
Interconnected Set of Enzymes Provide Lysine Biosynthetic Intermediates and Ornithine Derivatives as Key Precursors for the Biosynthesis of Bioactive Secondary Metabolites
Abstract
:
1. Introduction
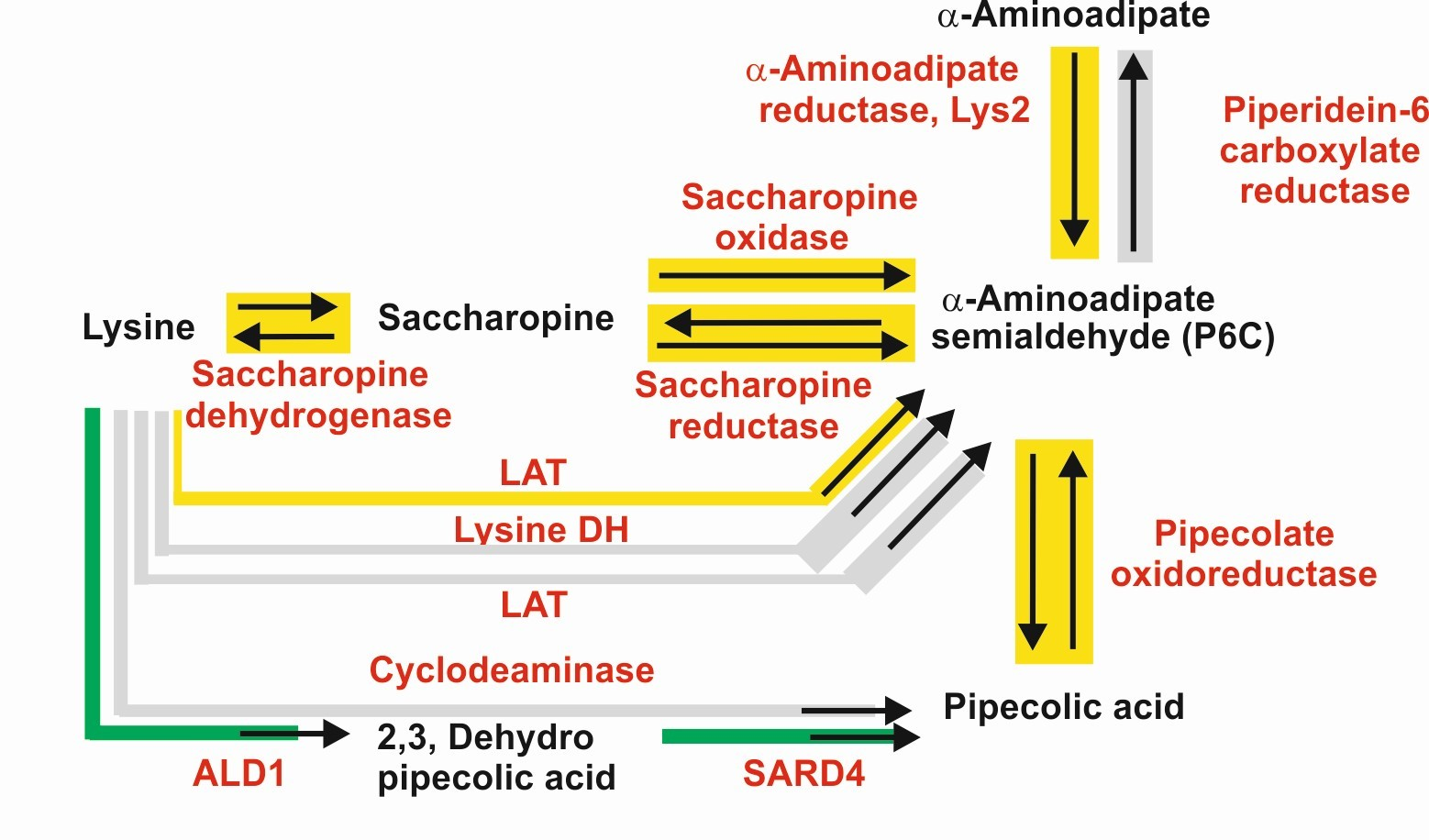
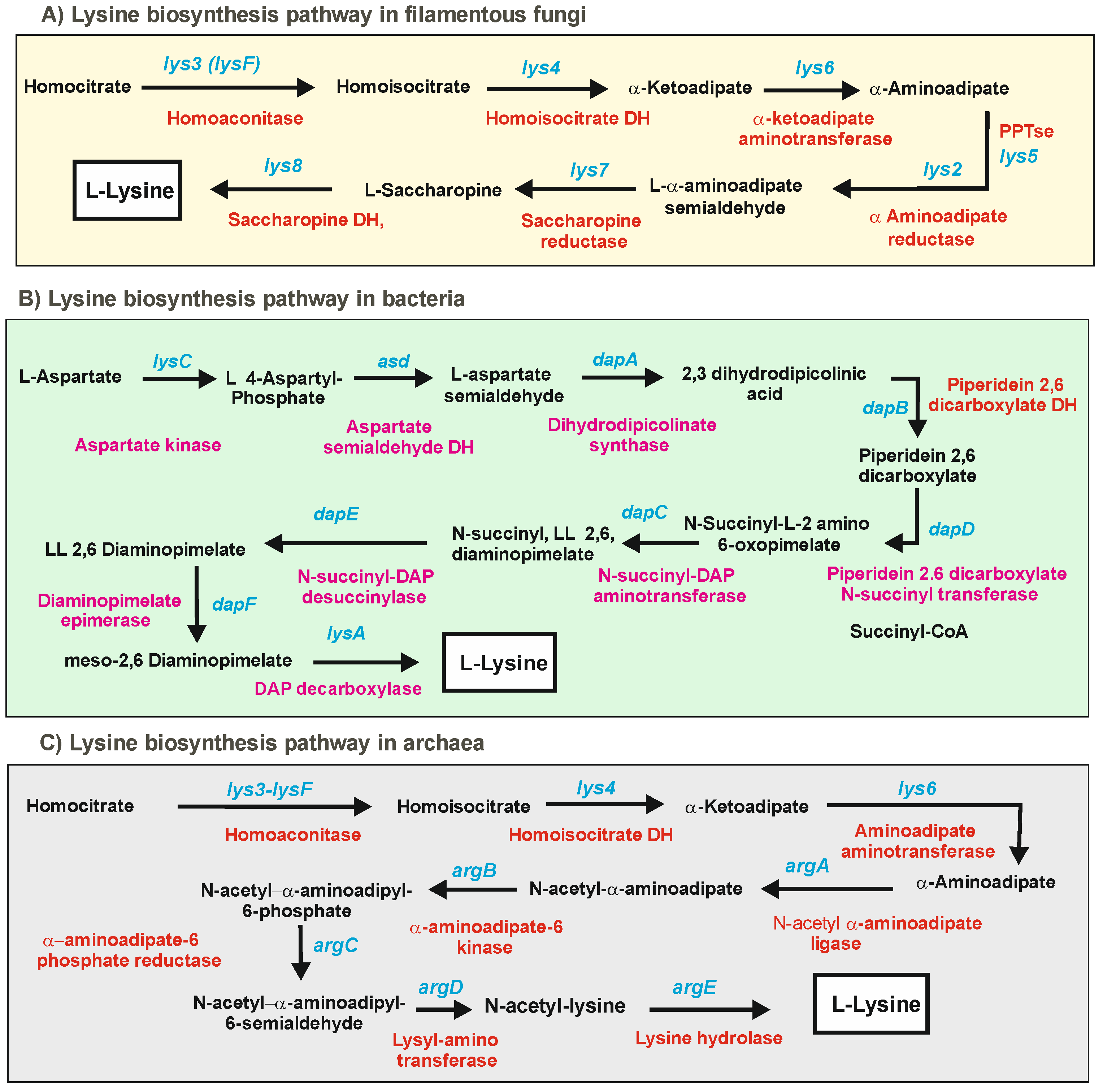
2. Biosynthesis of α-Aminoadipic Acid in Filamentous Fungi and Bacteria
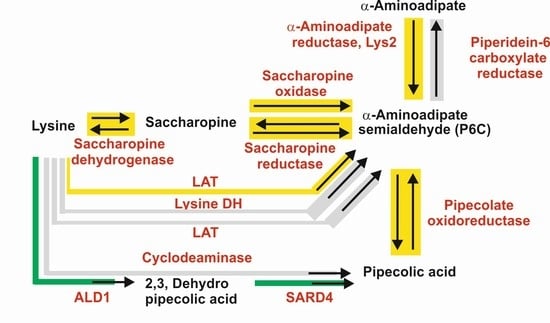
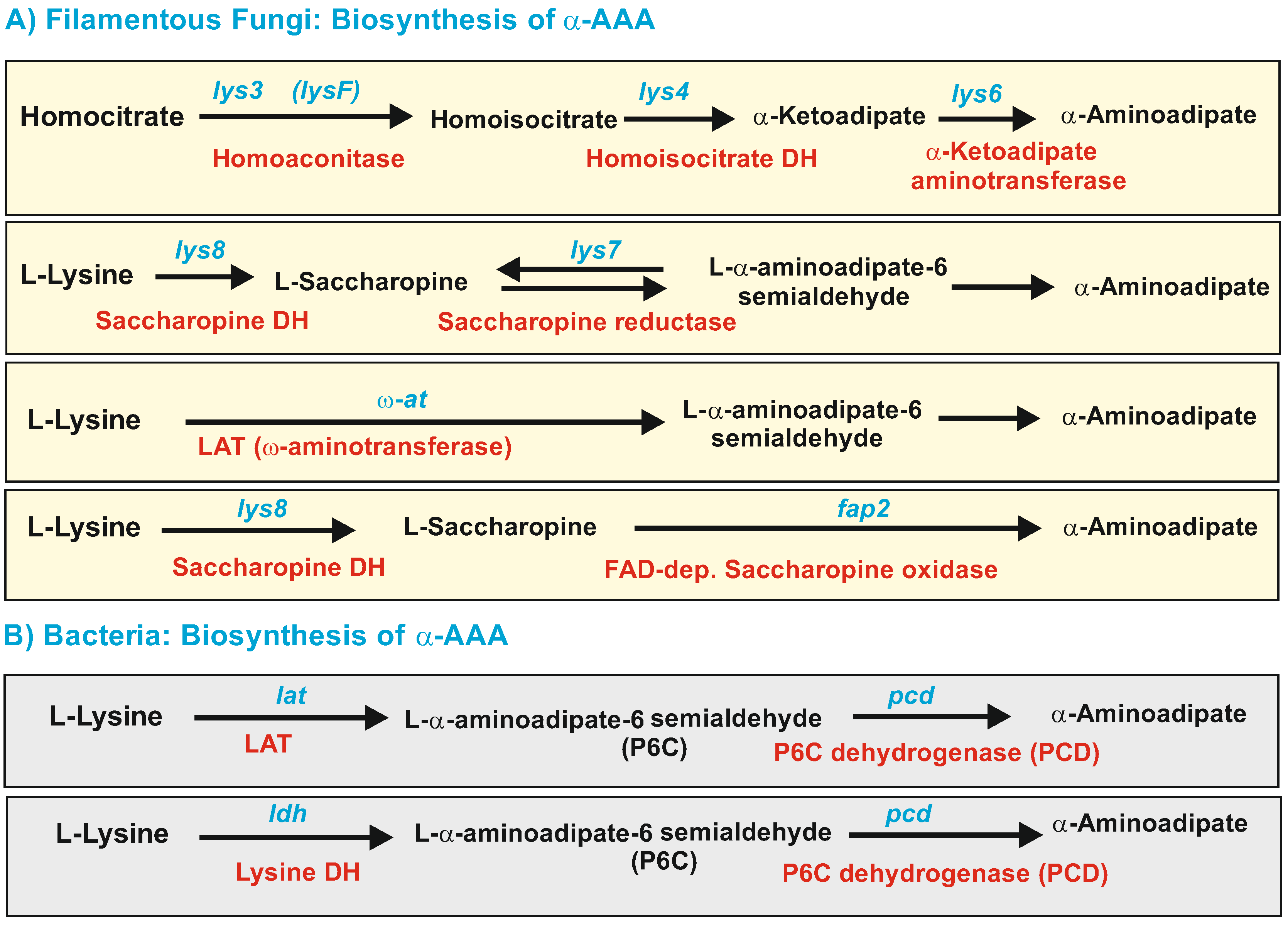
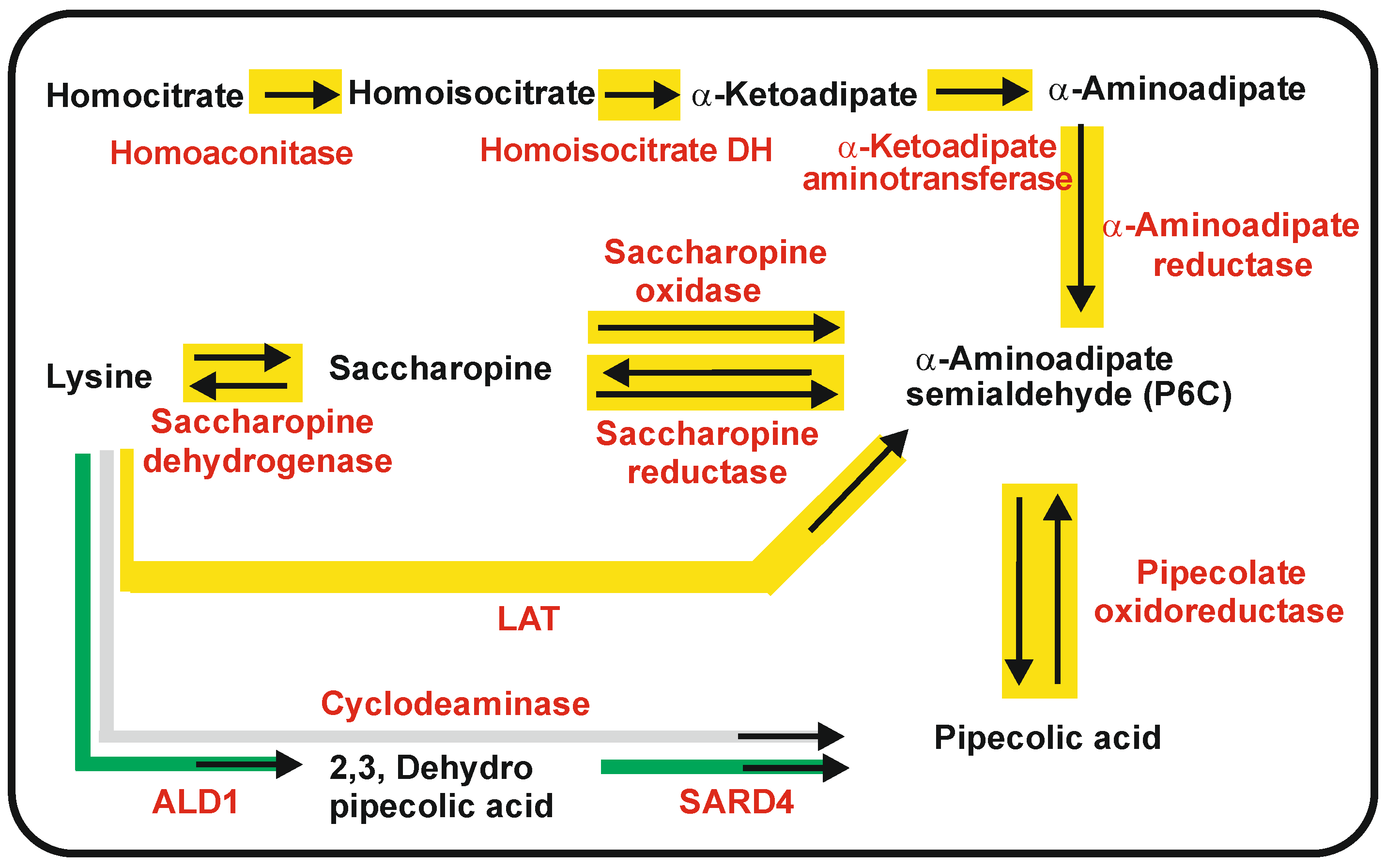
2.1. Biosynthesis of a-AAA and Interconversion of a-AAA and Lysine. Several Different Pathways Converge on the Biosynthesis of α-AAA in Filamentous Fungi (Figure 3)
2.1.1. α-AAA Is Formed in Fungi as an Intermediate of the Lysine Pathway
2.1.2. Five Genes Encode Enzymes Similar to the Saccharopine Reductase and Saccharopine Dehydrogenase in Filamentous Fungi: A Possible Role for These Multiple Enzymes
2.2. Formation of α-AAA by Catabolism of Lysine
2.2.1. LAT-Dependent Biosynthesis of α-AAA in Filamentous Fungi and Bacteria
2.2.2. Lysine 6-Dehydrogenase-Dependent Biosynthesis of α-AAA in Bacteria
3. Pipecolic Acid Biosynthesis in Fungi, Plants, and Bacteria
3.1. Biosynthetic Routes and Enzymes Involved in the Formation of Pipecolic Acid in Fungi: Interconversion of Lysine and Pipecolic Acid
3.2. Biosynthesis of Pipecolic Acid in Plants
3.3. One Step Biosynthesis of Pipecolic Acid in Bacteria: Lysine Cyclodeaminases
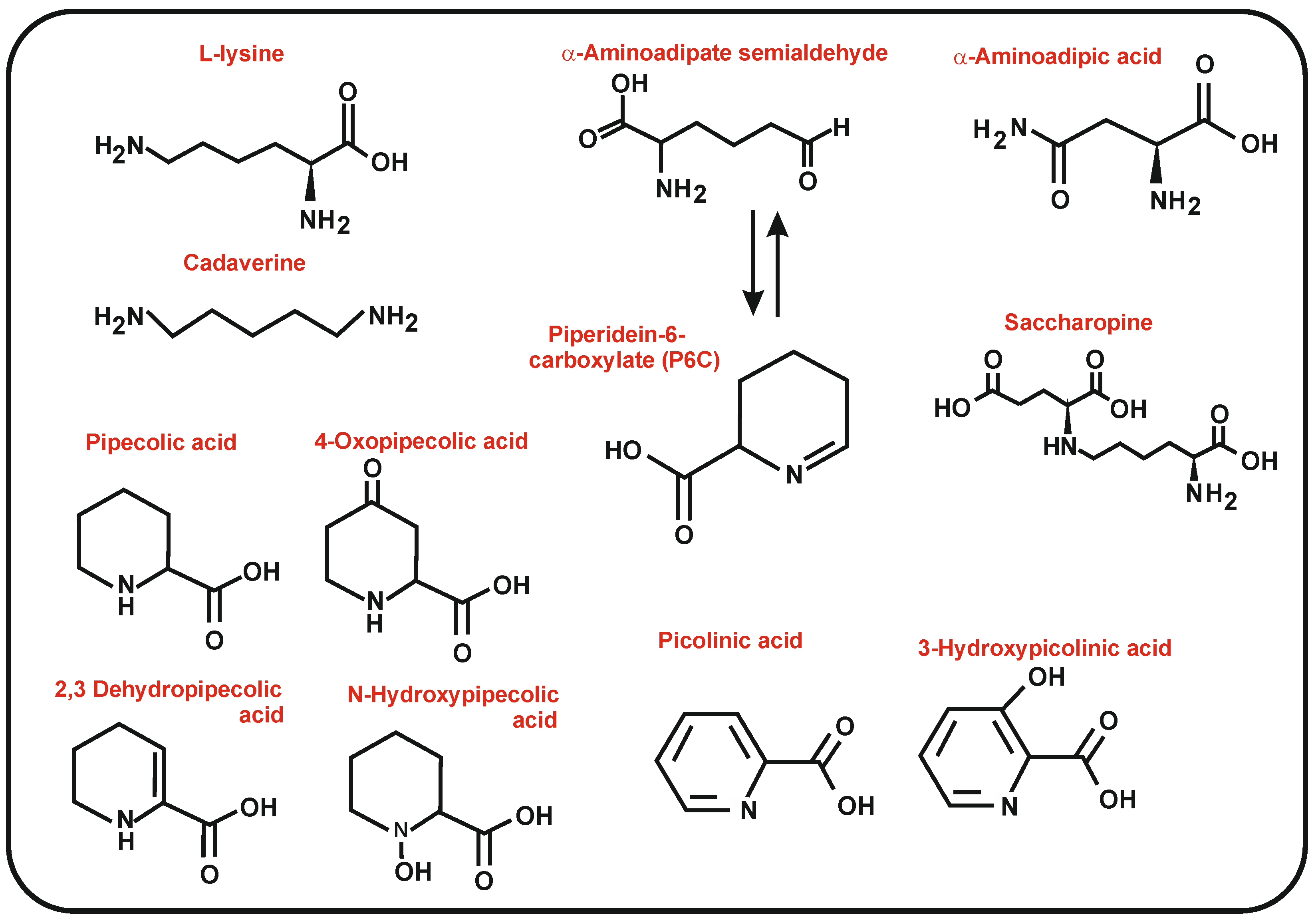
4. 4-Oxopipecolic Acid and 3-Hydroxypicolinic Acid Precursors Related to Pipecolic Acid
4.1. Biosynthesis of 4-Oxo-L-Pipecolic Acid
4.2. Biosynthesis of 3-Hydroxypicolinic Acid
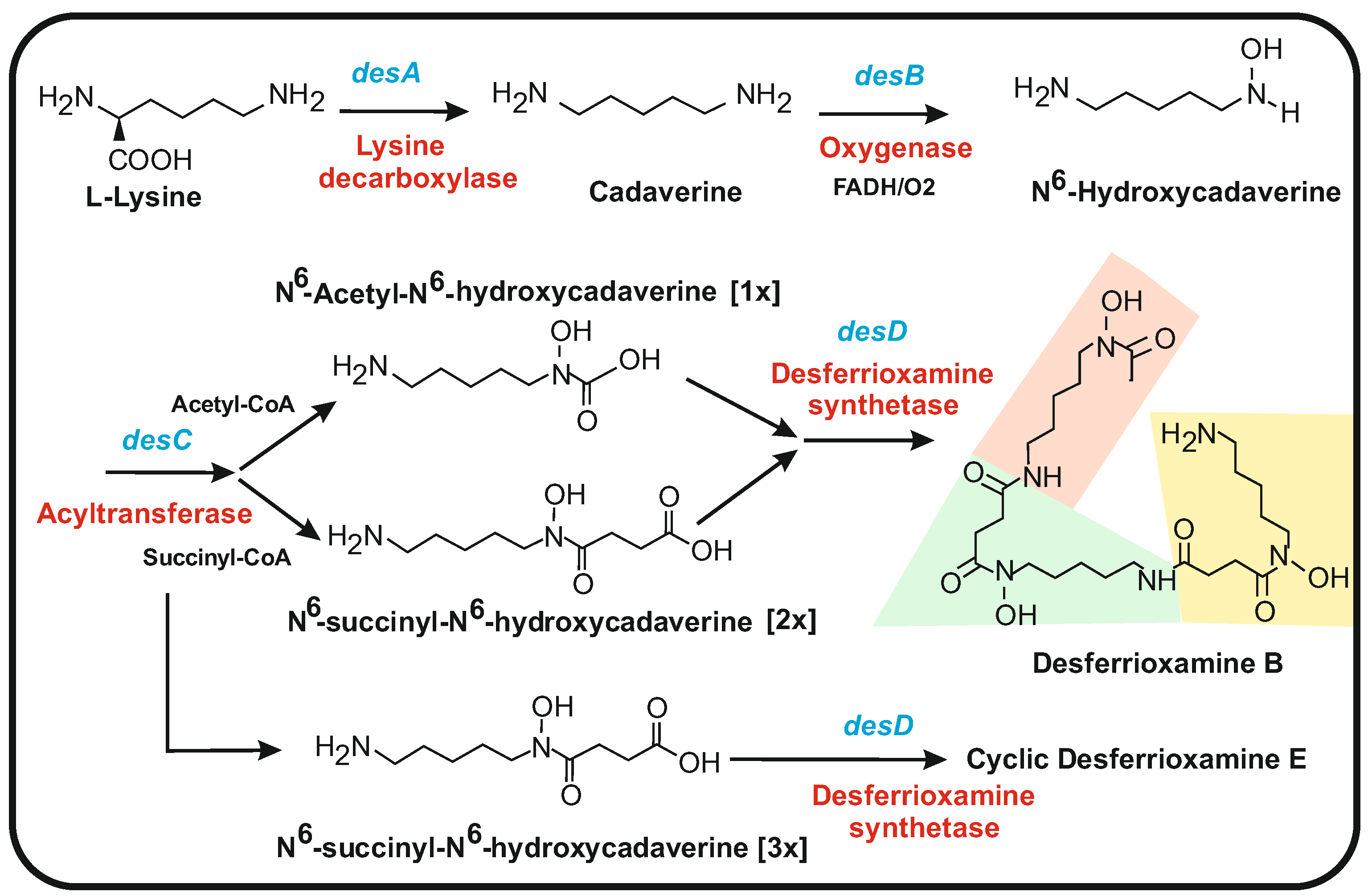
5. Cadaverine and Putrescine Precursors of Desferrioxamine-Type Siderophores
5.1. Biosynthesis of Desferrioxamines from Cadaverine
5.2. L-δ-N-Hydroxyornithine and D-δ-N-Formyl-N-Hydroxyornithine in the Biosynthesis of Bacterial Peptide Siderophores: Coelichelins
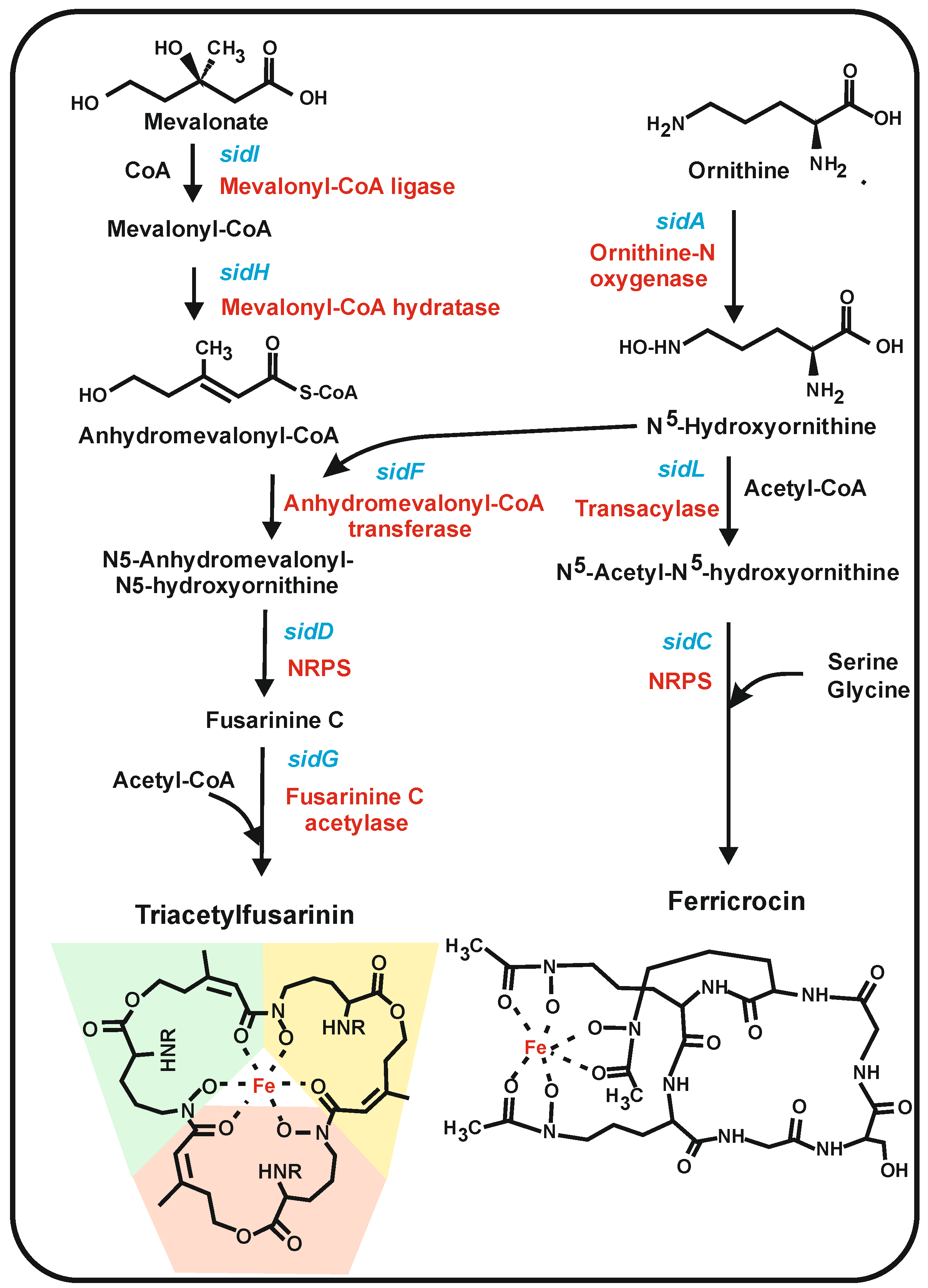
6. Siderophores Derived from Ornithine in Filamentous Fungi
6.1. Fungal Siderophores Synthesized by Condensation of Acylated N5-Hydroxylornithine Units: Fusarinins
6.2. Siderophores Synthesized by NRPSs in Filamentous Fungi: Ferrichromes
6.3. Coprogens
7. Role of Intracellular and Extracellular Siderophores
8. Summary and Future Outlook
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Davies, J. Small molecules: The lexicon of biodiversity. J. Biotechnol. 2007, 129, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Demain, A.L. Valuable Secondary Metabolites from Fungi. In Biosynthesis and Molecular Genetics of Fungal Secondary Metabolites; Martín, J.F., García-Estrada, C., Zeilinger, S., Eds.; Springer: New York, NY, USA, 2014; pp. 1–16. [Google Scholar]
- Zeilinger, S. Fungal Secondary Metabolites in the OMICS Era. In Biosynthesis and Molecular Genetics of Fungal Secondary Metabolites; Zeilinger, S., Martín, J.F., García-Estrada, C., Eds.; Springer: New York, NY, USA, 2014; pp. 1–12. [Google Scholar]
- Keller, N.P. Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol. 2019, 17, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Zabriskie, T.M.; Jackson, M.D. Lysine biosynthesis and metabolism in fungi. Prod. Rep. 2000, 17, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Andi, B.; Qian, J.; West, A.H.; Cook, P.F. The α-Aminoadipate Pathway for Lysine Biosynthesis in Fungi. Cell Biochem. Biophys. 2006, 46, 43–64. [Google Scholar] [CrossRef] [PubMed]
- Kosuge, T.; Hoshino, T. Lysine is synthesized through the α-aminoadipate pathway in Thermus thermophilus. FEMS Microbiol. Lett. 1998, 169, 361–367. [Google Scholar] [CrossRef]
- Kobashi, N.; Nishiyama, M.; Tanokura, M. Aspartate kinase-independent lysine synthesis in an extremely thermophilic bacterium, Thermus thermophilus: Lysine is synthesized via a–aminoadipic acid, not via diaminopimeric acid. J. Bacteriol. 1999, 181, 1713–1718. [Google Scholar] [CrossRef] [Green Version]
- Nishida, H.; Nishiyama, M. What Is Characteristic of Fungal Lysine Synthesis Through the a-Aminoadipate Pathway? J. Mol. Evol. 2000, 51, 299–302. [Google Scholar] [CrossRef]
- Suzuki, T.; Akiyama, N.; Yoshida, A.; Tomita, T.; Lassak, K.; Haurat, M.F.; Okada, T.; Takahashi, K.; Albers, S.-V.; Kuzuyama, T.; et al. Biochemical characterization of archaeal homocitrate synthase from Sulfolobus acidocaldarius. FEBS Lett. 2020, 594, 126–134. [Google Scholar] [CrossRef]
- Yoshida, A.; Tomita, T.; Atomi, H.; Kuzuyama, T.; Nishiyama, M. Lysine biosynthesis of Thermococcus kodakarensis with the capacity to function as an ornithine biosynthetic system. J. Biol. Chem. 2016, 291, 21630–21643. [Google Scholar] [CrossRef] [Green Version]
- Nishida, H.; Nishiyama, M.; Kobashi, N.; Kosuge, T.; Hoshino, T.; Yamane, H. A prokaryotic gene cluster involved in synthesis of lysine through the aminoadipate pathway: A key to the evolution of amino acid biosynthesis. Genome Res. 1999, 9, 1175–1183. [Google Scholar] [CrossRef]
- Miyazaki, T.; Miyazaki, J.; Yamane, H.; Nishiyama, M. alpha-Aminoadipate aminotransferase from an extremely thermophilic bacterium, Thermus thermophilus. Microbiology 2004, 150, 2327–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, J.F.; Liras, P. Insights into the Structure and Molecular Mechanisms of β-Lactam Synthesizing Enzymes in Fungi. In Biotechnology of Microbial Enzymes; Brahmachati, G., Demain, A.L., Adrio, J.L., Eds.; Elsevier: New York, NY, USA, 2016; pp. 215–241. [Google Scholar]
- Martín, J.F.; Alvarez-Alvarez, R.; Liras, P. Penicillin-Binding Proteins, β-Lactamases, and β-Lactamase Inhibitors in β-Lactam-Producing Actinobacteria: Self-Resistance Mechanisms. Int. J. Mol. Sci. 2022, 23, 5662. [Google Scholar] [CrossRef] [PubMed]
- Palmer, D.R.; Balogh, H.; Ma, G.; Zhou, X.; Marko, M.; Kaminskyj, S.G. Synthesis and antifungal properties of compounds which target the alpha-aminoadipate pathway. Pharmazie 2004, 59, 93–98. [Google Scholar]
- Aharonowitz, Y.; Cohen, G.; Martín, J.F. Penicillin and cephalosporin biosynthetic genes: Structure, organization, regulation and evolution. Ann. Rev. Microbiol. 1992, 46, 461–495. [Google Scholar] [CrossRef]
- Urrestarazu, A.; Vissers, S.; Iraqui, I.; Grenson, M. Phenylalanine- and tyrosine-auxotrophic mutants of Saccharomyces cerevisiae impaired in transamination. Mol. Gen. Genet. 1998, 257, 230–237. [Google Scholar] [CrossRef]
- Casqueiro, J.; Gutiérrez, S.; Bañuelos, O.; Fierro, F.; Velasco, J.; Martín, J.F. Characterization of the lys2 gene of Penicillium chrysogenum encoding a-aminoadipic acid reductase. Mol. Gen. Genet. 1998, 259, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Hijarrubia, M.J.; Aparicio, J.F.; Casqueiro, J.; Martín, J.F. Characterization of the lys2 gene of Acremonium chrysogenum encoding a functional α-aminoadipate activating and reducing enzyme. Mol. Gen. Genet. 2001, 264, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Coque, J.J.R.; Liras, P.; Láiz, L.; Martín, J.F. A gene encoding lysine 6-aminotransferase, which forms the b-lactam precursor β-aminoadipic acid, is located in the cluster of cephamycin biosynthetic genes in Nocardia lactamdurans. J. Bacteriol. 1991, 173, 6258–6264. [Google Scholar] [CrossRef] [Green Version]
- Naranjo, L.; Martín de Valmaseda, E.; Bañuelos, O.; López, P.; Riaño, J.; Casqueiro, J.; Martín, J.F. Conversion of Pipecolic Acid into Lysine in Penicillium chrysogenum Requires Pipecolate Oxidase and Saccharopine Reductase: Characterization of the lys7 Gene Encoding Saccharopine Reductase. J. Bacteriol. 2001, 183, 7165–7172. [Google Scholar] [CrossRef] [Green Version]
- Naranjo, L.; Martín de Valmaseda, E.; Casqueiro, J.; Ullán, R.V.; Lamas-Maceiras, M.; Bañuelos, O.; Martín, J.F. Inactivation of the lys7 Gene, Encoding Saccharopine Reductase in Penicillium chrysogenum, Leads to Accumulation of the Secondary Metabolite Precursors Piperideine-6-Carboxylic Acid and Pipecolic Acid from a-Aminoadipic Acid. Appl. Environ. Microbiol. 2004, 70, 1031–1039. [Google Scholar] [CrossRef] [Green Version]
- Johansson, E.; Steffens, J.J.; Emptage, M.; Lindqvist, Y.; Schneider, G. Cloning, expression, purification and crystallization of saccharopine reductase from Magnaporthe grisea. Acta Crystallogr. D Biol. Crystallogr. 2000, 56, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Mast, Y.; Weber, T.; Gölz, M.; Ort-Winklbauer, R.; Gondran, A.; Wohlleben, W.; Schinko, E. Characterization of the ‘pristinamycin supercluster’ of Streptomyces pristinaespiralis. Microbial Biotechnol. 2011, 4, 192–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molnar, I.; Aparicio, J.F.; Haydock, S.F.; Khaw, L.E.; Schwecke, T.; Konig, A.; Staunton, J.; Leadlay, P.F. Organisation of the biosynthetic gene cluster for rapamycin in Streptomyces hygroscopicus: Analysis of genes flanking the polyketide synthase. Gene 1996, 169, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Evans, S.A.; Wilkes, M.B.; Bhattacharjee, J.K.J. Novel posttranslational activation of the LYS2-encoded alpha-aminoadipate reductase for biosynthesis of lysine and site-directed mutational analysis of conserved amino acid residues in the activation domain of Candida albicans. J. Bacteriol. 2001, 183, 7120–7125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crecy-Laggard, V.; Blanc, V.; Gil, P.; Naudin, L.; Lorenzon, S.; Famechon, A.; Bamas-Jaques, N.; Crouzet, J.; Thibaut, D. Pristinamycin I Biosynthesis in Streptomyces pristinaespiralis: Molecular Characterization of the First Two Structural Peptide Synthetase Genes. J. Bacteriol. 1997, 179, 705–713. [Google Scholar] [CrossRef] [Green Version]
- Namwat, W.; Kamiokaa, Y.; Kinoshita, H.; Yamada, Y.; Nihira, T. Characterization of virginiamycin S biosynthetic genes from Streptomyces virginiae. Gene 2002, 286, 283–290. [Google Scholar] [CrossRef]
- Tunca, S.; Barreiro, C.; Sola-Landa, A.; Coque, J.J.R.; Martín, J.F. Transcriptional regulation of the desferrioxamine gene cluster of Streptomyces coelicolor is mediated by binding of DmdR1 to an iron box in the promoter of the desA gene. FEBS J. 2007, 274, 1110–1122. [Google Scholar] [CrossRef]
- Lautru, S.; Deeth, R.J.; Bailey, L.M.; Challis, G.L. Discovery of a new peptide natural product by Streptomyces coelicolor genome mining. Nat. Chem. Biol. 2005, 1, 265–269. [Google Scholar] [CrossRef]
- Barona-Gómez, F.; Wong, U.; Giannakopulos, A.E.; Derrick, P.J.; Challis, G.L. Identification of a cluster of genes that directs desferrioxamine biosynthesis in Streptomyces coelicolor M145. J. Am. Chem. Soc. 2004, 126, 16282–16283. [Google Scholar] [CrossRef]
- Barona-Gómez, F.; Lautru, S.; Francou, F.X.; Leblond, P.; Pernodet, J.L.; Challis, G.L. Multiple biosynthetic and uptake systems mediate siderophore-dependent iron acquisition in Streptomyces coelicolor A3(2) and Streptomyces ambofaciens ATCC 23877. Microbiology 2006, 152, 3355–3366. [Google Scholar] [CrossRef] [Green Version]
- Yasmin, S.; Alcazar-Fuoli, L.; Grundlinger, M.; Puempel, T.; Cairns, T.; Blatzer, M.; Lopez, J.F.; Grimalt, J.O.; Bignell, E.; Haas, H. Mevalonate governs interdependency of ergosterol and siderophore biosyntheses in the fungal pathogen Aspergillus fumigatus. Proc. Natl. Acad. Sci. USA 2011, 109, 497–504. [Google Scholar] [CrossRef]
- Eisendle, M.; Oberegger, H.; Zadra, I.; Haas, H. The siderophore system is essential for viability of Aspergillus nidulans: Functional analysis of two genes encoding l-ornithine N5-monooxygenase (sidA) and a non-ribosomal peptide synthetase (sidC). Mol. Microbiol. 2003, 49, 359–375. [Google Scholar] [CrossRef] [PubMed]
- Schrettl, M.; Bignell, E.; Kragl, C.; Joechl, C.; Rogers, T.; Arst, H.N., Jr.; Haynes, K.; Haas, H. Siderophore biosynthesis but not reductive iron assimilation is essential for Aspergillus fumigatus virulence. J. Exp. Med. 2004, 200, 1213–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olucha, J.; Meneely, K.M.; Chilton, A.S.; Lamb, A.L. Two structures of an N-hydroxylating flavoprotein monooxygenase: Ornithine hydroxylase from Pseudomonas aeruginosa. J. Biol. Chem. 2011, 286, 31789–31798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blatzer, M.; Schrettl, M.; Sarg, B.; Lindner, H.H.; Pfaller, K.; Haas, H. SidL, an Aspergillus fumigatus transacetylase involved in biosynthesis of the siderophores ferricrocin and hydroxyferricrocin. Appl. Environ. Microbiol. 2011, 77, 4959–4966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalal, M.A.; van der Helm, D. Siderophores of highly phytopathogenic Alternaria longipes. Structures of hydroxycoprogens. Biol. Met. 1989, 2, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Olney, W.; Labruyere, J.; Collins, J.F.; Curry, K. D-Aminophosphonovalerate is 100-fold more powerful than D-α-aminoadipate in blocking N-methylaspartate neurotoxicity. Brain Res. 1981, 221, 207–208. [Google Scholar] [CrossRef]
- Sadiq, A.; Sewald, N. 6-Alkynyl- and 6-aryl-substituted (R)-pipecolic acid derivatives. Org. Lett. 2013, 15, 2720–2722. [Google Scholar] [CrossRef]
- Buchli, R.; Alberati-Giani, D.; Malherbe, P.; Köhler, C.; Broger, C.; Cesura, A.M. Cloning and functional expression of a soluble form of kynurenine/alpha-aminoadipate aminotransferase from rat kidney. J. Biol. Chem. 1995, 270, 29330–29335. [Google Scholar] [CrossRef] [Green Version]
- Ehmann, D.E.; Gehring, A.M.; Walsh, C.T. Lysine Biosynthesis in Saccharomyces cerevisiae: Mechanism of R-Aminoadipate Reductase (Lys2) Involves Posttranslational Phosphopantetheinylation by Lys5. Biochemistry 1999, 38, 6171–6177. [Google Scholar] [CrossRef]
- Keszenman-Pereyra, D.; Lawrence, S.; Twfieg, M.-E.; Price, J.; Turner, G. The npgA/ cfwA gene encodes a putative 4′-phosphopantetheinyl transferase which is essential for penicillin biosynthesis in Aspergillus nidulans. Curr. Genet. 2003, 43, 186–190. [Google Scholar] [CrossRef]
- Márquez-Fernández, O.; Trigos, A.; Ramos-Balderas, J.L.; Viniegra-Gonzalez, G.; Deising, H.B.; Aguirre, J. Phosphopantetheinyl transferase CfwA/NpgA is required for Aspergillus nidulans secondary metabolism and asexual development. Eukaryot. Cell 2007, 6, 710–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Estrada, C.; Ullán, R.V.; Velasco-Conde, T.; Godio, R.P.; Teijeira, F.; Vaca, I.; Feltrer, R.; Kosalková, K.; Mauriz, E.; Martín, J.F. Post-translational enzyme modification by the phosphopantetheinyl transferase is required for lysine and penicillin biosynthesis but not for roquefortine or fatty acid formation in Penicillium chrysogenum. Biochem. J. 2008, 415, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, J.F.; Liras, P.; Sánchez, S. Modulation of Gene Expression in Actinobacteria by Translational Modification of Transcriptional Factors and Secondary Metabolite Biosynthetic Enzymes. Front. Microbiol. 2021, 12, 630694. [Google Scholar] [CrossRef]
- Jaklitsch, W.M.; Kubicek, C.P. Homocitrate synthase from Penicillium chrysogenum. Localization, purification of the cytosolic isoenzyme, and sensitivity to lysine. Biochem. J. 1990, 269, 247–253. [Google Scholar] [CrossRef] [Green Version]
- Bañuelos, O.; Casqueiro, J.; Fierro, F.; Hijarrubia, M.-J.; Gutiérrez, S.; Martín, J.F. Characterization and lysine control of expression of the lys1 gene of Penicillium chrysogenum encoding homocitrate synthase. Gene 1999, 226, 51–59. [Google Scholar] [CrossRef]
- Bañuelos, O.; Casqueiro, J.; Steidl, S.; Gutiérrez, S.; Brakhage, A.; Martín, J.F. Subcellular localization of the homocitrate synthase in Penicillium chrysogenum. Mol. Gen. Genet. 2002, 266, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Johansson, E.; Steffens, J.J.; Lindqvist, Y.; Schneider, G. Crystal structure of saccharopine reductase from Magnaporthe grisea, an enzyme of the alpha-aminoadipate pathway of lysine biosynthesis. Struct. Fold Des. 2000, 8, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Specht, T.; Dahlmann, T.A.; Zadra, I.; Kurnsteiner, H.; Kuck, U. Complete Sequencing and Chromosome-Scale Genome Assembly of the Industrial Progenitor Strain P2niaD18 from the Penicillin Producer Penicillium chrysogenum. Genome Announ. 2014, 2, e00577-14. [Google Scholar] [CrossRef] [Green Version]
- van den Berg, M.A.; Albang, R.; Albermann, K.; Badger, J.H.; Daran, J.M.; Driessen, A.J.; García-Estrada, C.; Fedorova, N.D.; Harris, D.M.; Heijne, W.H.; et al. Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum. Nat. Biotechnol. 2008, 26, 1161–1168. [Google Scholar] [CrossRef] [Green Version]
- Fierro, F.; García-Estrada, C.; Castillo, N.I.; Rodríguez, R.; Velasco-Conde, T.; Martín, J.F. Transcriptional and bioinformatic analysis of the 56.8kb DNA region amplified in tandem repeats containing the penicillin gene cluster in Penicillium chrysogenum. Fungal Genet. Biol. 2006, 43, 618–629. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, M.A.; Westerlaken, I.; Leeflang, C.; Kerkman, R.; Bovenberg, R.A. Functional characterization of the penicillin biosynthetic gene cluster of Penicillium chrysogenum Wis 54-1255. Fungal Genet. Biol. 2007, 44, 830–844. [Google Scholar] [CrossRef] [PubMed]
- Martín, J.F.; Liras, P. Transfer of Secondary Metabolite Gene Clusters: Assembly and Reorganization of the β-Lactam Gene Cluster from Bacteria to Fungi and Arthropods. In Horizontal Gene Transfer: Breaking Borders Between Living Kingdoms; Villa, T.G., Viñas, M., Eds.; Springer Nature: Berlin, Germany, 2019; pp. 337–361. [Google Scholar]
- Fierro, F.; Gutiérrez, S.; Díez, B.; Martín, J.F. Resolution of four chromosomes in penicillin-producing filamentous fungi: The penicillin gene cluster is located on chromosome II (9.6 Mb) in Penicillium notatum and chromosome I (10.4 Mb) in Penicillium chrysogenum. Mol. Gen. Genet. 1993, 241, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Esmahan, C.; Alvarez, E.; Montenegro, E.; Martín, J.F. Catabolism of lysine in Penicillium chrysogenum leads to formation of 2-aminoadipic acid, a precursor of penicillin biosynthesis. Appl. Environ. Microbiol. 1994, 60, 1705–1710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín de Valmaseda, E.M.; Campoy, S.; Naranjo, L.; Casqueiro, J.; Martín, J.F. Lysine is catabolized to 2-aminoadipic acid in Penicillium chrysogenum by an omega-aminotransferase and to saccharopine by a lysine 2-ketoglutarate reductase. Characterization of the omega-aminotransferase. Mol. Genet. Genom. 2005, 274, 272–282. [Google Scholar] [CrossRef]
- Madduri, K.; Stuttard, C.; Vining, L.C. Cloning and location of a gene governing lysine I-aminotransferase, an enzyme initiating b-lactam biosynthesis in Streptomyces spp. J. Bacteriol. 1991, 173, 985–988. [Google Scholar] [CrossRef] [Green Version]
- Barbe, V.; Bouzon, M.; Mangenot, S.; Badet, B.; Poulain, J.; Segurens, B.; Vallenet, D.; Marliere, P.; Weissenbach, J. Complete genome sequence of Streptomyces cattleya NRRL 8057, a producer of antibiotics and fluorometabolites. J. Bacteriol. 2011, 193, 5055–5056. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Llarena, J.P.; Rodríguez-García, A.; Enguita, F.J.; Martín, J.F.; Liras, P. The pcd gene encoding piperideine-6-carboxylate dehydrogenase involved in biosynthesis of α-aminoadipic acid is located in the cephamycin cluster of Streptomyces clavuligerus. J. Bacteriol. 1998, 180, 4753–4756. [Google Scholar] [CrossRef] [Green Version]
- de la Fuente, J.L.; Rumbero, A.; Martín, J.F.; Liras, P. Δ-1-Piperideine-6-carboxylate dehydrogenase, a new enzyme that forms α-aminoadipate in Streptomyces clavuligerus and other cephamycin C-producing actinomycetes. Biochem. J. 1997, 327, 59–64. [Google Scholar] [CrossRef]
- Fujii, T.; Narita, T.; Agematu, H.; Agata, N.; Isshiki, K. Characterization of L-lysine 6-aminotransferase and its structural gene from Flavobacterium lutescens IFO3084. J. Biochem. 2000, 128, 391–397. [Google Scholar] [CrossRef]
- Fujii, T.; Narita, T.; Agematu, H.; Agata, N.; Isshiki, K. Cloning and characterization of pcd encoding delta’-piperideine-6-carboxylate dehydrogenase from Flavobacterium lutescens IFO3084. J. Biochem. 2000, 128, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Hasse, D.; Hülsemann, J.; Carlsson, G.H.; Valegård, K.; Andersson, I. Structure and mechanism of piperideine-6-carboxylate dehydrogenase from Streptomyces clavuligerus. Acta Crystallogr. D Struct. Biol. 2019, 75, 1107–1118. [Google Scholar] [CrossRef]
- Sohng, Y.-S.; Nam, S.-H.; Ryu, D.D.Y. Biosynthetic Pathway of Cephabacins in Lysobacter lactamgenus: Molecular and Biochemical Characterization of the Upstream Region of the Gene Clusters for Engineering of Novel Antibiotics. Metab. Eng. 2001, 3, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Hijarrubia, M.J.; Aparicio, J.F.; Martín, J.F. Domain structure characterization of the mulfunctional α-aminoadipate reductase from Penicillium chrysogenum by limited proteolysis. J. Biol. Chem. 2003, 278, 8250–8256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misono, H.; Hashimoto, H.; Uehigashi, H.; Nagata, S.; Nagasaki, S.J. Properties of L-Lysine e-Dehydrogenase from Agrobacterium tumefaciens. J. Biochem. 1989, 105, 1002–1008. [Google Scholar] [CrossRef]
- Misono, H.; Yoshimura, T.; Nagasaki, S.; Soda, K. Stereospecific Abstraction of e-pro-R-Hydrogen of L-Lysine by L-Lysine-Dehydrogenase from Agrobacterium tumefaciens. J. Biochem. 1990, 107, 169–172. [Google Scholar] [CrossRef]
- Gómez, D.; Lucas-Elio, P.; Sanchez-Amat, A.; Solano, F. A novel type of lysine oxidase: L-Lysine-epsilon-oxidase. Biochim. Biophys. Acta 2006, 1764, 1577–1585. [Google Scholar] [CrossRef] [Green Version]
- Campillo-Brocal, J.C.; Lucas-Elío, P.; Sánchez-Amat, A. Distribution in Different Organisms of Amino Acid Oxidases with FAD or a Quinone as Cofactor and Their Role as Antimicrobial Proteins in Marine Bacteria. Drugs 2015, 13, 7403–7418. [Google Scholar] [CrossRef] [Green Version]
- Sim, K.L.; Perry, D. Analysis of swainsonine and its early metabolic precursors in cultures of Metarhizium anisopliae. Glycoconj. J. 1997, 14, 661–668. [Google Scholar] [CrossRef]
- Wickwire, B.M.; Harris, C.M.; Harris, T.M.; Broquist, H.P. Pipecolic acid biosynthesis in Rhizoctonia leguminicola. I. The lysine, saccharopine-piperideine-6-carboxylic acid pathway. J. Biol. Chem. 1990, 265, 14742–14747. [Google Scholar] [CrossRef]
- Wickwire, B.M.; Wagner, C.; Broquist, H.P. Pipecolic acid biosynthesis in Rhizoctonia leguminicola. II. Saccharopine oxidase: A unique flavine enzyme involved in pipecolic acid biosynthesis. J. Biol. Chem. 1990, 265, 14748–14753. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Dawe, A.L.; Creamer, R. Potential role for saccharopine reductase in swainsonine metabolism in endophytic fungus, Undifilum oxytropis. Fungal Biol. 2012, 116, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Müller, C.; Nolden, S.; Gebhardt, P.; Heinzelmann, E.; Lange, C.; Puk, O.; Welzel, K.; Wohlleben, W.; Schwartz, D. Sequencing and analysis of the biosynthetic gene cluster of the lipopeptide antibiotic Friulimicin in Actinoplanes friuliensis. Antimicrob. Agents Chemother. 2007, 51, 1028–1037. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Haltli, B.; Feng, X.; Cai, P.; Summers, M.J.; He, M. Investigation of the biosynthesis of the pipecolate moiety of neuroprotective polyketide meridamycin. J. Antibiot. 2011, 64, 533–538. [Google Scholar] [CrossRef] [Green Version]
- Gatto, G.J.; Boyne, M.T.; Kelleher, N.L.; Walsh, C.T. Biosynthesis of pipecolic acid by RapL, a lysine cyclodeaminase encoded in the rapamycin gene cluster. J. Am. Chem. Soc. 2006, 128, 3838–3847. [Google Scholar] [CrossRef]
- Turło, J.; Gajzlerska, W.; Klimaszewska, M.; Król, M.; Dawidowski, M.; Gutkowska, B. Enhancement of tacrolimus productivity in Streptomyces tsukubaensis by the use of novel precursors for biosynthesis. Enzyme Microb. Technol. 2012, 51, 388–395. [Google Scholar] [CrossRef]
- Bis, D.M.; Ban, Y.H.; James, E.D.; Alqahtani, N.; Viswanathan, R.; Lane, A.L. Characterization of the nocardiopsin biosynthetic gene cluster reveals similarities to and differences from the rapamycin and FK-506 pathways. Chembiochem 2015, 16, 990–997. [Google Scholar] [CrossRef]
- Steinmetz, H.; Glaser, N.; Herdtweck, E.; Sasse, F.; Reichenbach, H.; Höfle, G. Isolation, crystal and solution structure determination, and biosynthesis of tubulysins--powerful inhibitors of tubulin polymerization from myxobacteria. Angew Chem. Int. Ed. Engl. 2004, 43, 4888–4892. [Google Scholar] [CrossRef]
- He, M. Pipecolic acid in microbes: Biosynthetic routes and enzymes. J. Ind. Microbiol. Biotechnol. 2006, 33, 401–407. [Google Scholar] [CrossRef]
- Imazaki, A.; Tanaka, A.; Harimoto, Y.; Yamamoto, M.; Akimitsu, K.; Park, P.; Tsuge, T. Contribution of peroxisomes to secondary metabolism and pathogenicity in the fungal plant pathogen Alternaria alternata, Eukaryot. Cells 2010, 9, 682–694. [Google Scholar]
- Martín, J.F.; Ullán, R.V.; García-Estrada, C. Role of peroxisomes in the biosynthesis and secretion of b-lactams and other secondary metabolites. J. Ind. Microbiol. Biotechnol. 2012, 39, 367–382. [Google Scholar] [CrossRef]
- Fujii, T.; Mukaihara, M.; Agematu, H.; Tsunekawa, H. Biotransformation of L-lysine to L-pipecolic acid catalyzed by L-lysine 6-aminotransferase and pyrroline-5-carboxylate reductase. Biosci. Biotechnol. Biochem. 2002, 66, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Aspen, A.J.; Meister, A. Conversion of a-aminoadipic acid to L-pipecolic acid by Aspergillus nidulans. Biochemistry 1962, 1, 606. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.Q.; Dong, X. Systemic acquired resistance: Turning local infection into global defense. Annu. Rev. Plant Biol. 2013, 6, 839–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruner, K.; Griebel, T.; Navarova, H.; Attaran, E.; Zeier, J. Reprogramming of plants during systemic acquired resistance. Front. Plant Sci. 2013, 4, 252. [Google Scholar] [CrossRef] [Green Version]
- Fujioka, S.; Sakurai, A. Conversion of lysine to L-pipecolic acid induces flowering in Lemna paucicostata. Plant Cell Physiol. 1997, 38, 1278–1280. [Google Scholar] [CrossRef] [Green Version]
- Návarová, H.; Bernsdorff, F.; Döring, A.-C.; Zeier, J. Pipecolic acid, an endogenous mediator of defense amplification and priming, is a critical regulator of inducible plant immunity. Plant Cell 2012, 24, 5123–5141. [Google Scholar] [CrossRef] [Green Version]
- Ding, P.; Rekhter, D.; Ding, Y.; Feussner, K.; Busta, L.; Haroth, S.; Xu, S.; Li, X.; Jetter, R.; Feussner, I.; et al. Characterization of a pipecolic acid biosynthesis pathway required for systemic acquired resistance. Plant Cell 2016, 28, 2603–2615. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-C.; Holmes, E.C.; Rajniak, J.; Kim, J.-G.; Tang, S.; Fischer, C.R.; Mudgett, M.B.; Sattely, E.S. N-hydroxy-pipecolic acid is a mobile metabolite that induces systemic disease resistance in Arabidopsis. Proc. Natl Acad. Sci. USA 2018, 115, 4920–4929. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, M.; Kim, D.; Bernsdorff, F.; Ajami-Rashidi, Z.; Scholten, N.; Schreiber, S.; Zeier, T.; Schuck, S.; Reichel-Deland, V.; Zeier, J. Biochemical Principles and Functional Aspects of Pipecolic Acid Biosynthesis in Plant Immunity. Plant Physiol. 2017, 174, 124–153. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, M.; Zeier, T.; Bernsdorff, F.; Reichel-Deland, V.; Kim, D.; Hohman, M.; Scholten, N.; Schuck, S.; Bräutigan, A.; Hölzel, T.; et al. Flavin monooxygenase generated N-hydroxypipecolic acid is a critical element of plant systemic immunity. Cell 2018, 173, 456–469. [Google Scholar] [CrossRef] [Green Version]
- Germann, U.A.; Shlyakhter, D.; Mason, V.S.; Zelle, R.E.; Duffy, J.P.; Galullo, V.; Armistead, D.M.; Saunders, J.O.; Boger, J.; Harding, M.W. Cellular and biochemical characterization of VX-710 as a chemosensitizer: Reversal of P-glycoprotein-mediated multidrug resistance in vitro. Anticancer. Drugs 1997, 8, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Blažič, M.; Starcevic, A.; Lisfi, M.; Baranasic, D.; Goranovič, D.; Fujs, S.; Kuscer, E.; Kosec, G.; Petković, H.; Cullum, J.; et al. Annotation of the modular polyketide synthase and nonribosomal peptide synthetase gene clusters in the genome of Streptomyces tsukubaensis NRRL18488. Appl. Environ. Microbiol. 2012, 78, 8183–8190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ordóñez-Robles, M.; Rodríguez-García, A.; Martín, J.F. Target genes of the Streptomyces tsukubaensis FkbN regulator include most of the tacrolimus biosynthesis genes, a phosphopantetheinyl transferase and other PKS genes. Appl. Microbiol. Biotechnol. 2016, 100, 8091–8103. [Google Scholar] [CrossRef] [PubMed]
- García, P.F.; Wendisch, P.P.; Wendisch, V.F. Engineering Corynebacterium glutamicum for fast production of l-lysine and pipecolic acid. Appl. Microbiol. Biotechnol. 2016, 100, 8075–8090. [Google Scholar] [CrossRef]
- Ying, H.; Tao, S.; Wang, J.; Ma, W.; Chen, K.; Wang, X.; Ouyang, P. Expanding metabolic pathway for de novo biosynthesis of the chiral pharmaceutical intermediate L-pipecolic acid in Escherichia coli. Microb. Cell Fact. 2017, 16, 52. [Google Scholar] [CrossRef] [Green Version]
- Graupner, M.; White, R.H. Methanococcus jannaschii generates L-proline by cyclization of L-ornithine. J. Bacteriol. 2001, 183, 5203–5205. [Google Scholar] [CrossRef] [Green Version]
- Goodman, J.L.; Wang, S.; Alam, S.; Ruzicka, F.J.; Frey, P.A.; Wedekind, J.E. Ornithine cyclodeaminase: Structure, mechanism of action, and implications for the mu-crystallin family. Biochemistry 2004, 43, 13883–13891. [Google Scholar] [CrossRef]
- Tsotsou, G.E.; Barbirato, F. Biochemical characterisation of recombinant Streptomyces pristinaespiralis L-lysine cyclodeaminase. Biochimie 2007, 89, 591–604. [Google Scholar] [CrossRef]
- Min, K.; Yoon, H.-J.; Atsushi Matsuura, A.; Kim, Y.K.; Lee, H.H. Structural Basis for Recognition of L-lysine, L-ornithine, and L-2,4-diamino Butyric Acid by Lysine Cyclodeaminase. Mol. Cells 2018, 41, 331–341. [Google Scholar]
- Byun, S.M.; Jeong, S.W.; Cho, D.H.; Kim, Y.H. Optimized conversion of L-lysine to Lpipecolic acid using recombinant lysine cyclodeaminase from Streptomyces pristinaespiralis. Biotechnol Bioproc. 2015, 20, 73–78. [Google Scholar] [CrossRef]
- Ying, H.; Wang, J.; Shi, T.; Zhao, Y.; Wang, X.; Ouyang, P.; Chen, K. Studies of lysine cyclodeaminase from Streptomyces pristinaespiralis: Insights into the complex transition NAD+ state. Biochem. Biophys. Res. Commun. 2018, 495, 306–311. [Google Scholar] [CrossRef]
- Schulz, S.; Schall, C.; Stehle, T.; Breitmeyer, C.; Krysenko, S.; Bera, A.; Wohlleben, W. Optimization of the precursor supply for an enhanced FK506 production in Streptomyces tsukubaensis. Front. Biotechnol. Bioengeneer. 2022, 9, 729–734. [Google Scholar] [CrossRef]
- Thibaut, D.; Bisch, D.; Ratet, N.; Maton, L.; Couder, M.; Debussche, L.; Blanche, F. Purification of peptide synthetases involved in pristinamycin I biosynthesis. J. Bacteriol. 1997, 179, 697–704. [Google Scholar] [CrossRef] [Green Version]
- Blanc, V.; Gil, P.; Bamas, J.N.; Lorenzon, S.; Zagorec, M.; Schleuniger, J.; Bisch, D.; Blanche, F.; Debussche, L.; Crouzet, J.; et al. Identification and analysis of genes from Streptomyces pristinaespiralis encoding enzymes involved in the biosynthesis of the 4-dimethylamino-L-phenylalanine precursor of pristinamycin I. Mol. Microbiol. 1997, 23, 191–202. [Google Scholar] [CrossRef]
- Blanc, V.; Lagneaux, D.; Didier, P.; Gil, P.; Lacroix, P.; Crouzet, J. Cloning and analysis of structural genes from Streptomyces pristinaespiralis encoding enzymes involved in the conversion of pristinamycin IIB to pristinamycin IIA (PIIA): PIIA synthase and NADH:riboflavin 5′-phosphate oxidoreductase. J. Bacteriol. 1995, 177, 5206–5214. [Google Scholar] [CrossRef] [Green Version]
- Stassi, D.; Donadio, S.; Staver, M.J.; Katz, L. Identification of a Saccharopolyspora erythraea gene required for the final hydroxylation step in erythromycin biosynthesis. J. Bacteriol. 1993, 175, 182–189. [Google Scholar] [CrossRef] [Green Version]
- Reed, J.W.; Purvis, M.B.; Kingston, D.G.I.; Biot, A.; Gossele, F. Biosynthesis of antibiotics of the virginiamycin family. 7. Stereo- and regiochemical studies on the formation of the 3-hydroxypicolinic acid and pipecolic acid units. J. Org. Chem. 1989, 54, 1161–1165. [Google Scholar] [CrossRef]
- Terra, L.; Ratcliffe, N.; Castro, H.C.; Vicente, A.C.P.; Dyson, P. Biotechnological Potential of Streptomyces Siderophores as New Antibiotics. Curr. Med. Chem. 2021, 28, 1407–1421. [Google Scholar] [CrossRef]
- Traxler, M.F.; Watrous, J.D.; Alexandrov, T.; Dorrestein, P.C.; Kolter, R. Interspecies interactions stimulate diversification of the Streptomyces coelicolor secreted metabolome. MBio 2013, 4, e00459-13. [Google Scholar] [CrossRef] [Green Version]
- Schupp, T.; Toupet, C.; Divers, M. Cloning and expression of two genes of Streptomyces pilosus involved in the biosynthesis of the siderophore desferrioxamine B. Gene 1988, 64, 179–188. [Google Scholar] [CrossRef]
- Bentley, S.D.; Chater, K.F.; Cerdeño-Tárraga, A.M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.; Quail, M.A.; Kieser, H.; Harper, D.; et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar] [CrossRef]
- Burrell, M.; Hanfrey, C.C.; Kinch, L.N.; Elliott, K.A.; Michael, A.J. Evolution of a novel lysine decarboxylase in siderophore biosynthesis. Mol. Microbiol. 2012, 86, 485–499. [Google Scholar] [CrossRef]
- Olucha, J.; Lamb, A.L. Mechanistic and structural studies of the N- hydroxylating flavoprotein monooxygenases. Bioorg. Chem. 2011, 39, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Ronan, J.L.; Kadi, N.; McMahon, S.A.; Naismith, J.H.; Alkhalaf, L.M.; Challis, G.L. Desferrioxamine biosynthesis: Diverse hydroxamate assembly by substrate-tolerant acyl transferase DesC. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170068. [Google Scholar] [CrossRef]
- Park, W.; Woo, J.K.; Shin, J.; Oh, K.B. nonG, a constituent of the nonactin biosynthetic gene cluster, regulates nocardamine synthesis in Streptomyces albus J1074. Biochem. Biophys. Res. Commun. 2017, 490, 664–669. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, C.; Liu, C.; Ju, J.; Ma, J. Genome Sequencing of Streptomyces atratus SCSIOZH16 and Activation Production of Nocardamine via Metabolic Engineering. Front. Microbiol. 2018, 9, 1269. [Google Scholar] [CrossRef] [Green Version]
- Beckmann, N.; Schafferer, L.; Schrettl, M.; Binder, U.; Talasz, H.; Lindner, H.; Haas, H. Characterization of the Link between Ornithine, Arginine, Polyamine and Siderophore Metabolism in Aspergillus fumigatus. PLoS ONE 2013, 8, e67426. [Google Scholar] [CrossRef]
- Pérez-Redondo, R.; Rodríguez-Garcia, A.; Botas, A.; Santamarta, I.; Martín, J.F.; Liras, P. ArgR of Streptomyces coelicolor Is a Versatile. PLoS ONE 2012, 7, e3269. [Google Scholar] [CrossRef] [Green Version]
- Flores, F.J.; Martín, J.F. Iron-regulatory proteins DmdR1 and DmdR2 of Streptomyces coelicolor form two different DNA-protein complexes with iron boxes. Biochem. J. 2004, 380, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Flores, F.J.; Barreiro, C.; Coque, J.J.R.; Martín, J.F. Functional analysis of two divalent metal-dependent regulatory genes dmdR1 and dmdR2 in Streptomyces coelicolor and proteome changes in deletion mutants. FEBS J. 2005, 272, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Finking, R.; Marahiel, M.A. Biosynthesis of nonribosomal peptides. Annu. Rev. Microbiol. 2004, 58, 453–488. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, C.D.; Kaplan, J. Iron acquisition and transcriptional regulation. Chem. Rev. 2009, 109, 4536–4552. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.N.; Kim, J.; Kim, J.; Jung, W.H.; Lee, C.H. Influence of iron regulation on the metabolome of Cryptococcus neoformans. PLoS ONE 2012, 7, e41654. [Google Scholar] [CrossRef] [Green Version]
- Haas, H. Iron—A key nexus in the virulence of Aspergillus fumigatus. Front. Microbiol. 2012, 3, 28. [Google Scholar] [CrossRef] [Green Version]
- Haas, H.; Eisendle, M.; Turgeon, B.G. Siderophores in fungal physiology and virulence. Annu. Rev. Phytopathol. 2008, 46, 149–187. [Google Scholar] [CrossRef]
- Weinberg, E.D. Iron availability and infection. Biochim. Biophys. Acta. 2009, 1790, 600–605. [Google Scholar] [CrossRef] [Green Version]
- Renshaw, J.C.; Robson, G.D.; Trinci, A.P.J.; Wiebe, M.G.; Livens, F.R.; Collison, D.; Taylor, R.J. Fungal siderophores: Structures, functions and applications. Mycol. Res. 2002, 106, 1123–1142. [Google Scholar] [CrossRef]
- Johnson, L. Iron and siderophores in fungal–host interactions. Mycol. Res. 2008, 112, 170–183. [Google Scholar] [CrossRef]
- Oberegger, H.; Eisendle, M.; Schrettl, M.; Graessle, S.; Haas, H. 4′-Phosphopantetheinyl transferase-encoding npgA is essential for siderophore biosynthesis in Aspergillus nidulans. Curr. Genet. 2003, 44, 211–215. [Google Scholar] [CrossRef]
- Gründlinger, M.; Yasmin, S.; Lechner, B.E.; Geley, S.; Schrettl, M.; Hynes, M.; Haas, H. Fungal siderophore biosynthesis is partially localized in peroxisomes. Mol. Microbiol. 2013, 88, 862–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín, J.F. Transport systems, intracellular traffic of intermediates and secretion of β-lactam antibiotics in fungi. Fungal Biol. Biotechnol. 2020, 7, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiel, J.A.; Veenhuis, M.; van der Klei, I.J. PEX genes in fungal genomes: Common, rare or redundant. Traffic 2006, 7, 1291–1303. [Google Scholar] [CrossRef]
- Hof, C.; Eisfeld, K.; Welzel, K.; Antelo, L.; Foster, A.J.; Anke, H. Siderophore synthesis in Magnaporthe grisea is essential for vegetative growth, conidiation and resistance to oxidative stress. Fungal Genet. Biol. 2009, 46, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Asai, Y.; Hiratsuka, T.; Ueda, M.; Kawamura, Y.; Asamizu, S.; Onaka, H.; Arioka, M.; Nishimura, S.; Yoshida, M. Differential Biosynthesis and Roles of Two Ferrichrome-Type Siderophores, ASP2397/AS2488053 and Ferricrocin, in Acremonium persicinum. ACS Chem. Biol. 2022, 17, 207–216. [Google Scholar] [CrossRef]
- Yuan, W.M.; Gentil, G.D.; Budde, A.D.; Leong, S.A. Characterization of the Ustilago maydis sid2 Gene, Encoding a Multidomain Peptide Synthetase in the Ferrichrome Biosynthetic Gene Cluster. J. Bacteriol. 2001, 183, 4040–4051. [Google Scholar] [CrossRef] [Green Version]
- Oide, S.; Moeder, W.; Krasnoff, S.; Gibson, D.; Haas, H.; Yoshioka, K.; Turgeon, B.G. NPS6, encoding a 872 nonribosomal peptide synthetase involved in siderophore-mediated iron metabolism, is a conserved 873 virulence determinant of plant pathogenic ascomycetes. Plant Cell 2006, 18, 2836–2853. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.H.; Lin, C.H.; Chung, K.R. A nonribosomal peptide synthetase mediates siderophore production and virulence in the citrus fungal pathogen Alternaria alternata. Mol. Plant Pathol. 2013, 14, 497–505. [Google Scholar] [CrossRef]
- Lee, B.-N.; Kroken, S.; Chou, D.Y.T.; Robbertse, B.; Yoder, O.C.; Turgeon, B.G. Functional analysis of all nonribosomal peptide synthetases in Cochliobolus heterostrophus reveals a factor, NPS6, involved in virulence and resistance to oxidative stress. Eukaryot. Cell 2005, 4, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Wallner, A.; Blatzer, M.; Schrettl, M.; Sarg, B.; Lindner, H.; Haas, H. Ferricrocin, a siderophore involved in intra- and transcellular iron distribution in Aspergillus fumigatus. Appl. Environ. Microbiol. 2009, 75, 4194–4196. [Google Scholar] [CrossRef] [Green Version]
- Jami, M.S.; Barreiro, C.; García-Estrada, C.; Martín, J.F. Proteome analysis of the penicillin producer Penicillium chrysogenum: Characterization of protein changes during the industrial strain improvement. Mol. Cel. Proteom. 2010, 9, 1182–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Estrada, C.; Juan, F.; Martín, J.F.; Cueto, L.; Barreiro, C. Omics Approaches Applied to Penicillium chrysogenum and Penicillin Production: Revealing the Secrets of Improved Productivity. Genes 2020, 11, 712–738. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.J.; Burnham, M.K.; Edwards, J.; Earl, A.J.; Turner, G. Cloning and heterologous expression of the penicillin biosynthetic gene cluster from Penicillum chrysogenum. Biotechnology 1990, 8, 39–41. [Google Scholar] [PubMed]
- Awan, A.R.; Blount, B.A.; Bell, D.J.; Shaw, W.M.; Ho, J.C.H.; McKiernan, R.M.; Ellis, T. Biosynthesis of the antibiotic nonribosomal peptide penicillin in baker’s yeast. Nat. Commun. 2017, 8, 15202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Burgo, Y.; Álvarez-Álvarez, R.; Pérez-Redondo, R.; Liras, P. Heterologous expression of Streptomyces clavuligerus ATCC 27064 cephamycin C gene cluster. J. Biotechnol. 2014, 186, 21–29. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SM Precursor | Precursor Synthesizing Enzyme(s) | Secondary Metabolite (SM) | Producer Strain | Reference | |
|---|---|---|---|---|---|
| 1 | α-Aminoadipic acid (α-AAA) | α-Ketoadipate aminotransferase | Penicillins Cephalosporin C Cephamycins | Penicillium chrysogenum Acremonium chrysogenum Streptomyces clavuligerus | [5,18] |
| 2 | α-Aminoadipic semialdehyde (P6C) | α-AAA reductase Lysine-6-aminotransferase | Penicillins, Cephalosporin C Cephamycins | P. chrysogenum A. chrysogenum S. clavuligerus | [19,20,21] |
| 3 | Saccharopine | Saccharopine reductase and Pipecolate oxidoreductase | Pipecolic acid | Most fungi | [22,23,24] |
| 4 | 4-oxo-pipecolic acid | Pipecolate monoxygenase P450 | Swainsonine, Slaframine | Slafractonia leguminicola Metarhizium anisopliae Undifilum oxytropis | [5] |
| 5 | Pipecolic acid | Lysine cyclodeaminase | Rapamycin Pristinamycin I Tacrolimus | Streptomyces hygroscopicus Streptomyces pristinaespiralis Streptomyces tsukubaensis | [25,26,27] |
| 6 | 3-Hydroxypicolinic acid | Lysine-2-aminotransferase with dehydrase activity | Pristinamycin I Virginiamycin | S. pristinaespiralis Streptomyces virginiae | [28,29] |
| 7 | Cadaverine | Lysine decarboxylase | Desferrioxamines | Streptomyces coelicolor Streptomyces pilosus | [30] |
| 8 | N5-Hydroxyornithine N5-Formyl-N5-Hydroxy- ornithine | N5-ornithine monooxygenase N5-hydroxyornithine acyltransferase | Coelichelin | S. coelicolor Streptomyces ambofaciens | [31,32,33] |
| 9 | N5-anhydromevalonyl-N5-hydroxyornithine | N5-ornithine monooxygenase Mevalonil CoA ligase Mevalonyl-CoA dehydrase | Fusarinine, TAFC | Many fungi | [34,35] |
| 10 | N5-acetyl-N5-hydroxyornithine | N5-ornithine monooxygenase N5-hydroxyornithine acyltransferase | Ferrichrocin | Many fungi | [35,36,37,38] |
| 11 | N5-acetyl-N5-hydroxyornithine | N5-ornithine monooxygenase N5-hydroxyornithine acyltransferase | Coprogen Methyl-coprogen | Many fungi Alternaria alternata | [39] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liras, P.; Martín, J.F. Interconnected Set of Enzymes Provide Lysine Biosynthetic Intermediates and Ornithine Derivatives as Key Precursors for the Biosynthesis of Bioactive Secondary Metabolites. Antibiotics 2023, 12, 159. https://doi.org/10.3390/antibiotics12010159
Liras P, Martín JF. Interconnected Set of Enzymes Provide Lysine Biosynthetic Intermediates and Ornithine Derivatives as Key Precursors for the Biosynthesis of Bioactive Secondary Metabolites. Antibiotics. 2023; 12(1):159. https://doi.org/10.3390/antibiotics12010159
Chicago/Turabian StyleLiras, Paloma, and Juan Francisco Martín. 2023. "Interconnected Set of Enzymes Provide Lysine Biosynthetic Intermediates and Ornithine Derivatives as Key Precursors for the Biosynthesis of Bioactive Secondary Metabolites" Antibiotics 12, no. 1: 159. https://doi.org/10.3390/antibiotics12010159