
Exploring Alternative Pathways to Target Bacterial Type II Topoisomerases Using NBTI Antibacterials: Beyond Halogen-Bonding Interactions
 , , , and
, , , and
Abstract
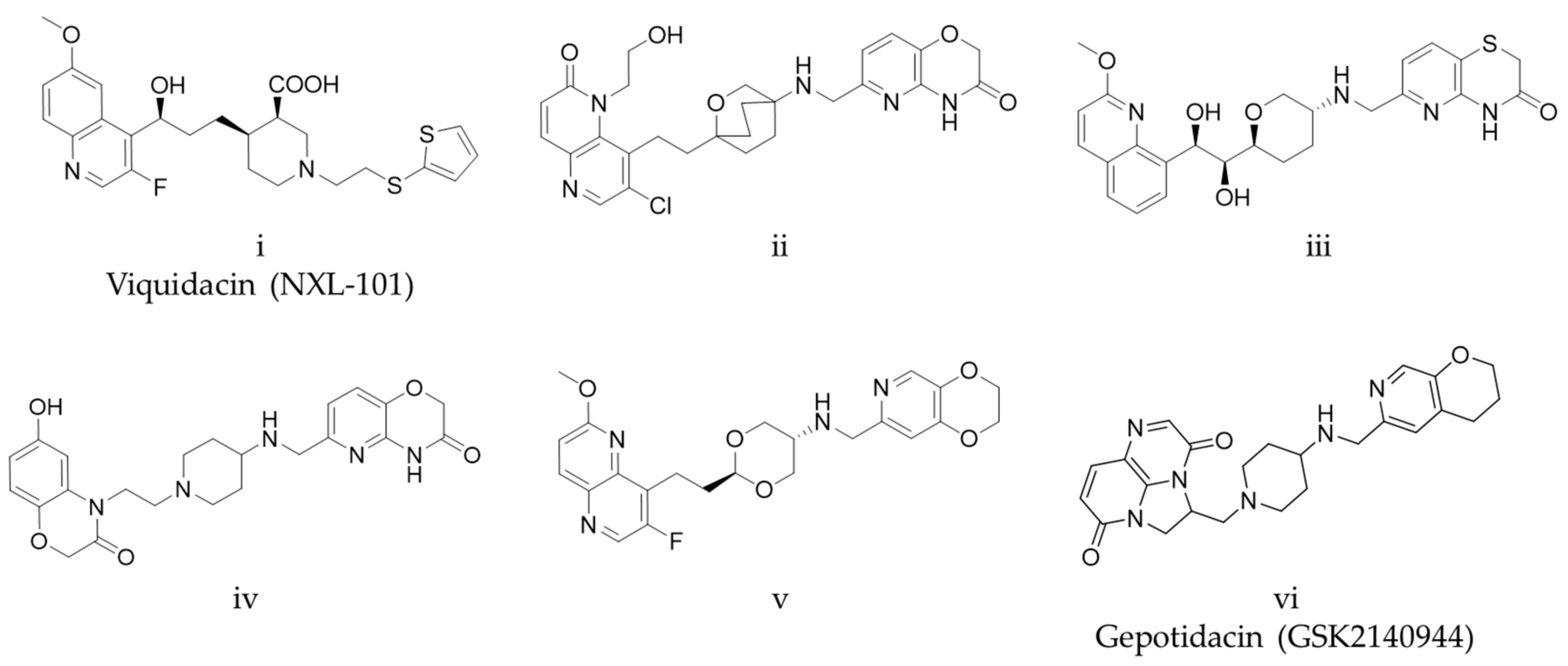
:1. Introduction
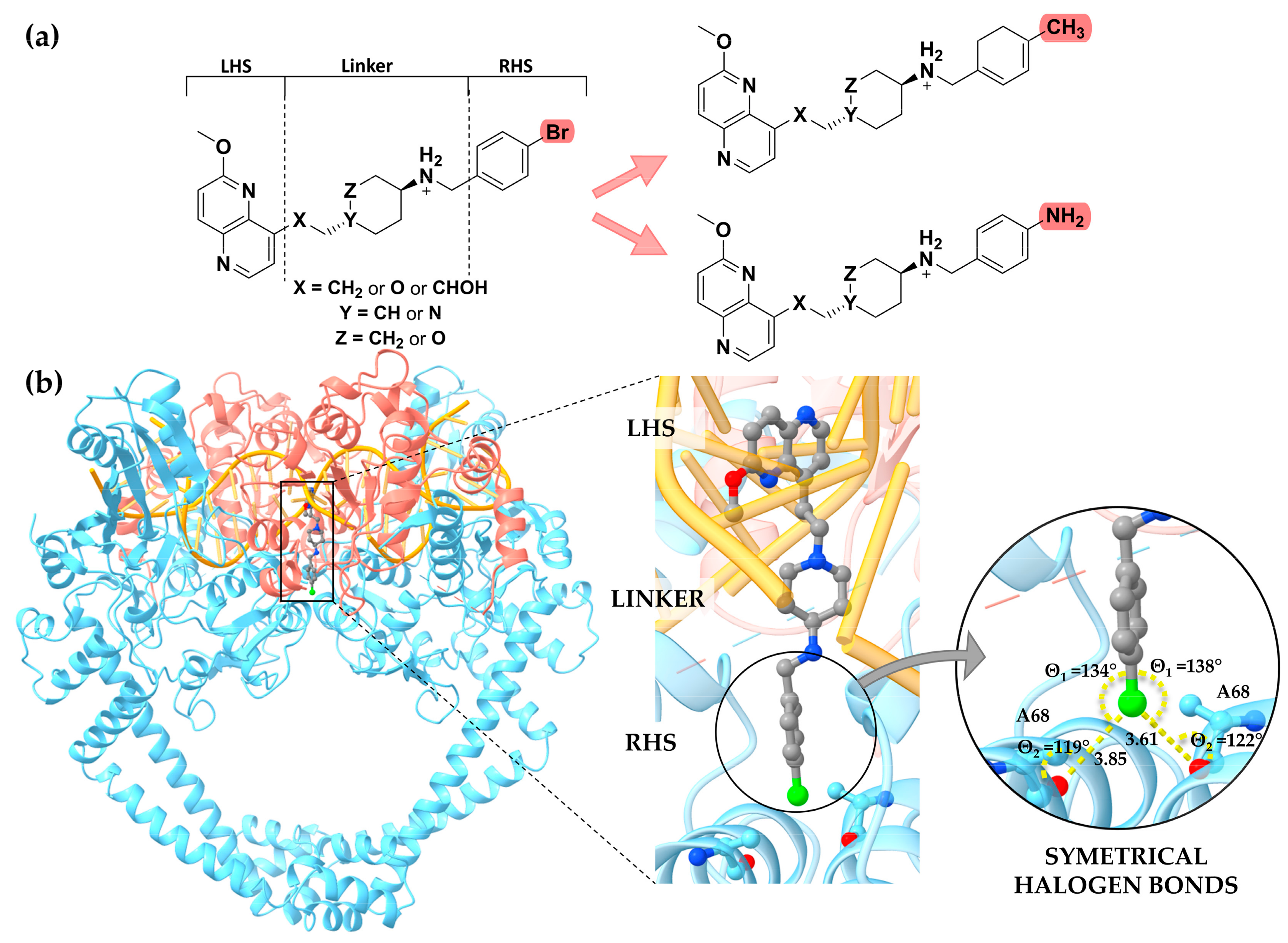
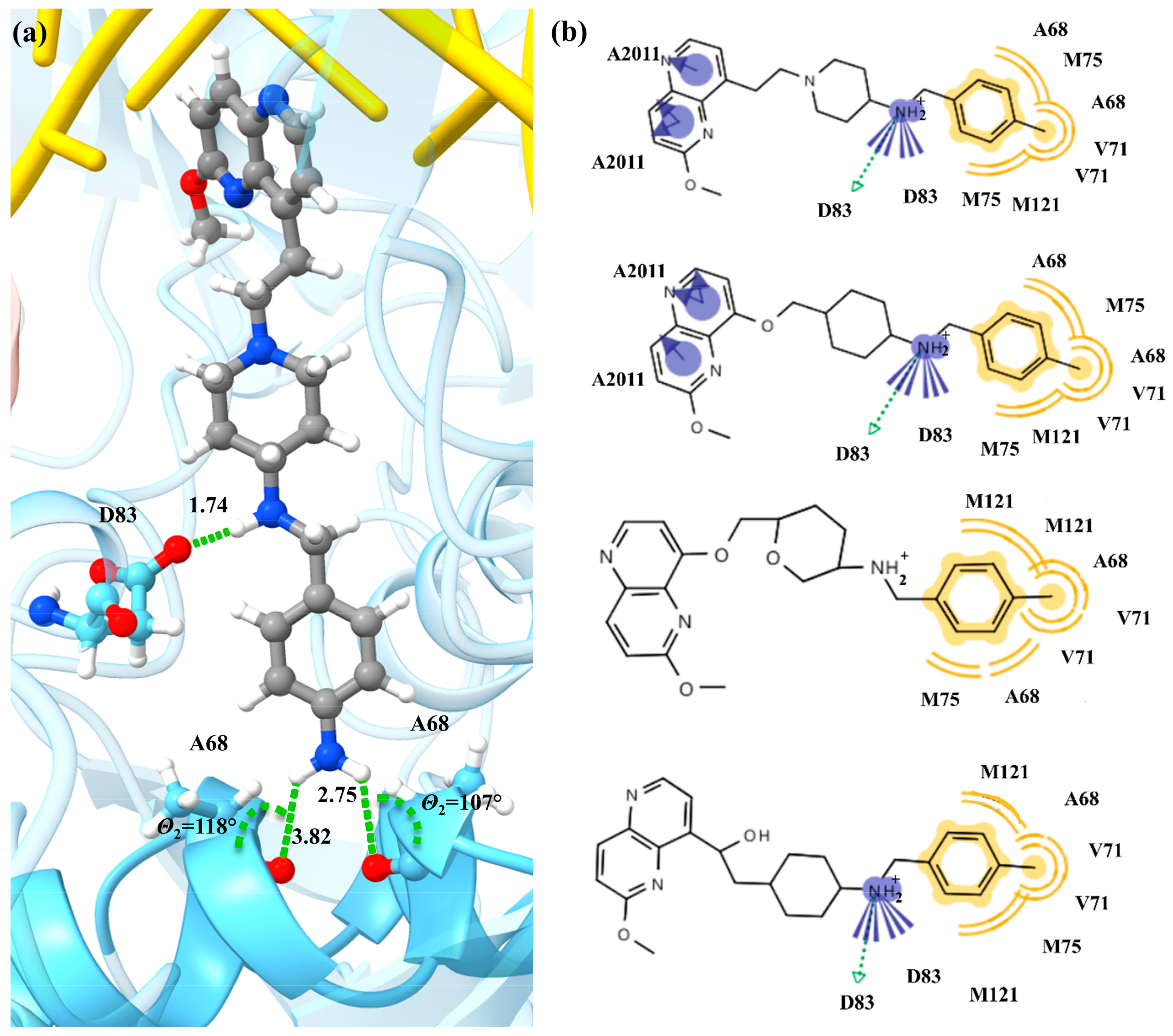
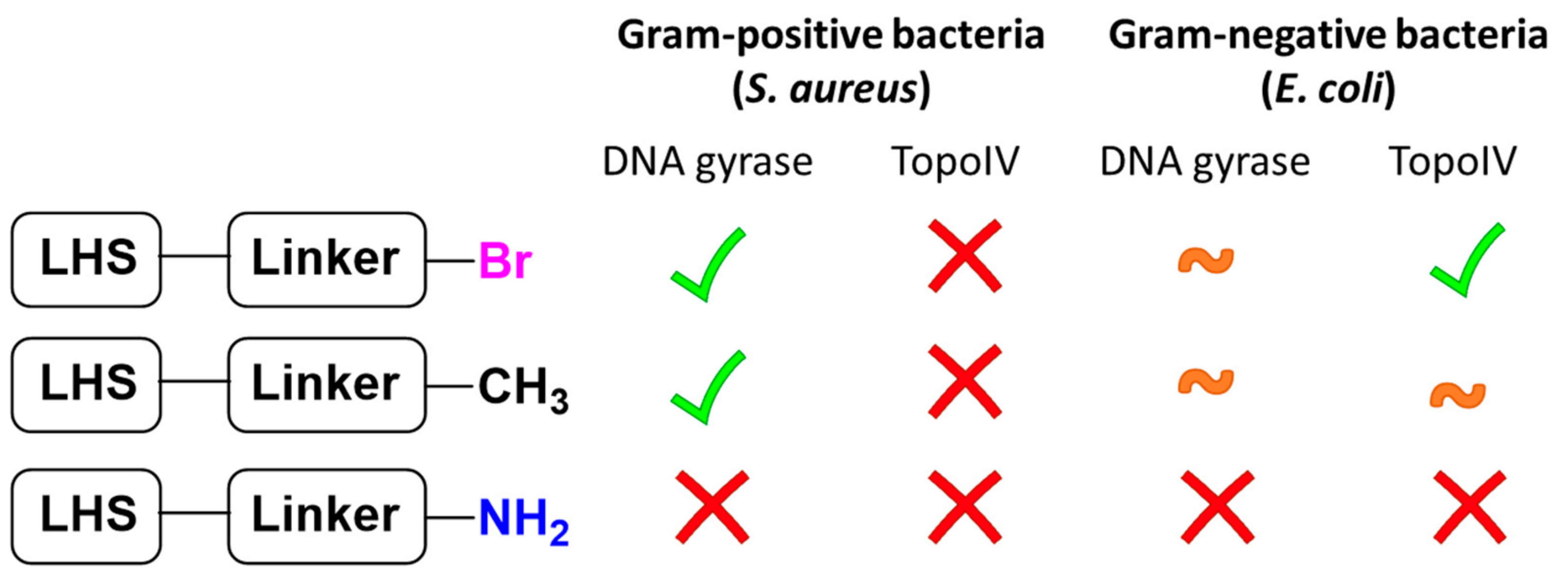
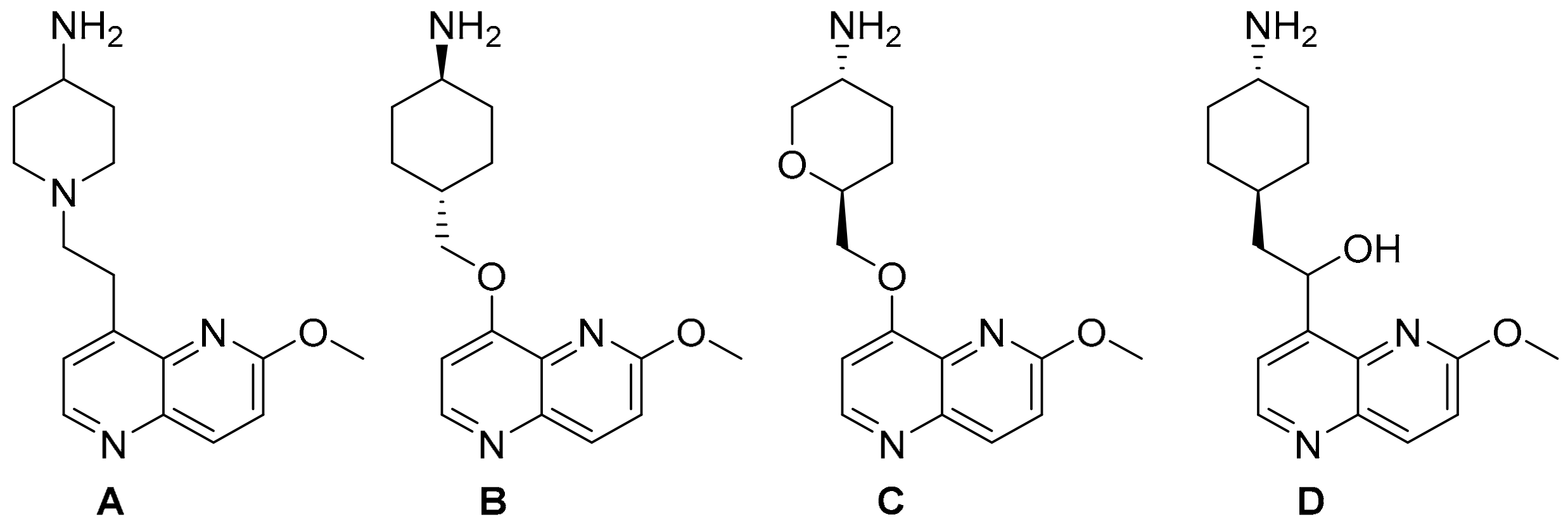
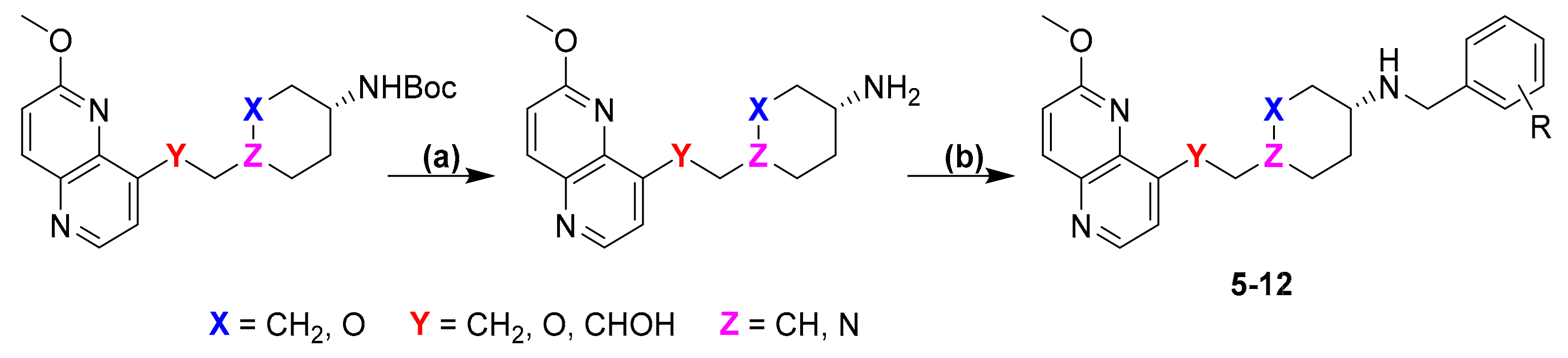
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cmpd | Structure | IC50 (µM) 1 | Human TopoIIα (% RA at 100 µM) 4 | |||
|---|---|---|---|---|---|---|
| DNA Gyrase 2 | topoIV 3 | |||||
| S. aureus | E. coli | S. aureus | E. coli | |||
| 1 * |  | 0.026 ± 0.010 | 0.570 ± 0.06 | 7.22 ± 0.55 | 0.042 ± 0.003 | 98 ± 3 |
| 2 ** |  | 0.372 ± 0.002 | 4.805 ± 0.774 | >100 | 0.495 ± 0.184 | 106 ± 3 |
| 3 ** |  | 0.294 ± 0.100 | 7.742 ± 2.308 | >100 | 0.131 ± 0.040 | 105 ± 1 |
| 4 ** |  | 0.065 ± 0.002 | 1.575 ± 0.404 | >100 | 0.052 ± 0.003 | 106 ± 0 |
| 5 |  | 0.145 ± 0.014 | 1.019 ± 0.005 | 1.226 ± 0.258 | 0.089 ± 0.019 | 103 ± 0 |
| 6 |  | 0.403 ± 0.020 | 1.519 ± 0.361 | >100 | 2.886 ± 0.880 | 101 ± 0 |
| 7 |  | 0.194 ± 0.018 | 1.309 ± 0.348 | >100 | 1.527 ± 0.043 | 101 ± 0 |
| 8 |  | 0.077 ± 0.010 | 1.315 ± 0.460 | 2.502 ± 0.252 | 0.083 ± 0.000 | 100 ± 1 |
| 9 |  | 1.383 ± 0.108 | >100 | >100 | 5.698 ± 0.014 | 100 ± 1 |
| 10 |  | >100 | >100 | >100 | >100 | 99 ± 1 |
| 11 |  | >100 | >100 | >100 | >100 | 100 ± 0 |
| 12 |  | 0.062 ± 0.000 | 0.084 ± 0.020 | 0.345 ± 0.025 | 0.023 ± 0.006 | 102 ± 0 |
| Gepo |  | 0.374 ± 0.019 | 0.244 ± 0.040 | 8.299 ± 0.361 | 0.049 ± 0.003 | ND |
| Cmpd | MIC (µg/mL) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 * | 2 ** | 3 ** | 4 ** | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | Gepo | |
| S. aureus (ATCC 29213) | 0.125 | 0.125 | 0.125 | 0.016 | 0.032 | 1 | 0.5 | 0.063 | 64 | >128 | >128 | 0.032 | 0.125 |
| MRSA (QA-11.7) 1 | 0.5 | 0.25 | 0.5 | 0.016 | 0.016 | 1 | 1 | 0.125 | ND | ND | ND | 0.063 | 0.063 |
| MRSA(QA-12.1) 2 | ND | 0.25 | 0.125 | 0.016 | 0.016 | 2 | 0.5 | 0.063 | ND | ND | ND | 0.032 | 0.125 |
| MRSA (QA-11.2) | ND | 0.5 | 0.25 | 0.031 | 0.032 | 0.5 | 0.25 | 0.032 | ND | ND | ND | 0.063 | 0.5 |
| E. coli (ATCC 25922) | 2 | 64 | 128 | 16 | 2 | >128 | >128 | 8 | >128 | >128 | >128 | 4 | 1 |
| E. coli D22 3 | 0.125 | 8 | 8 | 1 | 1 | 32 | 32 | 1 | >128 | >128 | >128 | 0.25 | 0.125 |
| E. coli N43 4 (CGSC# 5583) | 0.125 | 1 | 8 | 0.25 | 0.125 | 4 | 4 | 1 | 64 | >128 | >128 | 0.063 | 0.016 |
| E. coli ESBL QA-11.3 | ND | 128 | >128 | >128 | 4 | >128 | >128 | 8 | >128 | >128 | >128 | 4 | ND |
| K. pneumoniae | ND | 128 | 128 | 128 | 16 | >128 | >128 | 16 | >128 | >128 | >128 | 16 | 8 |
| P. aeruginosa RDK 184 | 128 | 32 | 128 | 128 | >128 | >128 | >128 | 128 | >128 | >128 | >128 | >128 | 8 |
| E. faecalis RDK 057 | 0.5 | 1 | 2 | 0.5 | 0.125 | 8 | 4 | 0.25 | >128 | >128 | >128 | 0.5 | 2 |
| VRE | 2 | 1 | 1 | 0.032 | 0.032 | 2 | 2 | 0.125 | ND | ND | ND | 0.25 | 1 |
| A. baumannii | ND | 128 | >128 | >128 | 1 | 64 | 64 | 2 | >128 | >128 | >128 | 2 | 8 |
| Cmpd | IC50 [μM] | |
|---|---|---|
| HUVEC | HepG2 | |
| 5 | 36.54 ± 9.82 | 19.99 ± 3.01 |
| 6 | 36.65 ± 7.91 | 22.46 ± 2.70 |
| 7 | >50 | >50 |
| 8 | 33.07 ± 6.13 | 22.28 ± 0.36 |
| 9 | 34.04 ± 16.87 | 26.14 ± 9.52 |
| 10 | >50 | >50 |
| 11 | >50 | >50 |
| 12 | ND | 12.00 ± 1.86 |
| Cmpd | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
|---|---|---|---|---|---|---|---|---|
| Formula | C24H30N4O | C24H29N3O2 | C23H29N5O | C25H31N3O2 | C23H29N5O | C23H28N4O2 | C22H26N4O3 | C24H26BrF3N4O |
| Molecular weight [g/mol] | 390.52 | 391.51 | 393.48 | 405.53 | 391.51 | 392.49 | 394.47 | 523.39 |
| Num. heavy atoms | 29 | 29 | 29 | 30 | 29 | 29 | 29 | 33 |
| Num. arom. heavy atoms | 16 | 16 | 16 | 16 | 16 | 16 | 16 | 16 |
| Fraction Csp3 | 0.42 | 0.42 | 0.39 | 0.44 | 0.39 | 0.39 | 0.36 | 0.42 |
| Num. rotatable bonds | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 8 |
| Num. H bonds acceptors | 5 | 5 | 6 | 5 | 5 | 5 | 6 | 8 |
| Num. H bonds donors | 1 | 1 | 1 | 2 | 2 | 2 | 2 | 1 |
| Molecular Refractivity | 121.60 | 116.31 | 112.59 | 120.75 | 121.04 | 115.75 | 112.03 | 129.33 |
| TPSA [Ų] | 50.28 | 56. 27 | 65.50 | 67.27 | 76.30 | 82.29 | 91.52 | 50.28 |
3. Materials and Methods
3.1. Molecular Docking Calculations
3.2. General Chemical Methods
3.3. Synthesis
3.3.1. Synthesis of Intermediates
3.3.2. General Procedure for Reductive Amination
3.3.3. Synthesis of the Final Compounds
1-(2-(6-Methoxy-1,5-naphthyridin-4-yl)ethyl)-N-(4-methylbenzyl)piperidin-4-amine (5)
(4-(((6-Methoxy-1,5-naphthyridin-4-yl)oxy)methyl)-N-(4-methylbenzyl)cyclohexan-1-amine (6)
6-(((6-Methoxy-1,5-naphthyridin-4-yl)oxy)methyl)-N-(4-methylbenzyl)tetrahydro-2H-pyran-3-amine (7)
1-(6-Methoxy-1,5-naphthyridin-4-yl)-2-(4-((4-methylbenzyl)amino)cyclohexyl)ethan-1-ol (8)
N-(4-Aminobenzyl)-1-(2-(6-methoxy-1,5-naphthyridin-4-yl)ethyl)piperidin-4-amine (9)
4-(((4-(((6-methoxy-1,5-naphthyridin-4-yl)oxy)methyl)cyclohexyl)amino)methyl)aniline (10)
N-(4-Aminobenzyl)-6-(((6-methoxy-1,5-naphthyridin-4-yl)oxy)methyl)tetrahydro-2H-pyran-3-amine (11)
N-(4-Bromo-3-(trifluoromethyl)benzyl)-1-(2-(6-methoxy-1,5-naphthyridin-4-yl)ethyl)piperidin-4-amine (12)
3.4. In Vitro DNA Gyrase and topoIV Inhibition
3.5. Human TopoIIα Selectivity Determination
3.6. Antimicrobial Activity Assay
3.7. Metabolic Activity Assessment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Champoux, J.J. DNA Topoisomerases: Structure, Function, and Mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Bush, N.G.; Evans-Roberts, K.; Maxwell, A. DNA Topoisomerases. EcoSal Plus 2015, 6, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.; Sankhe, K.; Suvarna, V.; Sherje, A.; Patel, K.; Dravyakar, B. DNA Gyrase Inhibitors: Progress and Synthesis of Potent Compounds as Antibacterial Agents. Biomed. Pharmacother. 2018, 103, 923–938. [Google Scholar] [CrossRef] [PubMed]
- Ehmann, D.E.; Lahiri, S.D. Novel Compounds Targeting Bacterial DNA Topoisomerase/DNA Gyrase. Curr. Opin. Pharmacol. 2014, 18, 76–83. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Ennis, K.; Vercaigne, L.; Walkty, A.; Gin, A.S.; Embil, J.; Smith, H.; Hoban, D.J. A Critical Review of the Fluoroquinolones. Drugs 2002, 62, 13–59. [Google Scholar] [CrossRef]
- Andriole, V.T. The Quinolones: Past, Present, and Future. Clin. Infect. Dis. 2005, 41, 113–119. [Google Scholar] [CrossRef]
- Piton, J.; Petrella, S.; Delarue, M.; André-Leroux, G.; Jarlier, V.; Aubry, A.; Mayer, C. Structural Insights into the Quinolone Resistance Mechanism of Mycobacterium tuberculosis DNA Gyrase. PLoS ONE 2010, 5, e12245. [Google Scholar] [CrossRef]
- Jacoby, G.A. Mechanisms of Resistance to Quinolones. Clin. Infect. Dis. 2005, 41, S120–S126. [Google Scholar] [CrossRef]
- Wiener, J.J.M.; Gomez, L.; Venkatesan, H.; Santillán, A.; Allison, B.D.; Schwarz, K.L.; Shinde, S.; Tang, L.; Hack, M.D.; Morrow, B.J.; et al. Tetrahydroindazole Inhibitors of Bacterial Type II Topoisomerases. Part 2: SAR Development and Potency against Multidrug-Resistant Strains. Bioorg. Med. Chem. Lett. 2007, 17, 2718–2722. [Google Scholar] [CrossRef]
- Black, M.T.; Stachyra, T.; Platel, D.; Girard, A.M.; Claudon, M.; Bruneau, J.M.; Miossec, C. Mechanism of Action of the Antibiotic NXL101, a Novel Nonfluoroquinolone Inhibitor of Bacterial Type II Topoisomerases. Antimicrob. Agents Chemother. 2008, 52, 3339–3349. [Google Scholar] [CrossRef]
- Bax, B.D.; Chan, P.F.; Eggleston, D.S.; Fosberry, A.; Gentry, D.R.; Gorrec, F.; Giordano, I.; Hann, M.M.; Hennessy, A.; Hibbs, M.; et al. Type IIA Topoisomerase Inhibition by a New Class of Antibacterial Agents. Nature 2010, 466, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E.G.; Bax, B.; Chan, P.F.; Osheroff, N. Mechanistic and Structural Basis for the Actions of the Antibacterial Gepotidacin against Staphylococcus aureus Gyrase. ACS Infect. Dis. 2019, 5, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Kokot, M.; Anderluh, M.; Hrast, M.; Minovski, N. The Structural Features of Novel Bacterial Topoisomerase Inhibitors That Define Their Activity on Topoisomerase IV. J. Med. Chem. 2022, 65, 6431–6440. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.M.; Gill, C.J.; Wu, J.; Toussaint, N.; Yin, J.; Tsuchiya, T.; Garlisi, C.G.; Kaelin, D.; Meinke, P.T.; Miesel, L.; et al. In Vitro and In Vivo Characterization of the Novel Oxabicyclooctane-Linked Bacterial Topoisomerase Inhibitor AM-8722, a Selective, Potent Inhibitor of Bacterial DNA Gyrase. Antimicrob. Agents Chemother. 2016, 60, 4830–4839. [Google Scholar] [CrossRef]
- Lu, Y.; Papa, J.L.; Nolan, S.; English, A.; Seffernick, J.T.; Shkolnikov, N.; Powell, J.; Lindert, S.; Wozniak, D.J.; Yalowich, J.; et al. Dioxane-Linked Amide Derivatives as Novel Bacterial Topoisomerase Inhibitors against Gram-Positive Staphylococcus aureus. ACS Med. Chem. Lett. 2020, 11, 2446–2454. [Google Scholar] [CrossRef]
- Geng, B.; Comita-Prevoir, J.; Eyermann, C.J.; Reck, F.; Fisher, S. Exploring Left-Hand-Side Substitutions in the Benzoxazinone Series of 4-Amino-Piperidine Bacterial Type IIa Topoisomerase Inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 5432–5435. [Google Scholar] [CrossRef]
- Li, L.; Okumu, A.A.; Nolan, S.; English, A.; Vibhute, S.; Lu, Y.; Hervert-Thomas, K.; Seffernick, J.T.; Azap, L.; Cole, S.L.; et al. 1,3-Dioxane-Linked Bacterial Topoisomerase Inhibitors with Enhanced Antibacterial Activity and Reduced HERG Inhibition. ACS Infect. Dis. 2019, 5, 1115–1128. [Google Scholar] [CrossRef] [PubMed]
- Black, M.T.; Coleman, K. New Inhibitors of Bacterial Topoisomerase GyrA/ParC Subunits. Curr. Opin. Investig. Drugs 2009, 10, 804–810. [Google Scholar]
- Vanscoy, B.D.; Scangarella-oman, N.E.; Fikes, S.; Min, S.; Huang, J.; Ingraham, K.; Bhavnani, S.M.; Conde, H.; Ambrose, G. Relationship between Gepotidacin Exposure and Prevention of On-Therapy Resistance Amplification in a Neisseria gonorrhoeae Hollow-Fiber In Vitro Infection Model. Antimicrob. Agents Chemother. 2020, 64, e00521-20. [Google Scholar] [CrossRef]
- Scangarella-Oman, N.E.; Hossain, M.; Hoover, J.L.; Perry, C.R.; Tiffany, C.; Barth, A.; Dumont, E.F. Dose Selection for Phase III Clinical Evaluation of Gepotidacin (GSK2140944) in the Treatment of Uncomplicated Urinary Tract Infections. Antimicrob. Agents Chemother. 2022, 66, e0149221. [Google Scholar] [CrossRef]
- Kolarič, A.; Germe, T.; Hrast, M.; Stevenson, C.E.M.; Lawson, D.M.; Burton, N.P.; Vörös, J.; Maxwell, A.; Minovski, N.; Anderluh, M. Potent DNA Gyrase Inhibitors Bind Asymmetrically to Their Target Using Symmetrical Bifurcated Halogen Bonds. Nat. Commun. 2021, 12, 150. [Google Scholar] [CrossRef] [PubMed]
- Kolarič, A.; Kokot, M.; Hrast, M.; Weiss, M.; Zdovc, I.; Trontelj, J.; Žakelj, S.; Anderluh, M.; Minovski, N. A Fine-Tuned Lipophilicity/Hydrophilicity Ratio Governs Antibacterial Potency and Selectivity of Bifurcated Halogen. Antibiotics 2021, 10, 862. [Google Scholar] [CrossRef]
- Görbitz, C.H. Hydrogen-bond Distances and Angles in the Structures of Amino Acids and Peptides. Acta Crystallogr. Sect. B 1989, 45, 390–395. [Google Scholar] [CrossRef]
- Kokot, M.; Weiss, M.; Zdovc, I.; Anderluh, M.; Hrast, M.; Minovski, N. Diminishing HERG Cardiotoxic Potential of Aminopiperidine-Naphthyridine Linked NBTI Antibacterials by Structural and Physico-Chemical Optimizations. Bioorg. Chem. 2022, 128, 106087. [Google Scholar] [CrossRef] [PubMed]
- Kokot, M.; Weiss, M.; Zdovc, I.; Hrast, M.; Anderluh, M.; Minovski, N. Structurally Optimized Potent Dual-Targeting NBTI Antibacterials with an Enhanced Bifurcated Halogen-Bonding Propensity. ACS Med. Chem. Lett. 2021, 12, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking Gareth. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved Protein-Ligand Docking Using GOLD. Proteins Struct. Funct. Genet. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- S. aureus Gyrase Supercoiling High Throughput Plate Assay. Available online: https://www.inspiralis.com/assets/TechnicalDocuments/Saureus-Gyrase-Supercoiling-Plate-Assay-Protocol.pdf (accessed on 16 April 2022).
- E. coli Gyrase Supercoiling High Throughput Plate Assay. Available online: https://www.inspiralis.com/assets/TechnicalDocuments/E-coli-Gyrase-Supercoiling-Plate-Assay-Protocol2.pdf (accessed on 16 April 2022).
- S. aureus TopoIV Relaxation High Throughput Plate Assay Introduction. Available online: https://www.inspiralis.com/assets/TechnicalDocuments/Saureus-TopoIV-Relaxation-Plate-Assay-Protocol2.pdf (accessed on 16 April 2022).
- E. coli TopoIV Relaxation High Throughput Plate Assay. Available online: https://www.inspiralis.com/assets/TechnicalDocuments/E-coli-TopoIV-Relaxation-Plate-Assay-Protocol3.pdf (accessed on 16 April 2022).
- Human Topoisomerase I Relaxation High Throughput Plate Assay. Available online: https://www.inspiralis.com/assets/TechnicalDocuments/Human-Topo-II-Relaxation-Plate-Assay-Protocol2.pdf (accessed on 16 April 2022).
- CLSI Document M07-A9; Clinical Laboratory Standards Institute Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Ninth Edition. Clinical Laboratory Standards Institute: Wayne, PA, USA, 2018.
- EUCAST Testing Breakpoint Tables for Interpretation of MICs and Zone Diameters. Available online: https://www.Eucast.Org/Ast_of_Bacteria/ (accessed on 1 April 2022).






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kokot, M.; Novak, D.; Zdovc, I.; Anderluh, M.; Hrast, M.; Minovski, N. Exploring Alternative Pathways to Target Bacterial Type II Topoisomerases Using NBTI Antibacterials: Beyond Halogen-Bonding Interactions. Antibiotics 2023, 12, 930. https://doi.org/10.3390/antibiotics12050930
Kokot M, Novak D, Zdovc I, Anderluh M, Hrast M, Minovski N. Exploring Alternative Pathways to Target Bacterial Type II Topoisomerases Using NBTI Antibacterials: Beyond Halogen-Bonding Interactions. Antibiotics. 2023; 12(5):930. https://doi.org/10.3390/antibiotics12050930
Chicago/Turabian StyleKokot, Maja, Doroteja Novak, Irena Zdovc, Marko Anderluh, Martina Hrast, and Nikola Minovski. 2023. "Exploring Alternative Pathways to Target Bacterial Type II Topoisomerases Using NBTI Antibacterials: Beyond Halogen-Bonding Interactions" Antibiotics 12, no. 5: 930. https://doi.org/10.3390/antibiotics12050930