
An Evolutionary Conservation and Druggability Analysis of Enzymes Belonging to the Bacterial Shikimate Pathway


Abstract
:1. Introduction
2. Results and Discussion
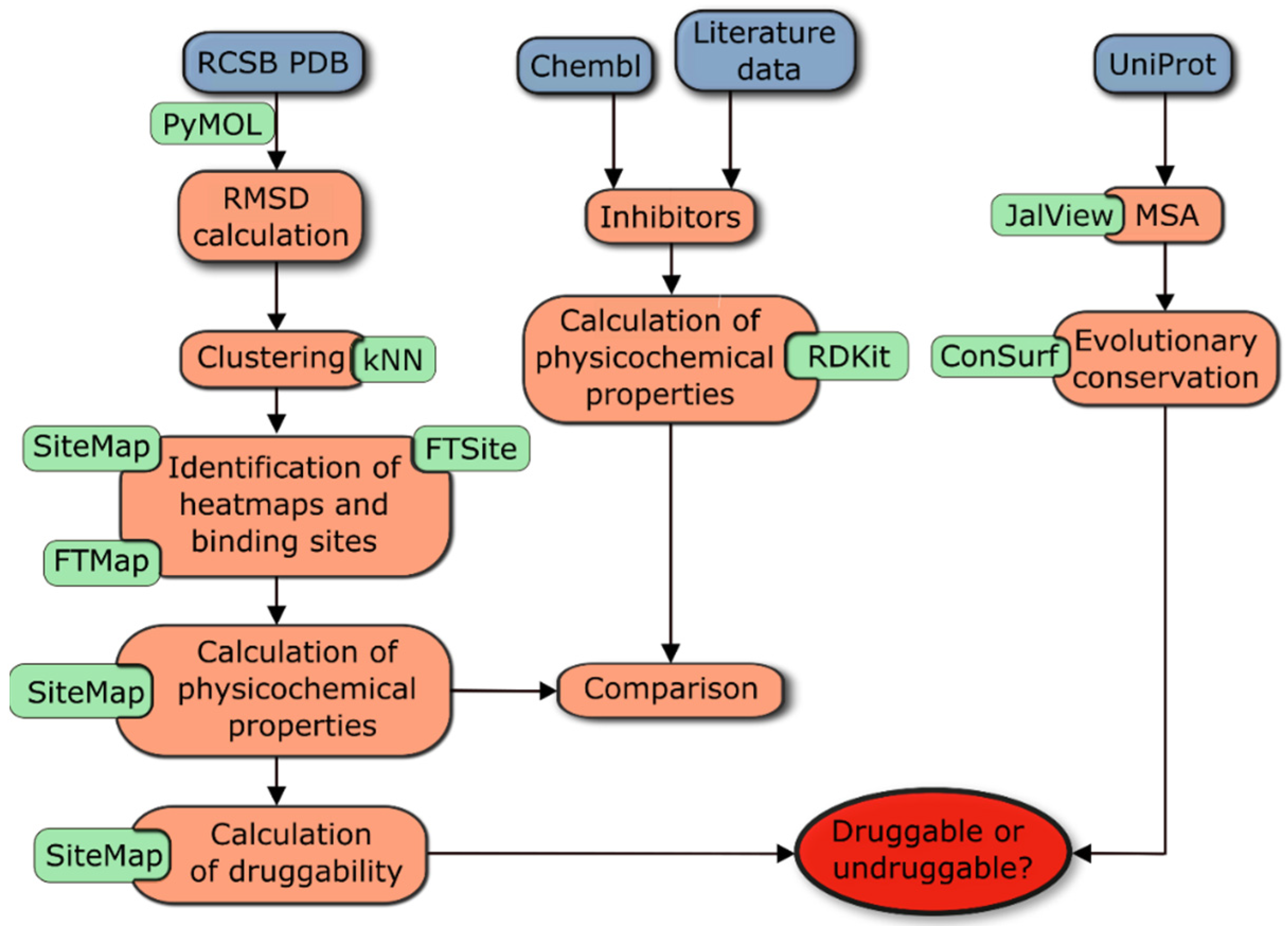
2.1. Study Design
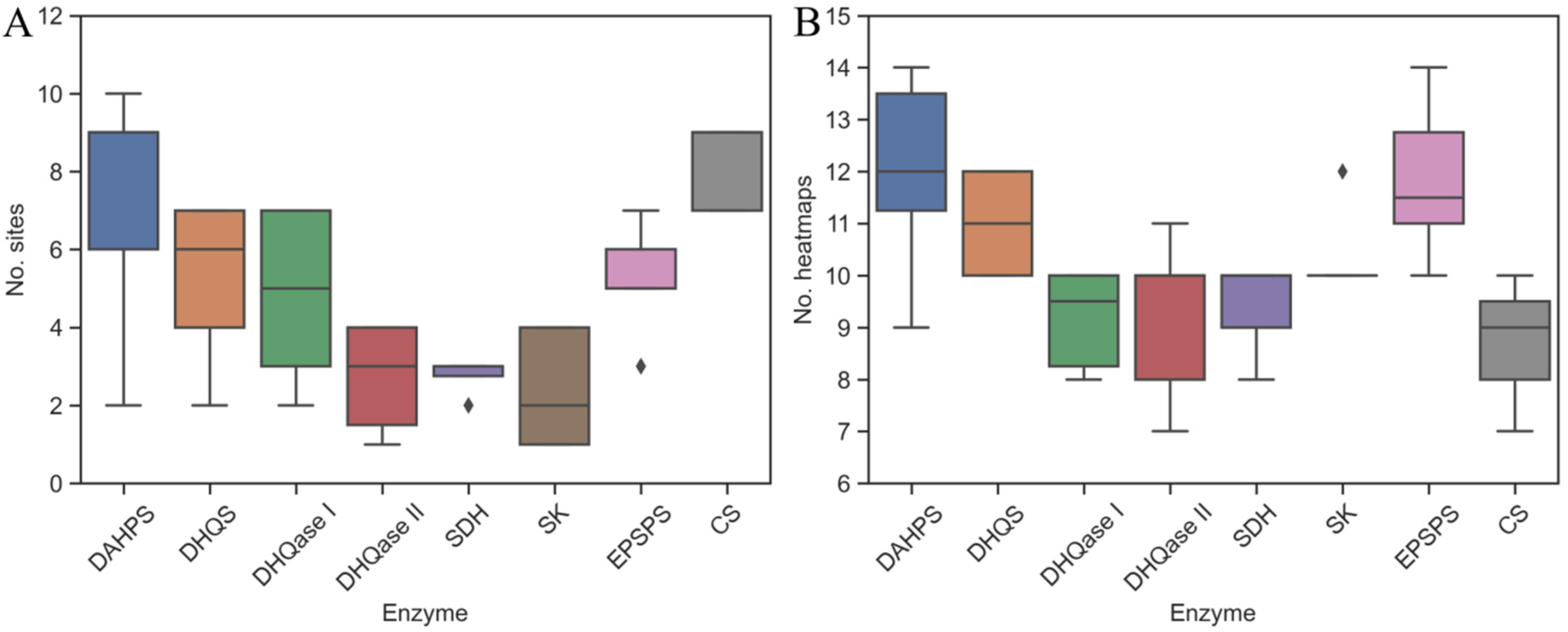
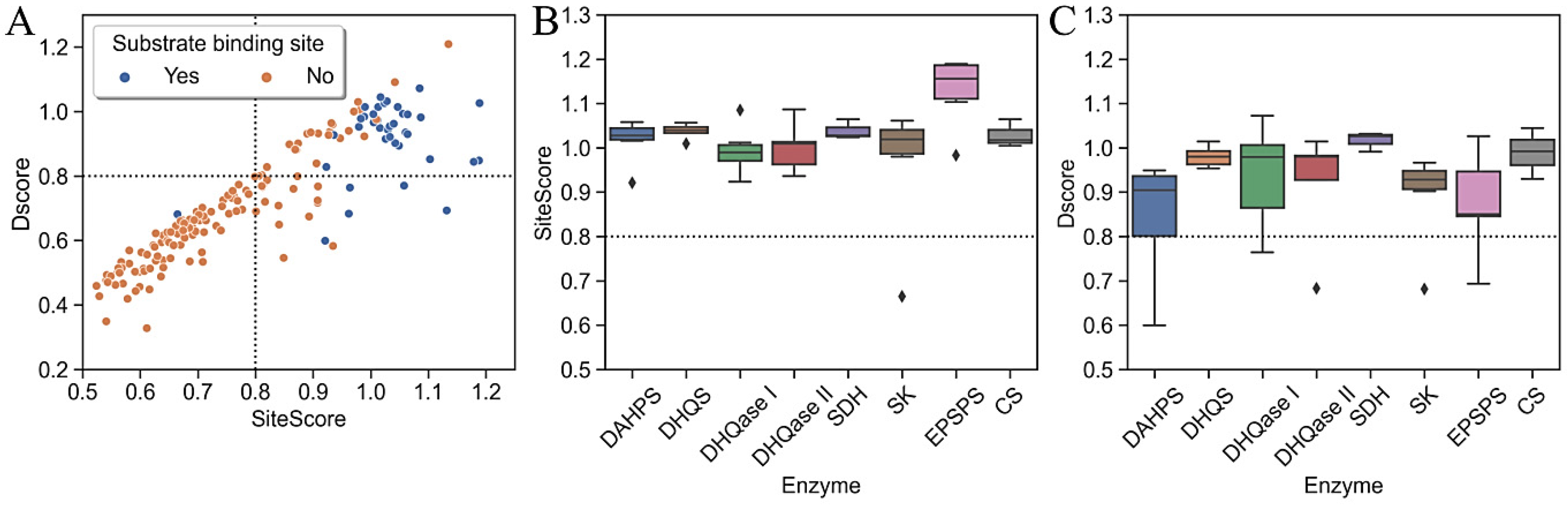
2.2. The Identification of Binding Sites
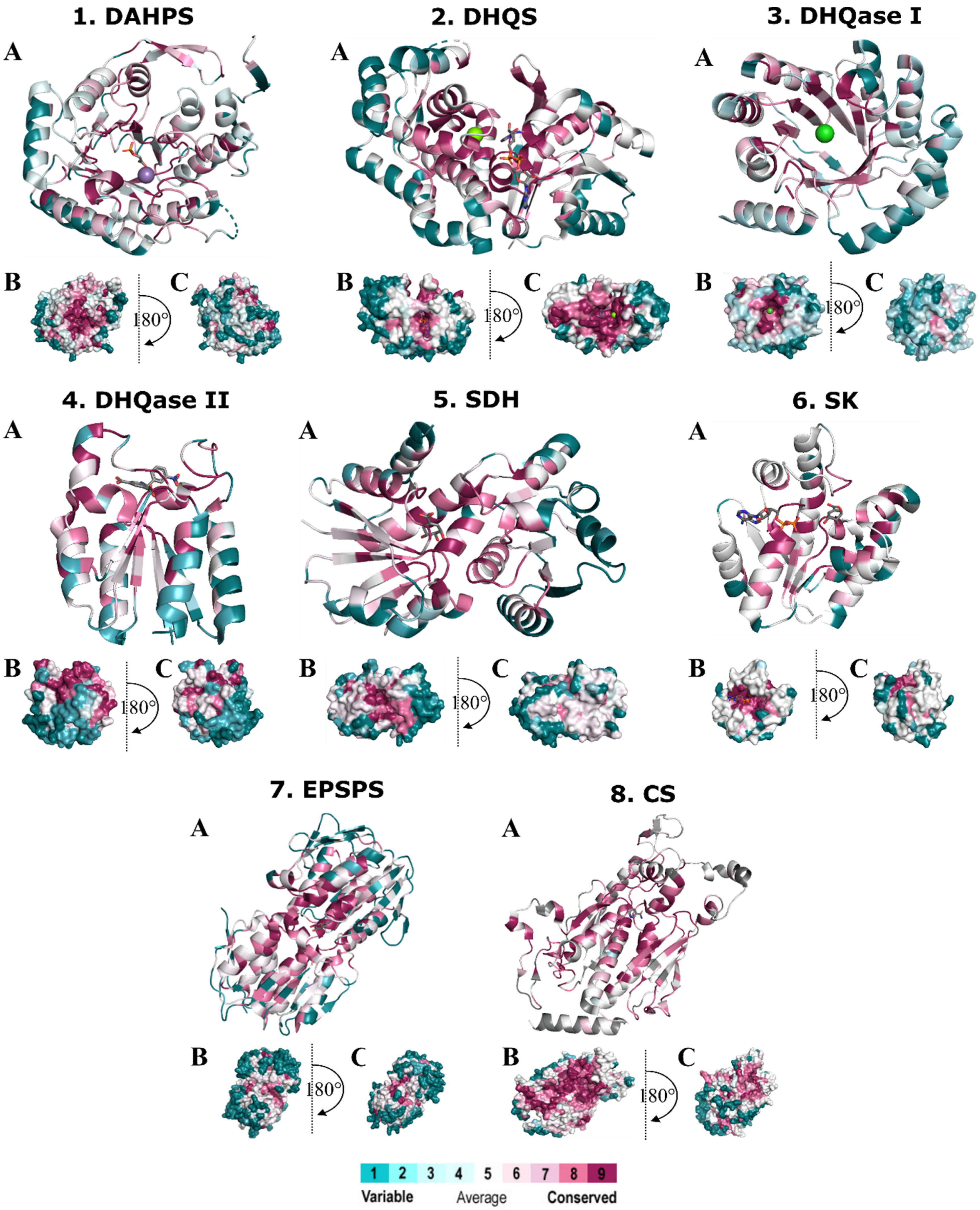
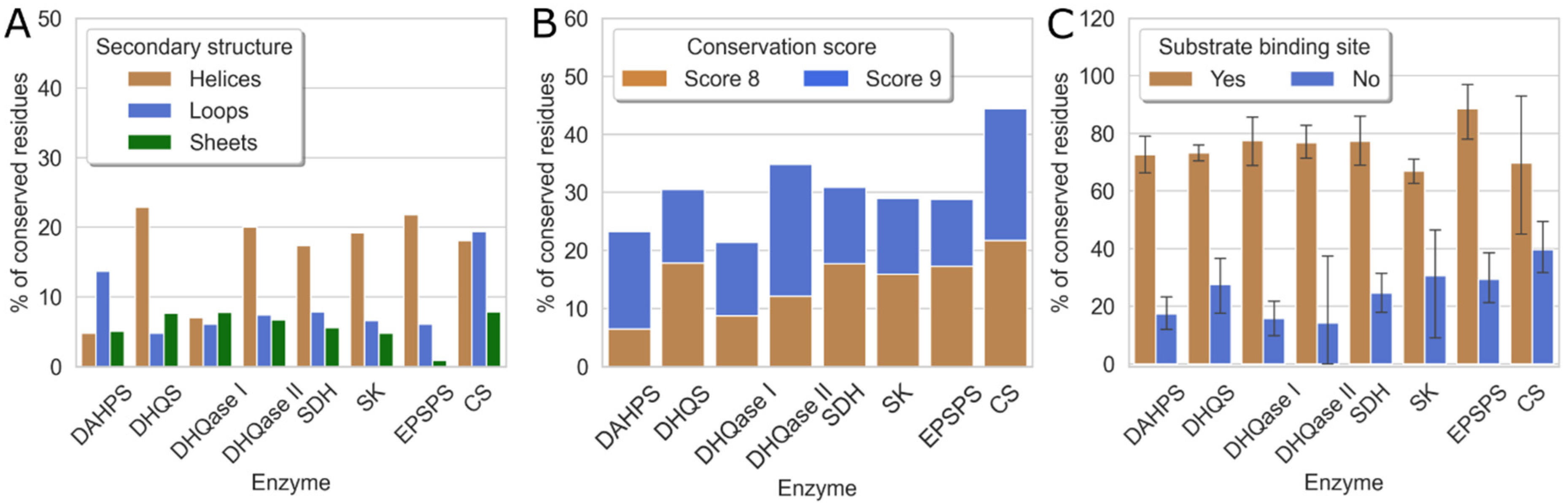
2.3. Evolutionary Conservation
2.4. Druggability Assessment
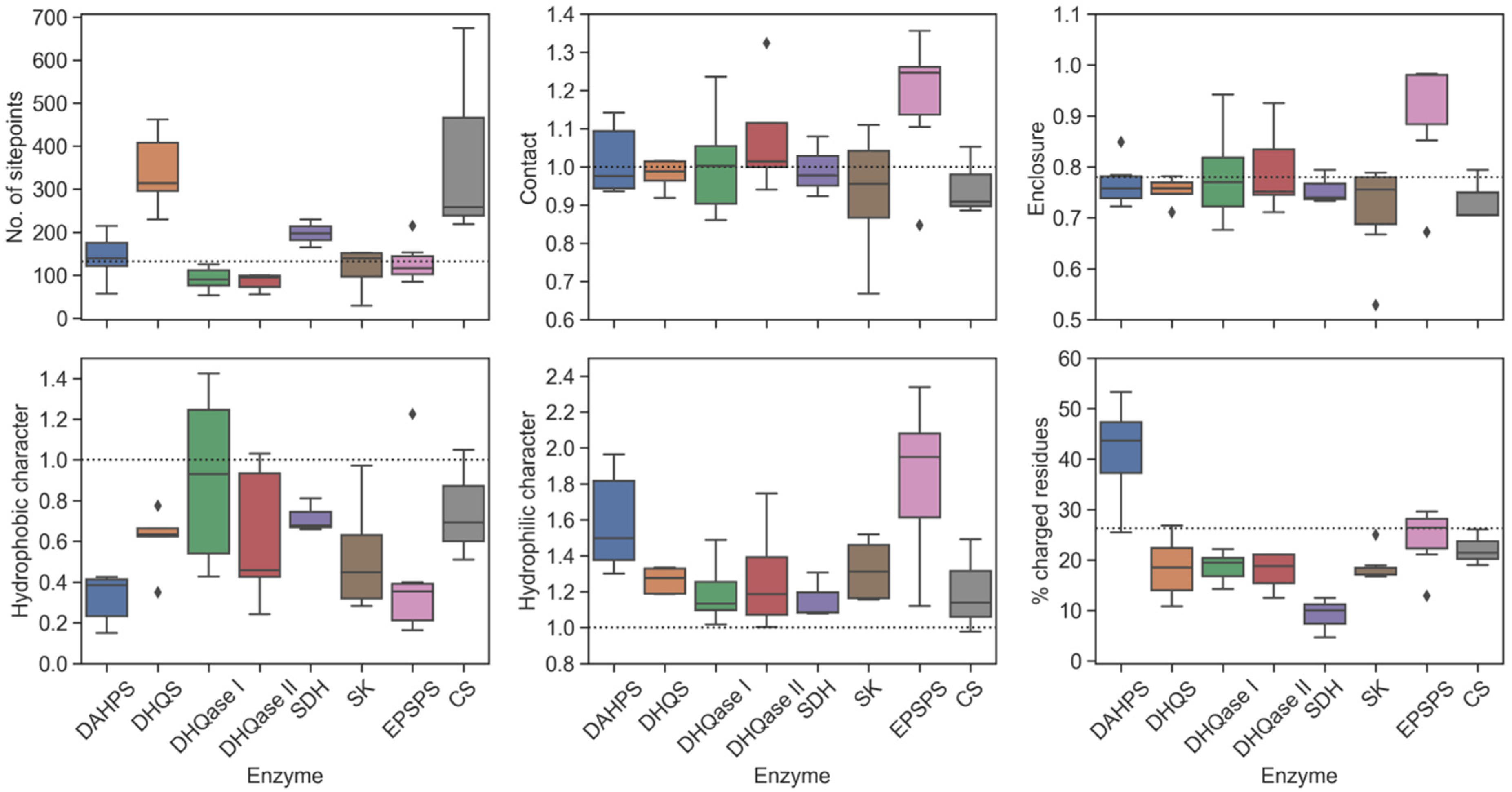
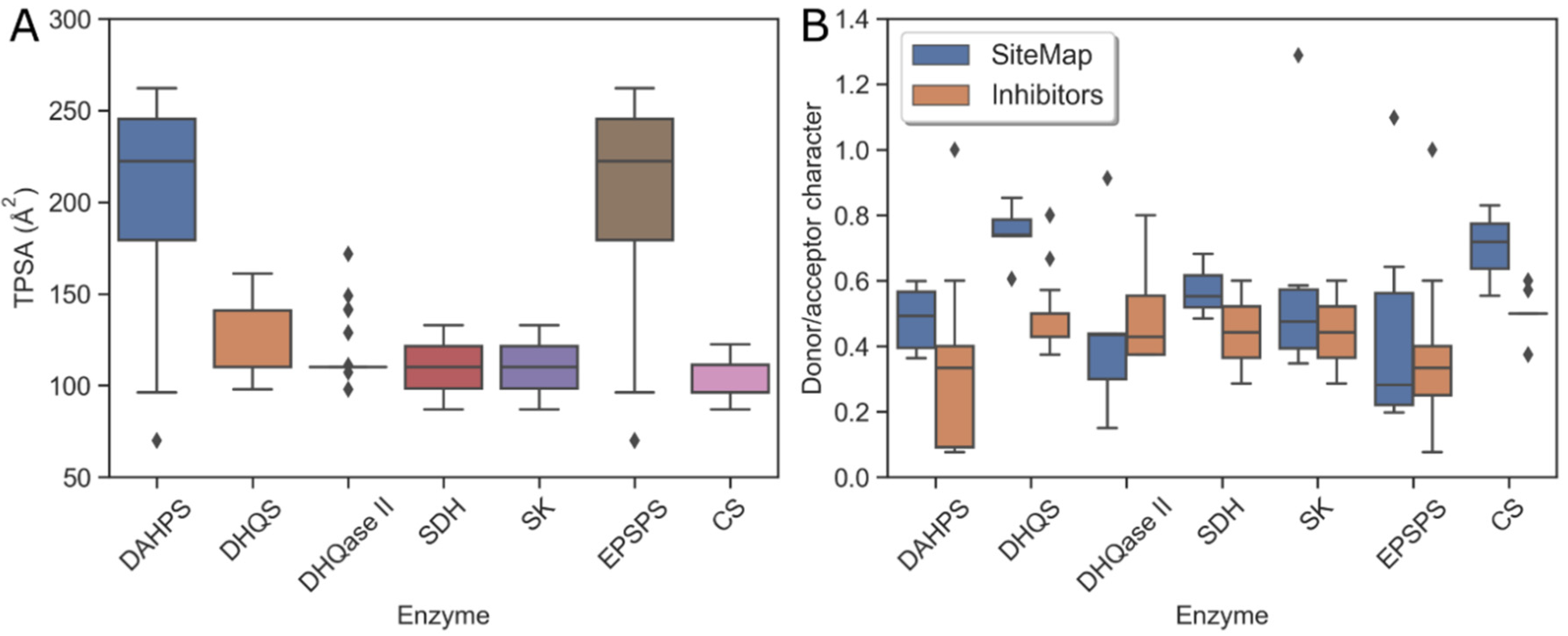
2.5. Evaluation of Physicochemical Properties of Binding Sites
2.6. Druggability of the Substrate-Binding Sites
2.7. Physicochemical Properties of Substrate-Binding Sites
3. Materials and Methods
3.1. Study Design
3.2. Identification of Hotspots and Binding Sites with FTMap and FTSite
3.3. Druggability Assessment with SiteMap
3.4. Protein Acquisition and Preparation
3.5. Superposition of 3D Structures and Selection of Representative Crystal Structures
3.6. Multiple Sequence Alignment
3.7. Calculation of Evolutionary Conservation
3.8. Calculation of TPSA
3.9. Statistical Analysis
3.10. Generation of Figures
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Uchil, R.R.; Kohli, G.S.; Katekhaye, V.M.; Swami, O.C. Strategies to combat antimicrobial resistance. J. Clin. Diagn. Res. JCDR 2014, 8, ME01–ME04. [Google Scholar] [CrossRef] [PubMed]
- Cattoir, V.; Felden, B. Future antibacterial strategies: From basic concepts to clinical challenges. J. Infect. Dis. 2019, 220, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Pati, N.B.; Vishwakarma, V.; Selvaraj, S.K.; Dash, S.; Saha, B.; Singh, N.; Suar, M. Salmonella typhimurium TTSS-2 deficient Mig-14 mutant shows attenuation in immunocompromised mice and offers protection against wild-type Salmonella typhimurium infection. BMC Microbiol. 2013, 13, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stritzker, J.; Janda, J.; Schoen, C.; Taupp, M.; Pilgrim, S.; Gentschev, I.; Schreier, P.; Geginat, G.; Goebel, W. Growth, virulence, and immunogenicity of Listeria monocytogenes aro mutants. Infect. Immun. 2004, 72, 5622–5629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishioka, Y.; Demerec, M.; Eisenstark, A. Genetic analysis of aromatic mutants of Salmonella typhimurium. Genetics 1967, 56, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Foulongne, V.; Walravens, K.; Bourg, G.; Boschiroli, M.L.; Godfroid, J.; Ramuz, M.; O’Callaghan, D. Aromatic compound-dependent Brucella suis is attenuated in both cultured cells and mouse models. Infect. Immun. 2001, 69, 547–550. [Google Scholar] [CrossRef] [Green Version]
- McArthur, J.D.; West, N.P.; Cole, J.N.; Jungnitz, H.; Guzman, C.A.; Chin, J.; Lehrbach, P.R.; Djordjevic, S.P.; Walker, M.J. An aromatic amino acid auxotrophic mutant of Bordetella bronchiseptica is attenuated and immunogenic in a mouse model of infection. Fems Microbiol. Lett. 2003, 221, 7–16. [Google Scholar] [CrossRef] [Green Version]
- Fittipaldi, N.; Harel, J.; D’Amours, B.; Lacouture, S.; Kobisch, M.; Gottschalk, M. Potential use of an unencapsulated and aromatic amino acid-auxotrophic Streptococcus suis mutant as a live attenuated vaccine in swine. Vaccine 2007, 25, 3524–3535. [Google Scholar] [CrossRef]
- Smith, D.A.; Parish, T.; Stoker, N.G.; Bancroft, G.J. Characterization of auxotrophic mutants of Mycobacterium tuberculosis and their potential as vaccine candidates. Infect. Immun. 2001, 69, 1142–1150. [Google Scholar] [CrossRef] [Green Version]
- Bentley, R. The shikimate pathway—A metabolic tree with many branches. Crit. Rev. Biochem. Mol. Biol. 1990, 25, 307–384. [Google Scholar] [CrossRef]
- Gibson, F.; Pittard, J. Pathways of biosynthesis of aromatic amino acids and vitamins and their control in microorganisms. Bacteriol. Rev. 1968, 32, 465–492. [Google Scholar] [CrossRef]
- Davis, B. Aromatic biosynthesis 1. The role of shikimic acid. J. Biol. Chem. 1951, 191, 315–325. [Google Scholar] [CrossRef]
- Roberts, F.; Roberts, C.W.; Johnson, J.J.; Kyle, D.E.; Krell, T.; Coggins, J.R.; Coombs, G.H.; Milhous, W.K.; Tzipori, S.; Ferguson, D.J.P.; et al. Evidence for the shikimate pathway in apicomplexan parasites (Vol 393, Pg 801, 1998). Nature 1998, 395, 306. [Google Scholar] [CrossRef]
- Herrmann, K.M.; Weaver, L.M. The shikimate pathway. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1999, 50, 473–503. [Google Scholar] [CrossRef] [PubMed]
- Coracini, J.D.; de Azevedo, W.F. Shikimate kinase, a protein target for drug design. Curr. Med. Chem. 2014, 21, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Steinrucken, H.; Amrhein, N. The Herbicide glyphosate is a potent inhibitor of 5-enolpyruvyl-shikimic-acid 3-phosphate synthase. Biochem. Biophys. Res. Commun. 1980, 94, 1207–1212. [Google Scholar] [CrossRef]
- Sikorski, J.A.; Gruys, K.J. Understanding glyphosate’s molecular mode of action with EPSP synthase: Evidence favoring an allosteric inhibitor model. Acc. Chem. Res. 1997, 30, 2–8. [Google Scholar] [CrossRef]
- Steinrucken, H.; Amrhein, N. 5-enolpyruvylshikimate-3-phosphate synthase of klebsiella-pneumoniae. 2. Inhibition by glyphosate [N-(phosphonomethyl)glycine]. Eur. J. Biochem. 1984, 143, 351–357. [Google Scholar] [CrossRef]
- Roberts, C.W.; Roberts, F.; Lyons, R.E.; Kirisits, M.J.; Mui, E.J.; Finnerty, J.; Johnson, J.J.; Ferguson, D.J.P.; Coggins, J.R.; Krell, T.; et al. The shikimate pathway and its branches in apicomplexan parasites. J. Infect. Dis. 2002, 185, S25–S36. [Google Scholar] [CrossRef] [Green Version]
- Mir, R.; Jallu, S.; Singh, T.P. The shikimate pathway: Review of amino acid sequence, function and three-dimensional structures of the enzymes. Crit. Rev. Microbiol. 2015, 41, 172–189. [Google Scholar] [CrossRef]
- Ducati, R.G.; Basso, L.A.; Santos, D.S. Mycobacterial shikimate pathway enzymes as targets for drug design. Curr. Drug Targets 2007, 8, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.E.S.; Duque, M.A.; de Freitas, T.F.; Galina, L.; Timmers, L.F.S.M.; Bizarro, C.V.; Machado, P.; Basso, L.A.; Ducati, R.G. Mycobacterium tuberculosis shikimate pathway enzymes as targets for the rational design of anti-tuberculosis drugs. Molecules 2020, 25, 1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogrodniczuk, M.; Motilall, A.; Fuanta, R. Shikimate kinase expression; towards screening of potential antibacterials from marine sponges. FASEB J. 2020, 34, 1. [Google Scholar] [CrossRef]
- Sivaranjani, P.; Naik, V.U.; Madhulitha, N.R.; Kumar, K.S.; Chiranjeevi, P.; Alexander, S.P.; Umamaheswari, A. Design of novel antimycobacterial molecule targeting shikimate pathway of Mycobacterium tuberculosis. Indian J. Pharm. Sci. 2019, 81, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Pereira, J.H.; Vasconcelos, I.B.; Oliveira, J.S.; Caceres, R.A.; de Azevedo, W.F.; Basso, L.A.; Santos, D.S. Shikimate kinase: A potential target for development of novel antitubercular agents. Curr. Drug Targets 2007, 8, 459–468. [Google Scholar] [CrossRef]
- Dias, M.V.B.; Ely, F.; Palma, M.S.; de Azevedo, W.F.; Basso, L.A.; Santos, D.S. Chorismate synthase: An attractive target for drug development against orphan diseases. Curr. Drug Targets 2007, 8, 437–444. [Google Scholar] [CrossRef]
- Khanapur, M.; Alvala, M.; Prabhakar, M.; Shiva Kumar, K.; Edwin, R.K.; Sri Saranya, P.S.V.K.; Patel, R.K.; Bulusu, G.; Misra, P.; Pal, M. Mycobacterium tuberculosis chorismate mutase: A potential target for TB. Bioorg. Med. Chem. 2017, 25, 1725–1736. [Google Scholar] [CrossRef]
- González-Bello, C. Specific chemical modification of bacterial type I dehydroquinase—Opportunities for drug discovery. Future Med. Chem. 2015, 7, 2371–2383. [Google Scholar] [CrossRef]
- De Avila, M.B.; Bitencourt-Ferreira, G.; de Azevedo, W.F., Jr. Structural basis for inhibition of enoyl-[acyl carrier protein] reductase (InhA) from Mycobacterium tuberculosis. Curr. Med. Chem. 2020, 27, 745–759. [Google Scholar] [CrossRef]
- Agoni, C.; Olotu, F.A.; Ramharack, P.; Soliman, M.E. Druggability and drug-likeness concepts in drug design: Are biomodelling and predictive tools having their say? J. Mol. Model. 2020, 26, 120. [Google Scholar] [CrossRef]
- Barril, X. Druggability predictions: Methods, limitations, and applications. WIREs Comput. Mol. Sci. 2013, 3, 327–338. [Google Scholar] [CrossRef]
- Macarron, R. Critical review of the role of HTS in drug discovery. Drug Discov. Today 2006, 11, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.; Homan, E.J.; Wiita, E.; Pedersen, K.; Almlof, I.; Gustavsson, A.-L.; Lundback, T.; Helleday, T.; Berglund, U.W. In silico druggability assessment of the NUDIX hydrolase protein family as a workflow for target prioritization. Front. Chem. 2020, 8, 443. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R. Target analysis: A priori assessment of druggability. In Proceedings of the Small Molecule—Protein Interactions; Waldmann, H., Koppitz, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2003; pp. 11–17. [Google Scholar]
- Hajduk, P.J.; Huth, J.R.; Fesik, S.W. Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 2005, 48, 2518–2525. [Google Scholar] [CrossRef]
- Vukovic, S.; Huggins, D.J. Quantitative metrics for drug–target ligandability. Drug Discov. Today 2018, 23, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Liu, T.; Baitaluk, M.; Nicola, G.; Hwang, L.; Chong, J. BindingDB in 2015: A public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2016, 44, D1045–D1053. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A web-accessible database of experimentally determined protein–ligand binding affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [Green Version]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap Family of web servers for determining and characterizing ligand binding hot spots of proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef] [Green Version]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A web server for automatic binding site prediction, analysis and druggability assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2021-3: SiteMap; Schrödinger, LLC.: New York, NY, USA, 2021.
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, M.; Ghersi, D.; Sanchez, R. SITEHOUND-Web: A server for ligand binding site identification in protein structures. Nucleic Acids Res. 2009, 37, W413–W416. [Google Scholar] [CrossRef]
- Liu, Q.; Li, L.; Xu, F. Systematic analysis and integrative discovery of active-site subpocket-specific dehydroquinate synthase inhibitors combating antibiotic-resistant Staphylococcus aureus infection. J. Bioinform. Comput. Biol. 2018, 16, 1850027. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhu, M.; Tang, Y.; Liu, J.; Yan, F.; Yu, Z.; Zhu, J. Integration of virtual screening and susceptibility test to discover active-site subpocket-specific biogenic inhibitors of helicobacter pylori shikimate dehydrogenase. Int. Microbiol. 2019, 22, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Gladki, A.; Kaczanowski, S.; Szczesny, P.; Zielenkiewicz, P. The evolutionary rate of antibacterial drug targets. BMC Bioinform. 2013, 14, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Gao, H.; Ji, K.; Yan, B.; Li, Q.; Mo, S.; Zheng, M.; Ou, Q.; Wu, B.; Li, N.; et al. Isolation and biochemical characterization of a metagenome-derived 3-deoxy-d-arabino-heptulosonate-7-phosphate synthase gene from subtropical marine mangrove wetland sediments. Amb Express 2019, 9, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiling, S.; Kelleher, A.; Matsumoto, M.M.; Robinson, G.; Asojo, O.A. Structure of type II dehydroquinase from Pseudomonas aeruginosa. Acta Crystallogr. Sect. F-Struct. Biol. Commun. 2014, 70, 1485–1491. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, N.; Kumar, M.; Sharma, P.; Yadav, S.P.; Kaur, P.; Sharma, S.; Singh, T.P. Binding studies and structure determination of the recombinantly produced type-II 3-dehydroquinate dehydratase from Acinetobacter baumannii. Int. J. Biol. Macromol. 2017, 94, 459–465. [Google Scholar] [CrossRef]
- Florova, G.; Denoya, C.D.; Morgenstern, M.R.; Skinner, D.D.; Reynolds, K.A. Cloning, expression, and characterization of a type II 3-dehydroquinate dehydratase gene from Streptomyces hygroscopicus. Arch. Biochem. Biophys. 1998, 350, 298–306. [Google Scholar] [CrossRef]
- Light, S.H.; Minasov, G.; Shuvalova, L.; Duban, M.-E.; Caffrey, M.; Anderson, W.F.; Lavie, A. Insights into the mechanism of type I dehydroquinate dehydratases from structures of reaction intermediates. J. Biol. Chem. 2011, 286, 3531–3539. [Google Scholar] [CrossRef] [Green Version]
- Peek, J.; Christendat, D. The shikimate dehydrogenase family: Functional diversity within a conserved structural and mechanistic framework. Arch. Biochem. Biophys. 2015, 566, 85–99. [Google Scholar] [CrossRef]
- Han, C.; Wang, L.; Yu, K.; Chen, L.; Hu, L.; Chen, K.; Jiang, H.; Shen, X. Biochemical characterization and inhibitor discovery of shikimate dehydrogenase from helicobacter pylori. Febs J. 2006, 273, 4682–4692. [Google Scholar] [CrossRef]
- Michel, G.; Roszak, A.W.; Sauve, V.; Maclean, J.; Matte, A.; Coggins, J.R.; Cygler, M.; Lapthorn, A.J. Structures of shikimate dehydrogenase AroE and its paralog YdiB—A common structural framework for different activities. J. Biol. Chem. 2003, 278, 19463–19472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagautdinov, B.; Kunishima, N. Crystal structures of shikimate dehydrogenase AroE from Thermus thermophilus HB8 and its cofactor and substrate complexes: Insights into the enzymatic mechanism. J. Mol. Biol. 2007, 373, 424–438. [Google Scholar] [CrossRef]
- Peek, J.; Lee, J.; Hu, S.; Senisterra, G.; Christendat, D. Structural and mechanistic analysis of a novel class of shikimate dehydrogenases: Evidence for a conserved catalytic mechanism in the shikimate dehydrogenase family. Biochemistry 2011, 50, 8616–8627. [Google Scholar] [CrossRef] [PubMed]
- Arcuri, H.A.; Borges, J.C.; Fonseca, I.O.; Pereira, J.H.; Ruggiero Neto, J.; Basso, L.A.; Santos, D.S.; de Azevedo, W.F. Structural studies of shikimate 5-dehydrogenase from Mycobacterium tuberculosis. Proteins-Struct. Funct. Bioinforma. 2008, 72, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Favela-Candia, A.; Tellez-Valencia, A.; Campos-Almazan, M.; Sierra-Campos, E.; Valdez-Solana, M.; Oria-Hernandez, J.; Castillo-Villanueva, A.; Najera, H.; Avitia-Dominguez, C. Biochemical, kinetic, and computational structural characterization of shikimate kinase from methicillin-resistant Staphylococcus aureus. Mol. Biotechnol. 2019, 61, 274–285. [Google Scholar] [CrossRef]
- Liu, Z.; Zhu, Q.; Li, Y.; Yu, J.; Wang, W.; Tan, R.; Zhou, J.; Liao, H. Isolation and in silico characterization of a shikimate kinase from Cassia obtusifolia. Acta Physiol. Plant. 2015, 37, 85. [Google Scholar] [CrossRef]
- Ahmad, S.; Baseer, S.; Navid, A.; Ahmad, F.; Azam, S.S. An integrated computational hierarchy for identification of potent inhibitors against shikimate kinase enzyme from Shigella sonnei, a major cause of global dysentery. Gene Rep. 2018, 11, 283–293. [Google Scholar] [CrossRef]
- Rainio, M.J.; Ruuskanen, S.; Helander, M.; Saikkonen, K.; Saloniemi, I.; Puigbò, P. Adaptation of bacteria to glyphosate: A microevolutionary perspective of the enzyme 5-enolpyruvylshikimate-3-phosphate synthase. Environ. Microbiol. Rep. 2021, 13, 309–316. [Google Scholar] [CrossRef]
- Leino, L.; Tall, T.; Helander, M.; Saloniemi, I.; Saikkonen, K.; Ruuskanen, S.; Puigbò, P. Classification of the glyphosate target enzyme (5-enolpyruvylshikimate-3-phosphate synthase) for assessing sensitivity of organisms to the herbicide. J. Hazard. Mater. 2021, 408, 124556. [Google Scholar] [CrossRef]
- Tapas, S.; Kumar, A.; Dhindwal, S.; Preeti; Kumar, P. Structural analysis of chorismate synthase from Plasmodium falciparum: A novel target for antimalaria drug discovery. Int. J. Biol. Macromol. 2011, 49, 767–777. [Google Scholar] [CrossRef]
- Neetu, N.; Sharma, M.; Mahto, J.K.; Kumar, P. Biophysical and in-silico studies of phytochemicals targeting chorismate synthase from drug-resistant Moraxella catarrhalis. Protein J. 2020, 39, 449–460. [Google Scholar] [CrossRef]
- Kukol, A.; Hughes, D.J. Large-scale analysis of influenza A virus nucleoprotein sequence conservation reveals potential drug-target sites. Virology 2014, 454–455, 40–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trigueiro-Louro, J.M.; Correia, V.; Santos, L.A.; Guedes, R.C.; Brito, R.M.M.; Rebelo-de-Andrade, H. To hit or not to hit: Large-scale sequence analysis and structure characterization of influenza A NS1 unlocks new antiviral target potential. Virology 2019, 535, 297–307. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.W.; Romero, P.; Uversky, V.N.; Dunker, A.K. Conservation of intrinsic disorder in protein domains and families: II. functions of conserved disorder. J. Proteome Res. 2006, 5, 888–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, B.; Li, M.; Li, T.; Yu, M.; Zhang, Y.; Liu, Z. Conservation of potentially druggable cavities in intrinsically disordered proteins. ACS Omega 2018, 3, 15643–15652. [Google Scholar] [CrossRef] [PubMed]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005, 33, W299–W302. [Google Scholar] [CrossRef]
- Jiao, W.; Blackmore, N.J.; Nazmi, A.R.; Parker, E.J. Quaternary structure is an essential component that contributes to the sophisticated allosteric regulation mechanism in a key enzyme from Mycobacterium tuberculosis. PLoS ONE 2017, 12, e0180052. [Google Scholar] [CrossRef] [Green Version]
- Jensen, R.A.; Xie, G.; Calhoun, D.H.; Bonner, C.A. The correct phylogenetic relationship of KdsA (3-deoxy-D-manno-octulosonate 8-phosphate synthase) with one of two independently evolved classes of AroA (3-deoxy-D-arabino-heptulosonate 7-phosphate synthase). J. Mol. Evol. 2002, 54, 416–423. [Google Scholar] [CrossRef]
- Webby, C.J.; Baker, H.M.; Lott, J.S.; Baker, E.N.; Parker, E.J. The structure of 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase from Mycobacterium tuberculosis reveals a common catalytic scaffold and ancestry for type I and type II enzymes. J. Mol. Biol. 2005, 354, 927–939. [Google Scholar] [CrossRef]
- Light, S.H.; Anderson, W.F. The diversity of allosteric controls at the gateway to aromatic amino acid biosynthesis. Protein Sci. 2013, 22, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramaniam, P.S.; Xie, G.; Xia, T.H.; Jensen, R.A. Substrate ambiguity of 3-deoxy-D-manno-octulosonate 8-phosphate synthase from Neisseria gonorrhoeae in the context of its membership in a protein family containing a subset of 3-deoxy-D-arabino-heptulosonate 7-phosphate synthases. J. Bacteriol. 1998, 180, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sterritt, O.W.; Kessans, S.A.; Jameson, G.B.; Parker, E.J. A pseudoisostructural type II DAH7PS enzyme from Pseudomonas aeruginosa: Alternative evolutionary strategies to control shikimate pathway flux. Biochemistry 2018, 57, 2667–2678. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.M.; GonzalezBello, C.; Kleanthous, C.; Hawkins, A.R.; Coggins, J.R.; Abell, C. Evidence from kinetic isotope studies for an enolate intermediate in the mechanism of type II dehydroquinases. Biochem. J. 1996, 319, 333–336. [Google Scholar] [CrossRef] [Green Version]
- TRomanowski, M.J.; Burley, S.K. Crystal structure of the Escherichia coli shikimate kinase I (AroK) that confers sensitivity to mecillinam. Proteins-Struct. Funct. Genet. 2002, 47, 558–562. [Google Scholar] [CrossRef]
- Kortemme, T.; Baker, D. A Simple physical model for binding energy hot spots in protein–protein complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 14116–14121. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. Unraveling hot spots in binding interfaces: Progress and challenges. Curr. Opin. Struct. Biol. 2002, 12, 14–20. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Sharir-Ivry, A.; Xia, Y. Quantifying evolutionary importance of protein sites: A tale of two measures. PLoS Genet. 2021, 17, e1009476. [Google Scholar] [CrossRef]
- Ma, B.; Elkayam, T.; Wolfson, H.; Nussinov, R. Protein–protein interactions: Structurally conserved residues distinguish between binding sites and exposed protein surfaces. Proc. Natl. Acad. Sci. USA 2003, 100, 5772–5777. [Google Scholar] [CrossRef] [Green Version]
- Slodkowicz, G.; Goldman, N. Integrated structural and evolutionary analysis reveals common mechanisms underlying adaptive evolution in mammals. Proc. Natl. Acad. Sci. USA 2020, 117, 5977–5986. [Google Scholar] [CrossRef] [PubMed]
- Bornemann, S.; Ramjee, M.; Balasubramanian, S.; Abell, C.; Coggins, J.; Lowe, D.; Thorneley, R. Escherichia coli chorismate synthase catalyzes the conversion of (6S)-6-fluoro-5-enolpyruvylshikimate-3-phosphate to 6-fluorochorismate—Implications for the enzyme mechanism and the antimicrobial action of (6S)-6-fluoroshikimate. J. Biol. Chem. 1995, 270, 22811–22815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasan, N.; Nester, E. Dehydroquinate synthase in bacillus-subtilis—Enzyme associated with chorismate synthase and flavin reductase. J. Biol. Chem. 1978, 253, 4999–5004. [Google Scholar] [CrossRef]
- Bornemann, S.; Lowe, D.J.; Thorneley, R.N.F. The transient kinetics of Escherichia coli chorismate synthase: Substrate, consumption, product formation, phosphate dissociation, and characterization of a flavin intermediate. Biochemistry 1996, 35, 9907–9916. [Google Scholar] [CrossRef] [PubMed]
- Macheroux, P.; Schonbrunn, E.; Svergun, D.I.; Volkov, V.V.; Koch, M.H.J.; Bornemann, S.; Thorneley, R.N.F. Evidence for a major structural change in Escherichia coli chorismate synthase induced by flavin and substrate binding. Biochem. J. 1998, 335, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Maclean, J.; Ali, S. The structure of chorismate synthase reveals a novel flavin binding to a unique chemical reaction. Structure 2003, 11, 1499–1511. [Google Scholar] [CrossRef] [Green Version]
- Capra, J.A.; Laskowski, R.A.; Thornton, J.M.; Singh, M.; Funkhouser, T.A. Predicting protein ligand binding sites by combining evolutionary sequence conservation and 3D structure. PLoS Comput. Biol. 2009, 5, e1000585. [Google Scholar] [CrossRef] [Green Version]
- Christopoulos, A. Allosteric binding sites on cell-surface receptors: Novel targets for drug discovery. Nat. Rev. Drug Discov. 2002, 1, 198–210. [Google Scholar] [CrossRef]
- Wenthur, C.J.; Gentry, P.R.; Mathews, T.P.; Lindsley, C.W. Drugs for allosteric sites on receptors. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 165–184. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.; Mamidi, A.S.; Rajmani, R.S.; Ray, A.; Roy, R.; Surolia, A. An allosteric inhibitor of Mycobacterium tuberculosis ArgJ: Implications to a novel combinatorial therapy. EMBO Mol. Med. 2018, 10, e8038. [Google Scholar] [CrossRef]
- Wellington, S.; Nag, P.P.; Michalska, K.; Johnston, S.E.; Jedrzejczak, R.P.; Kaushik, V.K.; Clatworthy, A.E.; Siddiqi, N.; McCarren, P.; Bajrami, B.; et al. A small-molecule allosteric inhibitor of Mycobacterium tuberculosis tryptophan synthase. Nat. Chem. Biol. 2017, 13, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Ivetac, A.; Andrew McCammon, J. Mapping the druggable allosteric space of G-protein coupled receptors: A fragment-based molecular dynamics approach. Chem. Biol. Drug Des. 2010, 76, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.C.; Hurley, N.C.; Tortorici, M.; Ciossani, G.; Borrello, M.T.; Vellore, N.A.; Ganesan, A.; Mattevi, A.; Baron, R. Expanding the druggable space of the LSD1/CoREST epigenetic target: New potential binding regions for drug-like molecules, peptides, protein partners, and chromatin. PLoS Comput. Biol. 2013, 9, e1003158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vittorio, S.; Seidel, T.; Garon, A.; Gitto, R.; Langer, T.; De Luca, L. In silico identification of potential druggable binding sites on CIN85 SH3 domain. Int. J. Mol. Sci. 2021, 22, 534. [Google Scholar] [CrossRef]
- Borota, A.; Halip, L.; Curpan, R.; Bora, A.; Avram, S.; Mracec, M.; Mracec, M. Structure- and ligand- based studies to gain insight into the pharmacological implications of histamine H-3 receptor. Struct. Chem. 2021, 32, 1141–1149. [Google Scholar] [CrossRef]
- Veeravarapu, H.; Malkhed, V.; Mustyala, K.K.; Vadija, R.; Malikanti, R.; Vuruputuri, U.; Muthyala, M.K.K. Structure-based drug design, synthesis and screening of MmaA1 inhibitors as novel anti-TB agents. Mol. Divers. 2021, 25, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Muddagoni, N.; Bathula, R.; Dasari, M.; Potlapally, S.R. Homology modeling, virtual screening, prime-MMGBSA, AutoDock-identification of inhibitors of FGR protein. Biointerface Res. Appl. Chem. 2021, 11, 11088–11103. [Google Scholar] [CrossRef]
- Roky, M.; Trent, J.O.; Demuth, D.R. Identification of functional domains of the minor fimbrial antigen involved in the interaction of Porphyromonas gingivalis with oral streptococci. Mol. Oral Microbiol. 2020, 35, 66–77. [Google Scholar] [CrossRef] [Green Version]
- Bathula, R.; Lanka, G.; Muddagoni, N.; Dasari, M.; Nakkala, S.; Bhargavi, M.; Somadi, G.; Sivan, S.K.; Potlapally, S.R. Identification of potential aurora kinase-C protein inhibitors: An amalgamation of energy minimization, virtual screening, prime MMGBSA and AutoDock. J. Biomol. Struct. Dyn. 2020, 38, 2314–2325. [Google Scholar] [CrossRef]
- Zhou, W.; Lu, S.; Zhang, Y.; Yin, L.; Zhu, L.; Zhao, J.; Lu, T.; Chen, Y.; Liu, H. In silico design of new B-raf kinase type-II inhibitors through combined molecular modeling studies. Lett. Drug Des. Discov. 2019, 16, 570–583. [Google Scholar] [CrossRef]
- Verma, P.; Tiwari, V. Targeting outer membrane protein component AdeC for the discovery of efflux pump inhibitor against AdeABC efflux pump of multidrug resistant Acinetobacter baumannii. Cell Biochem. Biophys. 2018, 76, 391–400. [Google Scholar] [CrossRef]
- Usman, M.M.S.; Bharbhuiya, T.K.; Mondal, S.; Rani, S.; Kyal, C.; Kumari, R. Combined protein and ligand based physicochemical aspects of molecular recognition for the discovery of CDK9 inhibitor. Gene Rep. 2018, 13, 212–219. [Google Scholar] [CrossRef]
- Bowkett, D.; Talon, R.; Tallant, C.; Schofield, C.; von Delft, F.; Knapp, S.; Bruton, G.; Brennan, P.E. Identifying small-molecule binding sites for epigenetic proteins at domain-domain interfaces. Chemmedchem 2018, 13, 1051–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adeniji, E.A.; Olotu, F.A.; Soliman, M.E.S. Exploring the lapse in druggability: Sequence analysis, structural dynamics and binding site characterization of K-Ras(G12C) variant, a feasible oncotherapeutics target. Anticancer Agents Med. Chem. 2018, 18, 1540–1550. [Google Scholar] [CrossRef]
- Olotu, F.A.; Omolabi, K.F.; Soliman, M.E.S. Leaving no stone unturned: Allosteric targeting of SARS-CoV-2 spike protein at putative druggable sites disrupts human angiotensin-converting enzyme interactions at the receptor binding domain. Inform. Med. Unlocked 2020, 21, 100451. [Google Scholar] [CrossRef] [PubMed]
- David-Eden, H.; Mankin, A.S.; Mandel-Gutfreund, Y. Structural signatures of antibiotic binding sites on the ribosome. Nucleic Acids Res. 2010, 38, 5982–5994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayyildiz, M.; Celiker, S.; Ozhelvaci, F.; Akten, E.D. Identification of alternative allosteric sites in glycolytic enzymes for potential use as species-specific drug targets. Front. Mol. Biosci. 2020, 7, 88. [Google Scholar] [CrossRef]
- Sheik Amamuddy, O.; Veldman, W.; Manyumwa, C.; Khairallah, A.; Agajanian, S.; Oluyemi, O.; Verkhivker, G.M.; Tastan Bishop, Ö. Integrated computational approaches and tools for allosteric drug discovery. Int. J. Mol. Sci. 2020, 21, 847. [Google Scholar] [CrossRef] [Green Version]
- Perola, E.; Herman, L.; Weiss, J. Development of a rule-based method for the assessment of protein druggability. J. Chem. Inf. Model. 2012, 52, 1027–1038. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Huth, J.R.; Tse, C. Predicting protein druggability. Drug Discov. Today 2005, 10, 1675–1682. [Google Scholar] [CrossRef]
- Gao, M.; Skolnick, J. A Comprehensive survey of small-molecule binding pockets in proteins. PLoS Comput. Biol. 2013, 9, e1003302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, R.D.; Lu, J.; Carlson, H.A. Are there physicochemical differences between allosteric and competitive ligands? PLoS Comput. Biol. 2017, 13, e1005813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; He, X.; Ni, D.; Zhang, J. Allosteric modulator discovery: From serendipity to structure-based design. J. Med. Chem. 2019, 62, 6405–6421. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, H.; Zhu, L.; Wu, Z.; Li, W.; Tang, Y.; Liu, G. In silico prediction of human renal clearance of compounds using quantitative structure-pharmacokinetic relationship models. Chem. Res. Toxicol. 2020, 33, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.C.; Coleman, R.G.; Smyth, K.T.; Cao, Q.; Soulard, P.; Caffrey, D.R.; Salzberg, A.C.; Huang, E.S. Structure-based maximal affinity model predicts small-molecule druggability. Nat. Biotechnol. 2007, 25, 71–75. [Google Scholar] [CrossRef]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME—The konstanz information miner: Version 2.0 and beyond. ACM SIGKDD Explor. Newsl. 2009, 11, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating Evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef] [Green Version]
- Celniker, G.; Nimrod, G.; Ashkenazy, H.; Glaser, F.; Martz, E.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf: Using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem. 2013, 53, 199–206. [Google Scholar] [CrossRef]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An Improved Methodology to Estimate and Visualize Evolutionary Conservation in Macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. Available online: https://academic.oup.com/nar/article/44/W1/W344/2499373 (accessed on 21 June 2021). [CrossRef] [Green Version]
- The PyMOL Molecular Graphics System, Version 2.3.3; Schrödinger, LLC.: New York, NY, USA, 2019.
- Gonçalves, W.R.S.; Gonçalves-Almeida, V.M.; Arruda, A.L.; Meira, W., Jr.; da Silveira, C.H.; Pires, D.E.V.; de Melo-Minardi, R.C. PDBest: A user–friendly platform for manipulating and enhancing protein structures. Bioinformatics 2015, 31, 2894–2896. [Google Scholar] [CrossRef] [Green Version]
- KNIME: The Konstanz Information Miner. SpringerLink. Available online: https://link.springer.com/chapter/10.1007/978-3-540-78246-9_38 (accessed on 21 June 2021).
- Schrödinger Release 2021-3: Maestro; Schrödinger, LLC.: New York, NY, USA, 2021.
- UniProt consortium UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [CrossRef] [PubMed]
- KEGG: Kyoto Encyclopedia of Genes and Genomes. Available online: http://www.kegg.jp/ (accessed on 14 October 2021).
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henikoff, S.; Henikoff, J.G. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef] [Green Version]
- Mayrose, I.; Graur, D.; Ben-Tal, N.; Pupko, T. Comparison of site-specific rate-inference methods for protein sequences: Empirical bayesian methods are superior. Mol. Biol. Evol. 2004, 21, 1781–1791. [Google Scholar] [CrossRef] [Green Version]
- Pupko, T.; Graur, D. Fast computation of maximum likelihood trees by numerical approximation of amino acid replacement probabilities. Comput. Stat. Data Anal. 2002, 40, 285–291. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for PK(a) prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Schrödinger Release 2022-1: Epik; Schrödinger, LLC.: New York, NY, USA, 2021.
- Waskom, M.L. Seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.; Bussonnier, M.; Frederic, J.; Kelley, K.; Hamrick, J.; Grout, J.; Corlay, S.; et al. Jupyter notebooks—A publishing format for reproducible computational workflows. Position. Power Acad. Publ. Play. Agents Agendas 2016, 87–90. [Google Scholar] [CrossRef]
- Blanco, B.; Prado, V.; Lence, E.; Otero, J.M.; Garcia-Doval, C.; van Raaij, M.J.; Llamas-Saiz, A.L.; Lamb, H.; Hawkins, A.R.; Gonzalez-Bello, C. Mycobacterium tuberculosis Shikimate Kinase Inhibitors: Design and Simulation Studies of the Catalytic Turnover. J. Am. Chem. Soc. 2013, 135, 12366–12376. [Google Scholar] [CrossRef]
- Balachandran, N.; Heimhalt, M.; Liuni, P.; To, F.; Wilson, D.J.; Junop, M.S.; Berti, P.J. Potent Inhibition of 3-Deoxy-D-Arabinoheptulosonate-7-Phosphate (DAHP) Synthase by DAHP Oxime, a Phosphate Group Mimic. Biochemistry 2016, 55, 6617–6629. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.R.; Jiao, W.; Parker, E.J. Synthesis and Evaluation of Dual Site Inhibitors of 3-Deoxy-D-Arabino-Heptulosonate 7-Phosphate Synthase. Bioorg. Med. Chem. Lett. 2011, 21, 5092–5097. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.L.; Corey, S.D.; Font, J.L.; Walker, M.C.; Sikorski, J.A. New Simplified Inhibitors of EPSP Synthase: The Importance of Ring Size for Recognition at the Shikimate 3-Phosphate Site. Bioorg. Med. Chem. Lett. 1996, 6, 2853–2858. [Google Scholar] [CrossRef]
- Zhu, N.; Wang, X.; Li, D.; Lin, Y.; You, X.; Jiang, J.; Xu, Y.; Jiang, W.; Si, S. IMB-T130 Targets 3-Dehydroquinate Synthase and Inhibits Mycobacterium tuberculosis. Sci. Rep. 2018, 8, 17439. [Google Scholar] [CrossRef]
- Montchamp, J.L.; Frost, J.W. Cyclohexenyl and Cyclohexylidene Inhibitors of 3-Dehydroquinate Synthase: Active Site Interactions Relevant to Enzyme Mechanism and Inhibitor Design. J. Am. Chem. Soc. 1997, 119, 7645–7653. [Google Scholar] [CrossRef]
- Prado, V.; Lence, E.; Maneiro, M.; Vazquez-Ucha, J.C.; Beceiro, A.; Thompson, P.; Hawkins, A.R.; Gonzalez-Bello, C. Targeting the Motion of Shikimate Kinase: Development of Competitive Inhibitors That Stabilize an Inactive Open Conformation of the Enzyme. J. Med. Chem. 2016, 59, 5471–5487. [Google Scholar] [CrossRef]
- Gonzalez-Bello, C.; Manthey, M.K.; Harris, J.H.; Hawkins, A.R.; Coggins, J.R.; Abell, C. Synthesis of 2-Bromo- and 2-Fluoro-3-Dehydroshikimic Acids and 2-Bromo- and 2-Fluoroshikimic Acids Using Synthetic and Enzymatic Approaches. J. Org. Chem. 1998, 63, 1591–1597. [Google Scholar] [CrossRef]
- Lence, E.; Tizon, L.; Otero, J.M.; Peon, A.; Prazeres, V.F.V.; Llamas-Saiz, A.L.; Fox, G.C.; van Raaij, M.J.; Lamb, H.; Hawkins, A.R.; et al. Mechanistic Basis of the Inhibition of Type II Dehydroquinase by (2S)- and (2R)-2-Benzyl-3-Dehydroquinic Acids. Acs Chem. Biol. 2013, 8, 568–577. [Google Scholar] [CrossRef]
- Thomas, M.G.; Lawson, C.; Allanson, N.M.; Leslie, B.W.; Bottomley, J.R.; McBride, A.; Olusanya, O.A. A Series of 2(Z)-2-Benzylidene-6,7-Dihydroxybenzofuran-3[2H]-Ones as Inhibitors of Chorismate Synthase. Bioorg. Med. Chem. Lett. 2003, 13, 423–426. [Google Scholar] [CrossRef]
- Peek, J.; Shi, T.; Christendat, D. Identification of Novel Polyphenolic Inhibitors of Shikimate Dehydrogenase (AroE). J. Biomol. Screen. 2014, 19, 1090–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Lower Limit 1 |
|---|---|
| Number of site points | 132 |
| Site score | 1 (0.8) |
| DScore | 1 (0.8) |
| Exposure | 0.49 |
| Enclosure | 0.78 |
| Contact | 1.0 |
| Phobic, Philic 2 | 1.0 |
| HL balance 3 | 1.6 |
| Donor/acceptor | 0.76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frlan, R. An Evolutionary Conservation and Druggability Analysis of Enzymes Belonging to the Bacterial Shikimate Pathway. Antibiotics 2022, 11, 675. https://doi.org/10.3390/antibiotics11050675
Frlan R. An Evolutionary Conservation and Druggability Analysis of Enzymes Belonging to the Bacterial Shikimate Pathway. Antibiotics. 2022; 11(5):675. https://doi.org/10.3390/antibiotics11050675
Chicago/Turabian StyleFrlan, Rok. 2022. "An Evolutionary Conservation and Druggability Analysis of Enzymes Belonging to the Bacterial Shikimate Pathway" Antibiotics 11, no. 5: 675. https://doi.org/10.3390/antibiotics11050675