetG | P. multocida | Core genome | Gal residues of LPS | Unknown | Unknown | [18] |
| XI | PptA | N. meningitidis | Core genome | Pilin subunit PilE | Unknown | Influence pilin structure and antigenicity | [29] |
| N. gonorrhoeae | Core genome | Pilin subunit PilE | Unknown | Influence pilin structure and antigenicity | [30,31] | ||
| N/A | PtdA | H. ducreyi | Core genome | Unknown | Unknown | Resistance to CAMPs | [53] |
| N/A | PtdB | H. ducreyi | Core genome | Unknown | Unknown | Resistance to CAMPs | [53] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thai, V.C.; Stubbs, K.A.; Sarkar-Tyson, M.; Kahler, C.M. Phosphoethanolamine Transferases as Drug Discovery Targets for Therapeutic Treatment of Multi-Drug Resistant Pathogenic Gram-Negative Bacteria. Antibiotics 2023, 12, 1382. https://doi.org/10.3390/antibiotics12091382
Thai VC, Stubbs KA, Sarkar-Tyson M, Kahler CM. Phosphoethanolamine Transferases as Drug Discovery Targets for Therapeutic Treatment of Multi-Drug Resistant Pathogenic Gram-Negative Bacteria. Antibiotics. 2023; 12(9):1382. https://doi.org/10.3390/antibiotics12091382
Chicago/Turabian StyleThai, Van C., Keith A. Stubbs, Mitali Sarkar-Tyson, and Charlene M. Kahler. 2023. "Phosphoethanolamine Transferases as Drug Discovery Targets for Therapeutic Treatment of Multi-Drug Resistant Pathogenic Gram-Negative Bacteria" Antibiotics 12, no. 9: 1382. https://doi.org/10.3390/antibiotics12091382