
Improvement of the Antibacterial Activity of Phage Lysin-Derived Peptide P87 through Maximization of Physicochemical Properties and Assessment of Its Therapeutic Potential
 , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
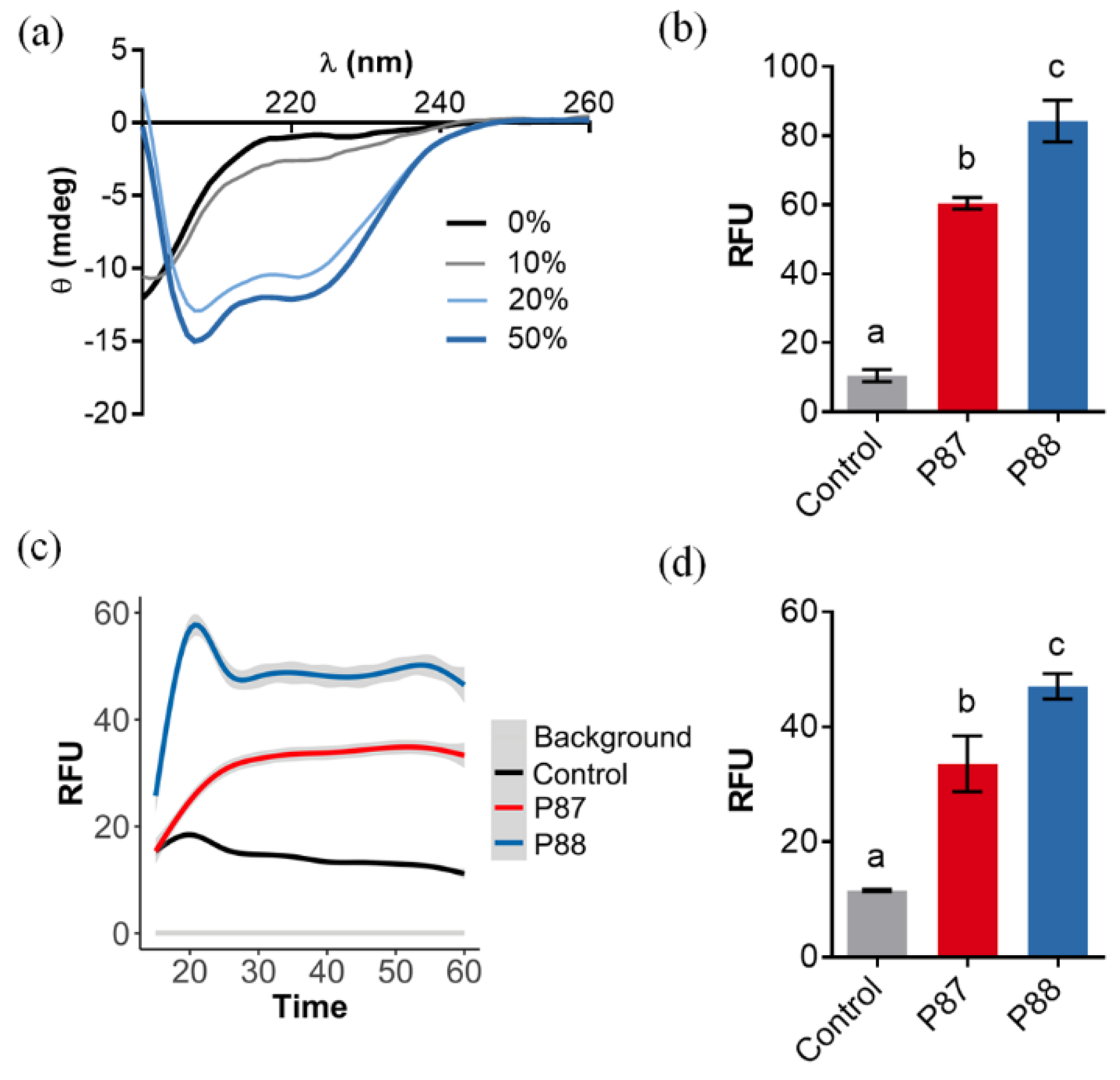
2.1. Design of an Optimized AMP Based on P87
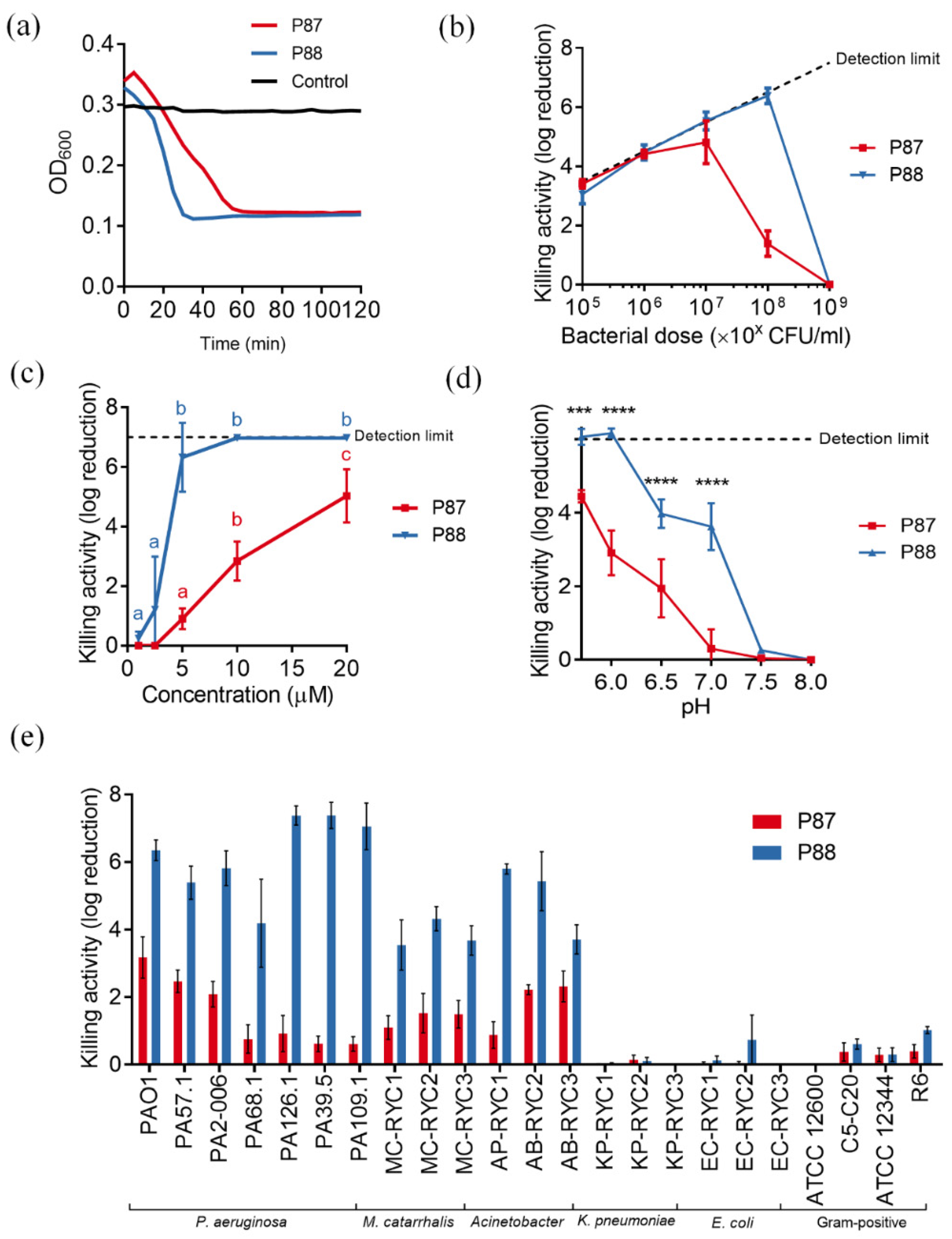
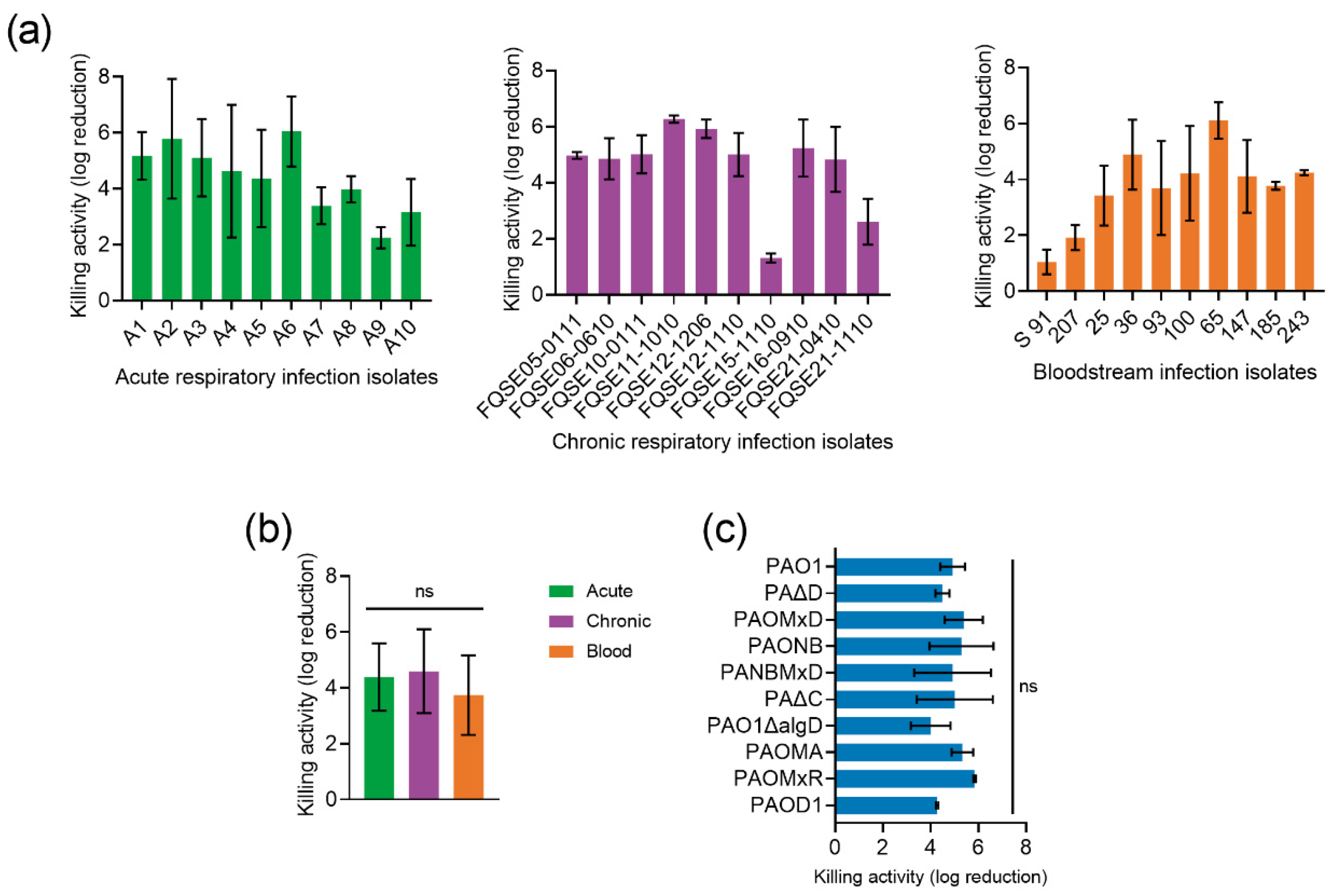
2.2. Compared Antibacterial Activity of the Novel Peptide P88
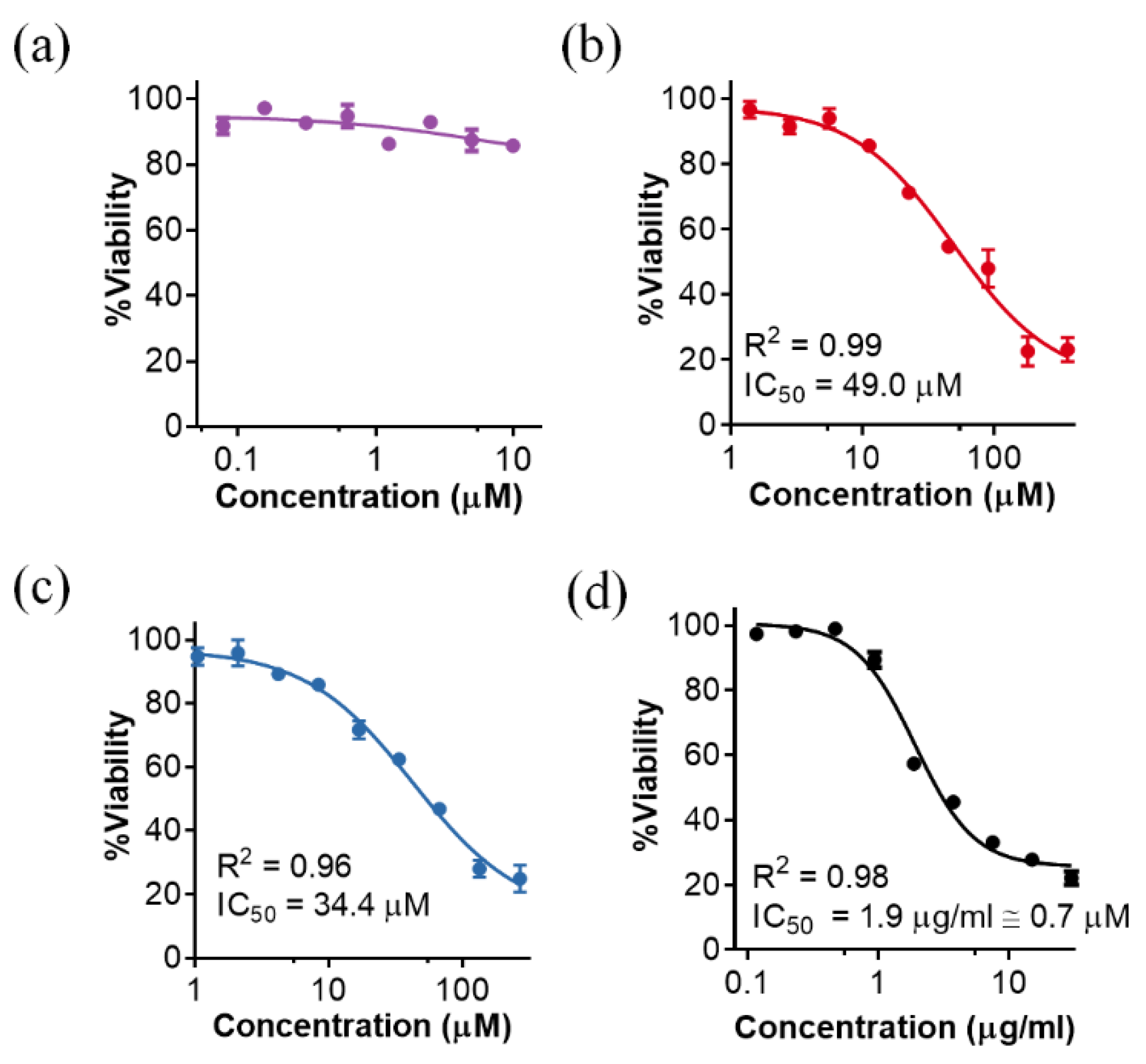
2.3. Cytotoxicity of P88
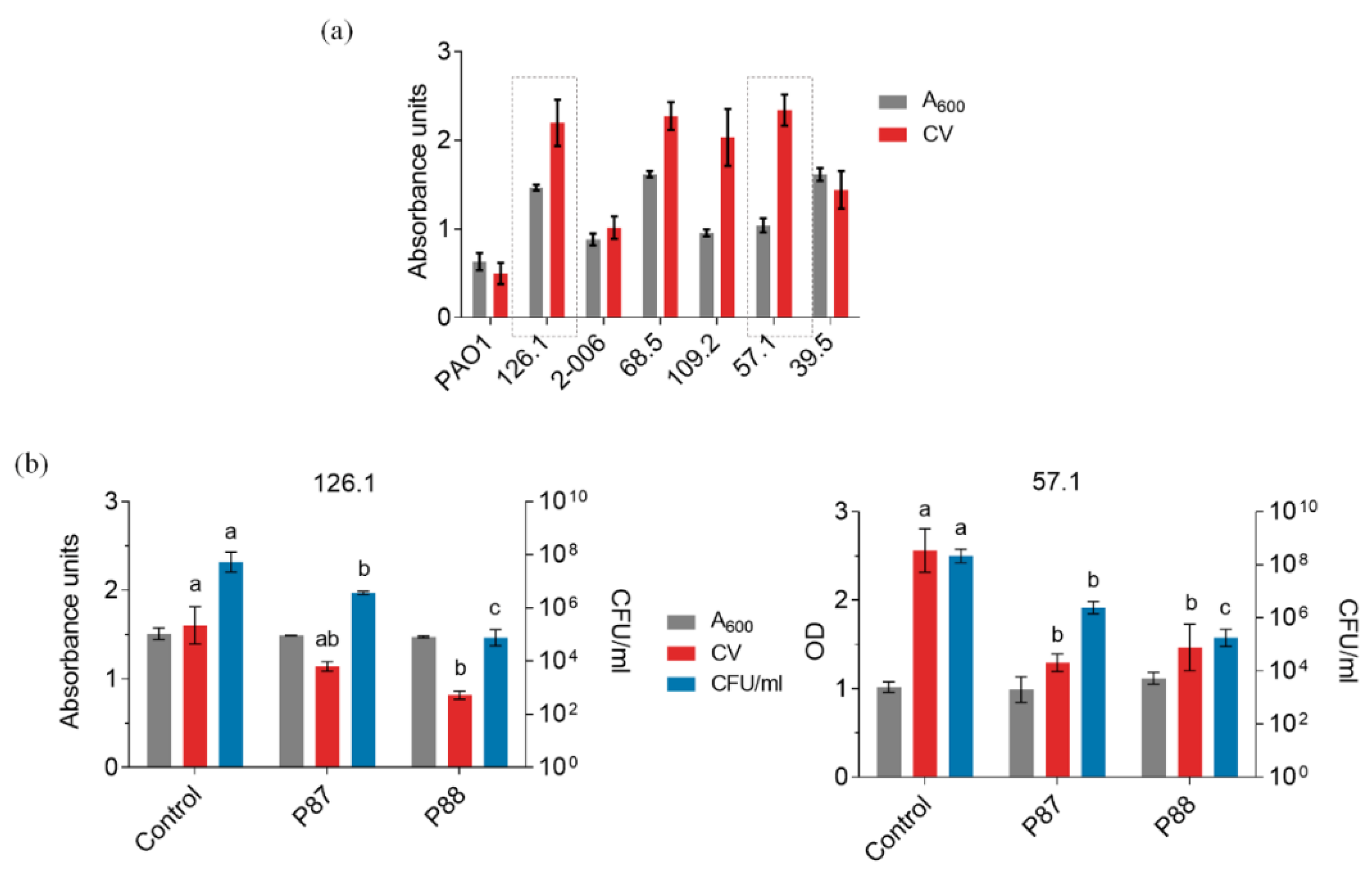
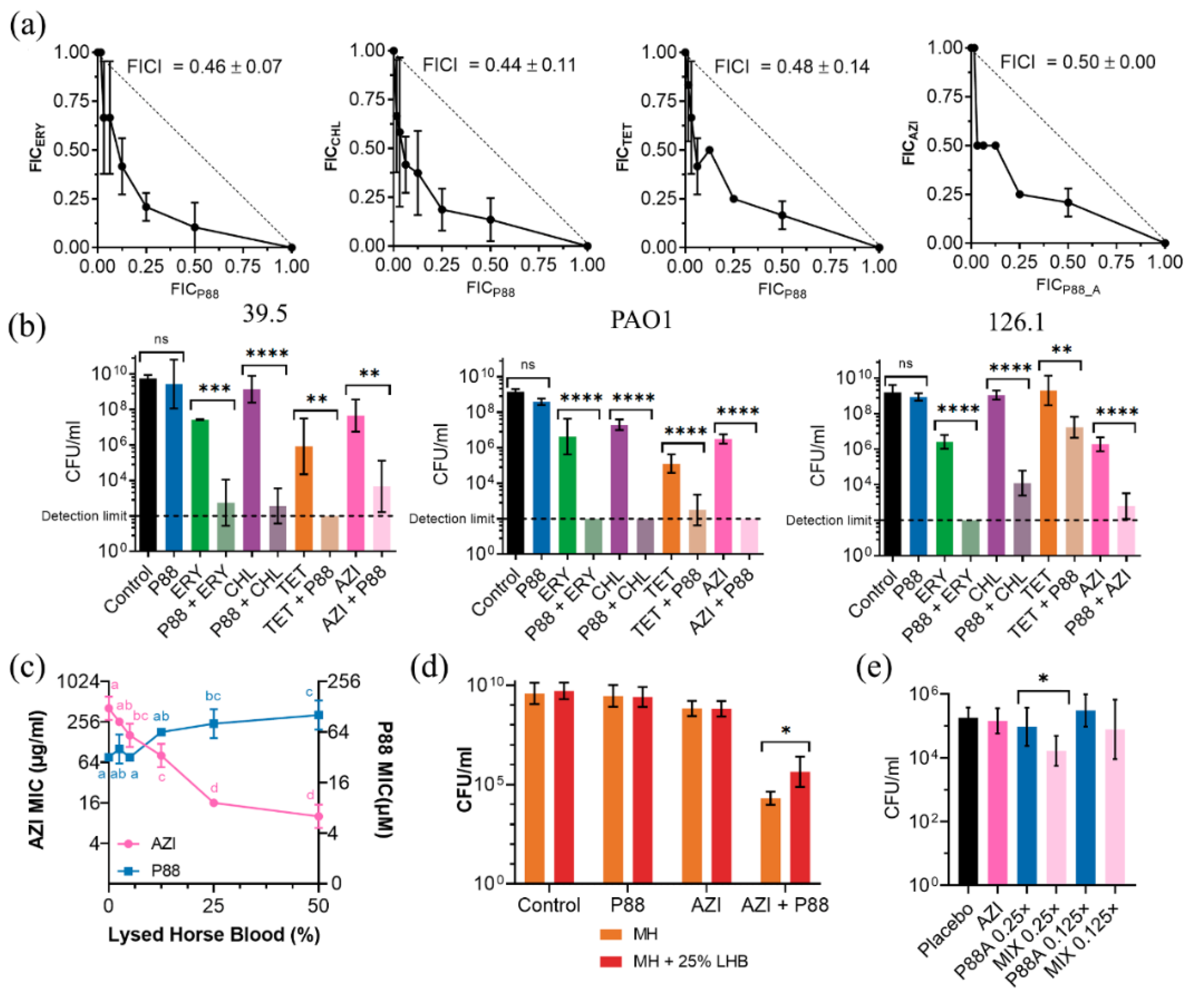
2.4. Synergy of P88 with Antibiotics
3. Conclusions
4. Materials and Methods
4.1. Bacterial Strains and Culture Conditions
4.2. Proteins and Peptides Obtention
4.3. Circular Dichroism
4.4. Cell Permeability Fluorescence Assays
4.5. Antibacterial Activity Resting Cells Assays
4.6. Cytotoxicity
4.7. Minimum Inhibitory Concentrations and Synergy
4.8. Biofilms
4.9. In Vivo Mouse Pneumonia Models
4.10. Bioinformatic Analyses
4.11. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dams, D.; Briers, Y. Enzybiotics: Enzyme-based antibacterials as therapeutics. Adv. Exp. Med. Biol. 2019, 1148, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Abdelkader, K.; Gerstmans, H.; Saafan, A.; Dishisha, T.; Briers, Y. The preclinical and clinical progress of bacteriophages and their lytic enzymes: The parts are easier than the whole. Viruses 2019, 11, 96. [Google Scholar] [CrossRef] [Green Version]
- Linden, S.B.; Alreja, A.B.; Nelson, D.C. Application of bacteriophage-derived endolysins to combat streptococcal disease: Current state and perspectives. Curr. Opin. Biotechnol. 2021, 68, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, D.; Briers, Y. Lysins breaking down the walls of Gram-negative bacteria, no longer a no-go. Curr. Opin. Biotechnol. 2021, 68, 15–22. [Google Scholar] [CrossRef]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Lee, E.Y.; Wong, G.C.L.; Ferguson, A.L. Machine learning-enabled discovery and design of membrane-active peptides. Bioorg. Med. Chem. 2018, 26, 2708–2718. [Google Scholar] [CrossRef]
- Wang, G. Improved methods for classification, prediction, and design of antimicrobial peptides. Methods Mol. Biol. 2015, 1268, 43–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porto, W.F.; Irazazabal, L.; Alves, E.S.F.; Ribeiro, S.M.; Matos, C.O.; Pires, A.S.; Fensterseifer, I.C.M.; Miranda, V.J.; Haney, E.F.; Humblot, V.; et al. In silico optimization of a guava antimicrobial peptide enables combinatorial exploration for peptide design. Nat. Commun. 2018, 9, 1490. [Google Scholar] [CrossRef] [Green Version]
- Rotem, S.; Radzishevsky, I.; Inouye, R.T.; Samore, M.; Mor, A. Identification of antimicrobial peptide regions derived from genomic sequences of phage lysins. Peptides 2006, 27, 18–26. [Google Scholar] [CrossRef]
- Thandar, M.; Lood, R.; Winer, B.Y.; Deutsch, D.R.; Euler, C.W.; Fischetti, V.A. Novel engineered peptides of a phage lysin as effective antimicrobials against multidrug-resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 2016, 60, 2671–2679. [Google Scholar] [CrossRef]
- Peng, S.Y.; You, R.I.; Lai, M.J.; Lin, N.T.; Chen, L.K.; Chang, K.C. Highly potent antimicrobial modified peptides derived from the Acinetobacter baumannii phage endolysin LysAB2. Sci. Rep. 2017, 7, 11477. [Google Scholar] [CrossRef] [Green Version]
- Vázquez, R.; García, E.; García, P. Sequence-function relationships in phage-encoded bacterial cell wall lytic enzymes and their implications for phage-derived products design. J. Virol. 2021, 95, e0032121. [Google Scholar] [CrossRef]
- Ghose, C.; Euler, C.W. Gram-Negative Bacterial Lysins. Antibiotics 2020, 9, 74. [Google Scholar] [CrossRef] [Green Version]
- Sykilinda, N.N.; Nikolaeva, A.Y.; Shneider, M.M.; Mishkin, D.V.; Patutin, A.A.; Popov, V.O.; Boyko, K.M.; Klyachko, N.L.; Miroshnikov, K.A. Structure of an Acinetobacter broad-range prophage endolysin reveals a C-terminal alpha-helix with the proposed role in activity against live bacterial cells. Viruses 2018, 10, 309. [Google Scholar] [CrossRef] [Green Version]
- Vázquez, R.; Blanco-Gañán, S.; Ruiz, S.; García, P. Mining of Gram-negative surface-active enzybiotic candidates by sequence-based calculation of physicochemical properties. Front. Microbiol. 2021, 12, 660403. [Google Scholar] [CrossRef]
- Vázquez, R.; Seoane-Blanco, M.; Rivero-Buceta, V.; Ruiz, S.; van Raaij, M.J.; García, P. Monomodular Pseudomonas aeruginosa phage JG004 lysozyme (Pae87) contains a bacterial surface-active antimicrobial peptide-like region and a possible substrate-binding subdomain. Acta Crystallogr. D Struct. Biol. 2022, 78, 435–454. [Google Scholar] [CrossRef] [PubMed]
- Zelezetsky, I.; Tossi, A. Alpha-helical antimicrobial peptides--using a sequence template to guide structure-activity relationship studies. Biochim. Biophys. Acta 2006, 1758, 1436–1449. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta 2009, 1788, 1687–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saralegui, C.; Herencias, C.; Halperin, A.V.; de Dios-Caballero, J.; Perez-Viso, B.; Salgado, S.; Lanza, V.F.; Canton, R.; Baquero, F.; Prieto, M.A.; et al. Strain-specific predation of Bdellovibrio bacteriovorus on Pseudomonas aeruginosa with a higher range for cystic fibrosis than for bacteremia isolates. Sci. Rep. 2022, 12, 10523. [Google Scholar] [CrossRef] [PubMed]
- Salas, M.; Wernecki, M.; Fernandez, L.; Iglesias, B.; Gutierrez, D.; Alvarez, A.; Garcia, L.; Prieto, E.; Garcia, P.; Rodriguez, A. Characterization of Clinical MRSA Isolates from Northern Spain and Assessment of Their Susceptibility to Phage-Derived Antimicrobials. Antibiotics 2020, 9, 447. [Google Scholar] [CrossRef]
- Mulcahy, L.R.; Isabella, V.M.; Lewis, K. Pseudomonas aeruginosa biofilms in disease. Microb. Ecol. 2014, 68, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fricks-Lima, J.; Hendrickson, C.M.; Allgaier, M.; Zhuo, H.; Wiener-Kronish, J.P.; Lynch, S.V.; Yang, K. Differences in biofilm formation and antimicrobial resistance of Pseudomonas aeruginosa isolated from airways of mechanically ventilated patients and cystic fibrosis patients. Int. J. Antimicrob. Agents 2011, 37, 309–315. [Google Scholar] [CrossRef] [Green Version]
- Head, N.E.; Yu, H. Cross-sectional analysis of clinical and environmental isolates of Pseudomonas aeruginosa: Biofilm formation, virulence, and genome diversity. Infect. Immun. 2004, 72, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raz, A.; Serrano, A.; Hernandez, A.; Euler, C.W.; Fischetti, V.A. Isolation of phage lysins that effectively kill Pseudomonas aeruginosa in mouse models of lung and skin infection. Antimicrob. Agents Chemother. 2019, 63, e00024-19. [Google Scholar] [CrossRef] [Green Version]
- Dosler, S.; Karaaslan, E. Inhibition and destruction of Pseudomonas aeruginosa biofilms by antibiotics and antimicrobial peptides. Peptides 2014, 62, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Picoli, T.; Peter, C.M.; Zani, J.L.; Waller, S.B.; Lopes, M.G.; Boesche, K.N.; Vargas, G.D.A.; Hubner, S.O.; Fischer, G. Melittin and its potential in the destruction and inhibition of the biofilm formation by Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa isolated from bovine milk. Microb. Pathog. 2017, 112, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Bouzas, V.; Haller, T.; Hobi, N.; Felder, E.; Pastoriza-Santos, I.; Perez-Gil, J. Nontoxic impact of PEG-coated gold nanospheres on functional pulmonary surfactant-secreting alveolar type II cells. Nanotoxicology 2014, 8, 813–823. [Google Scholar] [CrossRef]
- Sierra, J.M.; Fuste, E.; Rabanal, F.; Vinuesa, T.; Vinas, M. An overview of antimicrobial peptides and the latest advances in their development. Expert. Opin. Biol. Ther. 2017, 17, 663–676. [Google Scholar] [CrossRef]
- Giacometti, A.; Cirioni, O.; Del Prete, M.S.; Paggi, A.M.; D’Errico, M.M.; Scalise, G. Combination studies between polycationic peptides and clinically used antibiotics against Gram-positive and Gram-negative bacteria. Peptides 2000, 21, 1155–1160. [Google Scholar] [CrossRef]
- Buyck, J.M.; Plesiat, P.; Traore, H.; Vanderbist, F.; Tulkens, P.M.; Van Bambeke, F. Increased susceptibility of Pseudomonas aeruginosa to macrolides and ketolides in eukaryotic cell culture media and biological fluids due to decreased expression of oprM and increased outer-membrane permeability. Clin. Infect. Dis. 2012, 55, 534–542. [Google Scholar] [CrossRef]
- Lopez-Causape, C.; Rojo-Molinero, E.; Mulet, X.; Cabot, G.; Moya, B.; Figuerola, J.; Togores, B.; Perez, J.L.; Oliver, A. Clonal dissemination, emergence of mutator lineages and antibiotic resistance evolution in Pseudomonas aeruginosa cystic fibrosis chronic lung infection. PLoS ONE 2013, 8, e71001. [Google Scholar] [CrossRef] [PubMed]
- Barbier, M.; Oliver, A.; Rao, J.; Hanna, S.L.; Goldberg, J.B.; Alberti, S. Novel phosphorylcholine-containing protein of Pseudomonas aeruginosa chronic infection isolates interacts with airway epithelial cells. J. Infect. Dis. 2008, 197, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Barbier, M.; Martinez-Ramos, I.; Townsend, P.; Alberti, S. Surfactant protein A blocks recognition of Pseudomonas aeruginosa by CKAP4/P63 on airway epithelial cells. J. Infect. Dis. 2012, 206, 1753–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Ramos, I.; Mulet, X.; Moya, B.; Barbier, M.; Oliver, A.; Alberti, S. Overexpression of MexCD-OprJ reduces Pseudomonas aeruginosa virulence by increasing its susceptibility to complement-mediated killing. Antimicrob. Agents Chemother. 2014, 58, 2426–2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically, 11th ed.; CLSI: Wayne, PA, USA, 2018. [Google Scholar]
- CLSI. Methods for Determining Bactericidal Activity of Antimicrobial Agents, 1st ed.; CLSI: Wayne, PA, USA, 1999. [Google Scholar]
- Vázquez, R.; García, P. Synergy between two chimeric lysins to kill Streptococcus pneumoniae. Front. Microbiol. 2019, 10, 1251. [Google Scholar] [CrossRef] [Green Version]
- Odds, F.C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003, 52, 1. [Google Scholar] [CrossRef]
- Ren, H.; Liu, Y.; Zhou, J.; Long, Y.; Liu, C.; Xia, B.; Shi, J.; Fan, Z.; Liang, Y.; Chen, S.; et al. Combination of Azithromycin and Gentamicin for Efficient Treatment of Pseudomonas aeruginosa Infections. J. Infect. Dis. 2019, 220, 1667–1678. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2 Elegant Graphics for Data Analysis, 2nd ed.; Springer International Publishing: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. HELIQUEST: A web server to screen sequences with specific alpha-helical properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drozdetskiy, A.; Cole, C.; Procter, J.; Barton, G.J. JPred4: A protein secondary structure prediction server. Nucleic Acids Res. 2015, 43, W389–W394. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial | PAO1 | 39.5 | 126.1 |
|---|---|---|---|
| P88 | 20 (68) 2 | 50 (170) | 50 (170) |
| LVX | 0.25 | 0.13 | 0.25 |
| ERY | 512 | 512 | 256 |
| KAN | 16 | 256 | 256 |
| CHL | 512 | 512 | 128 |
| TET | 64 | 1024 | 32 |
| GEN | 0.20 | >16.00 | 2.00 |
| AZI | 128 | 512 | 256 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez, R.; Doménech-Sánchez, A.; Ruiz, S.; Sempere, J.; Yuste, J.; Albertí, S.; García, P. Improvement of the Antibacterial Activity of Phage Lysin-Derived Peptide P87 through Maximization of Physicochemical Properties and Assessment of Its Therapeutic Potential. Antibiotics 2022, 11, 1448. https://doi.org/10.3390/antibiotics11101448
Vázquez R, Doménech-Sánchez A, Ruiz S, Sempere J, Yuste J, Albertí S, García P. Improvement of the Antibacterial Activity of Phage Lysin-Derived Peptide P87 through Maximization of Physicochemical Properties and Assessment of Its Therapeutic Potential. Antibiotics. 2022; 11(10):1448. https://doi.org/10.3390/antibiotics11101448
Chicago/Turabian StyleVázquez, Roberto, Antonio Doménech-Sánchez, Susana Ruiz, Julio Sempere, Jose Yuste, Sebastián Albertí, and Pedro García. 2022. "Improvement of the Antibacterial Activity of Phage Lysin-Derived Peptide P87 through Maximization of Physicochemical Properties and Assessment of Its Therapeutic Potential" Antibiotics 11, no. 10: 1448. https://doi.org/10.3390/antibiotics11101448