
How to Combat Gram-Negative Bacteria Using Antimicrobial Peptides: A Challenge or an Unattainable Goal?

 , , and
, , and
Abstract
:1. Introduction
2. Which AMPs Are Available for Combating Gram-Negative Bacteria?
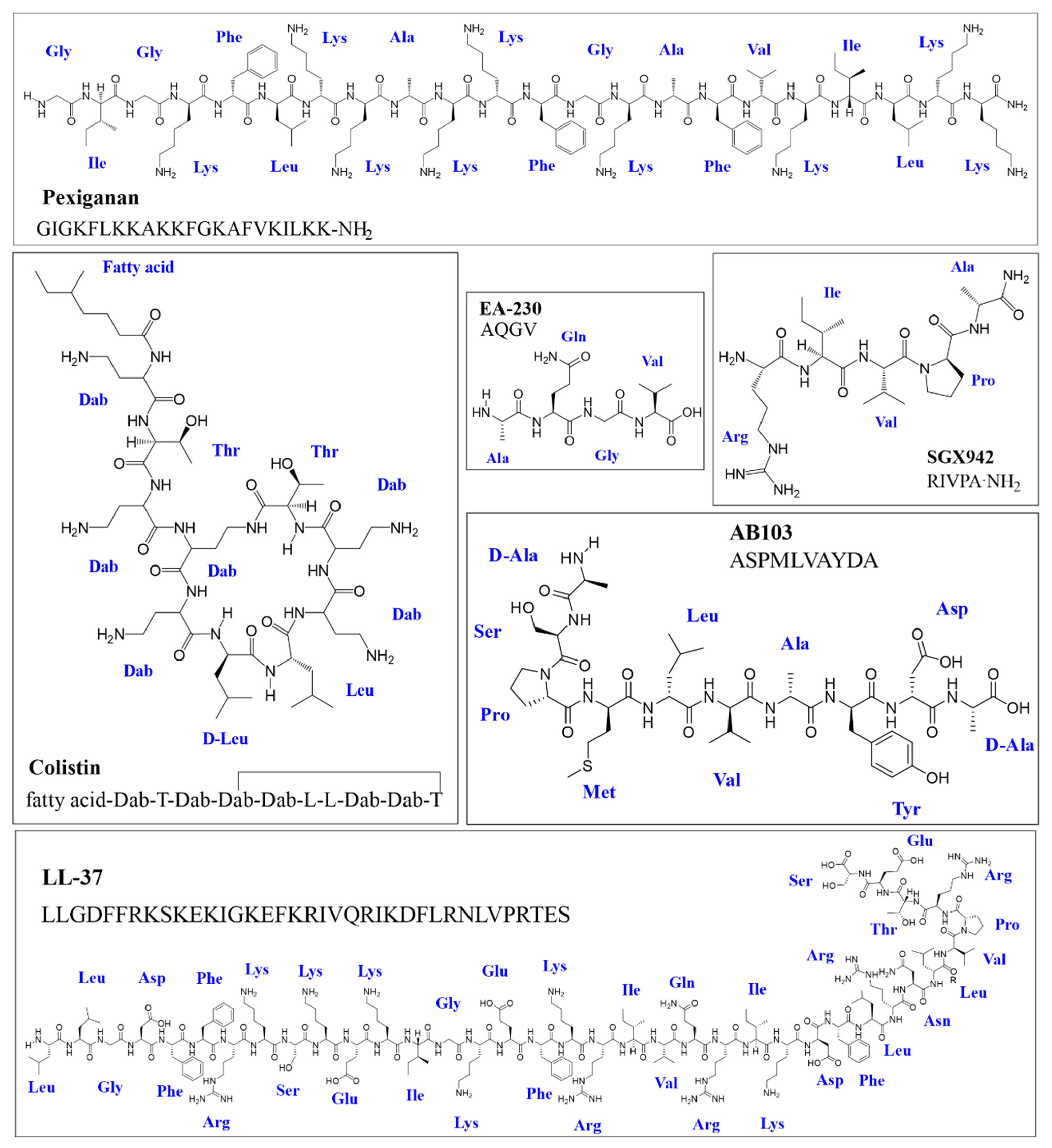
- Colistin or polymyxin (natural): Colistin is a cyclic decapeptide incorporating a fatty acid chain at the N-terminus with six Dab (an unusual amino acid called L-2,4-diaminobutyric acid) (Figure 1). It has a +5 net charge at physiological pH and has direct membranolytic antibacterial activity and potent anti-endotoxin activity due to its direct interaction with lipopolysaccharide (LPS) [40,41]. Colistin was discovered in 1947, FDA approved and used for more than a decade (since 1960); however, in the 1970s it became overlooked due to its adverse effects. Nonetheless, due to increasing preoccupation regarding the lack of effective new antimicrobials, its controlled use has been reconsidered and is now regarded as a safe drug [42]. Colistin is currently on the World Health Organisation’s model list of essential medicines (21st list, 2019) as a last-resort antibiotic and is the only AMP approved that is selective against Gram-negative bacteria [43].
- Pexiganan or MSI 78 (designed): Pexiganan was the first animal-derived peptide (isolated from frog skin) to reach phase III clinical studies. It is a 22 residue-long molecule which was synthesised with the amide group at the C-terminus extreme and is currently in phase III clinical studies (Figure 1) [19]. This molecule disrupts the membrane via toroidal-type pore formation thereby enabling it to have a bactericidal effect against Gram-negative and Gram-positive bacteria [44]. The best results have been obtained with this molecule regarding a cream for treating diabetic foot infections [27,28]; however, it is not FDA approved because it has not been shown to have advantages over currently available agents in terms of safety and therapeutic effectiveness [19].
- LL-37 (natural): LL-37 is a natural human cathelicidin hCAP18-derived from a 37 amino acid-long peptide (Figure 1). LL-37 is currently in phase II trials regarding its topical use for treating bacterial colonisation, inflammation response and diabetic foot ulcer healing rate (NCT04098562). This peptide has been one of the most extensively studied AMPs due to its diverse biological properties as it has an LPS neutralising, immunomodulatory and chemiotactic effect in addition to having direct antimicrobial activity against a broad range of microorganisms by pore formation [30,45]. However, its toxicity has led to problems regarding protease stability and production costs associated with its length; multiple shorter derivatives have been evaluated, demonstrating improved properties which would probably have greater therapeutic scope than LL-37 [46].
- Other AMPs in clinical stage trials: Broad-spectrum AMPs (i.e., those derived from melittin and protamine, 29 amino acid-long melimine (ACTRN12613000369729) and its 15 amino acid-long derivative mel4 (ACTRN1261500072556)) are being evaluated in clinical phase as alternatives for preventing Gram-negative and Gram-positive microbial colonisation of contact lenses [32,33]. Engineered cationic antimicrobial peptide (eCAP) WLBU2 (PLG0206) is in phase I clinical trials (ACTRN12618001920280); it has been projected for the treatment of prosthetic joint infections due to its anti-Gram-positive bacteria activity [34]. However, this broad-spectrum peptide could be useful for treating Gram-negative bacteria infections such as P. aeruginosa and other multi-resistant bacteria [39]. WLBU2 has proven to be superior to natural peptides such as colistin and LL-37 due to it having less potential for inducing resistance and greater stability in in vivo conditions [36,37]. In spite of this, WLBU2′s poor selectivity continues to be a limiting factor [39].
- Anti-Gram-negative AMPs with an uncertain therapeutic future: A peptide may have promising characteristics in in vitro conditions, or even in animal model trials; however, this does not guarantee success along the long road to approval by the FDA [14,47]. AMPs seeming to be a promising alternative for combating Gram-negative bacterial infections have sometimes had unexpected adverse effects (AF) during more advanced stages. For example, it emerged during phase II and III studies that the use of talactoferrin (TLF, rhLF) for treating severe sepsis was prejudicial, as mortality was higher in the group of patients receiving this AMP than in the control group [48]. Another example concerns the protegrin 1 derivate called murepavadin (POL7080); its phase III study regarding treatment of Pseudomonas aeruginosa-related hospital-acquired pneumonia was terminated before time due to safety problems connected with renal failure (ClinicalTrials.gov Identifier: NCT03582007). Another scenario deals with the difficulty of demonstrating promising results during the clinical phase suggested by in vitro assays; a clear example of this would be iseganan (another protegrin 1 derivative) which did not prove effective during phase III trials for preventing ventilator-associated pneumonia or as mouthwash for reducing mucositis and stomatitis (oral mucositis) in chemotherapy patients via oral administration [49,50].
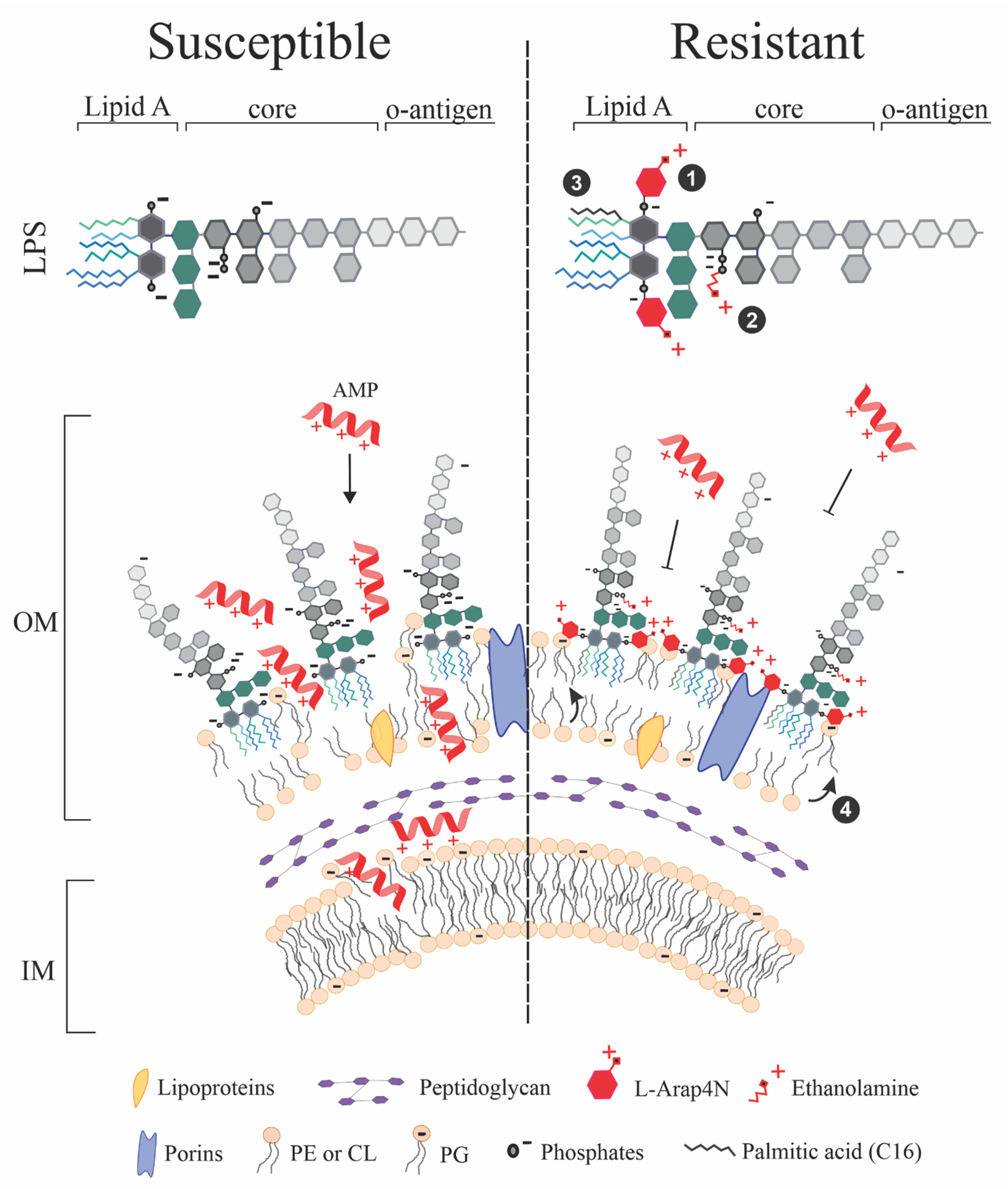
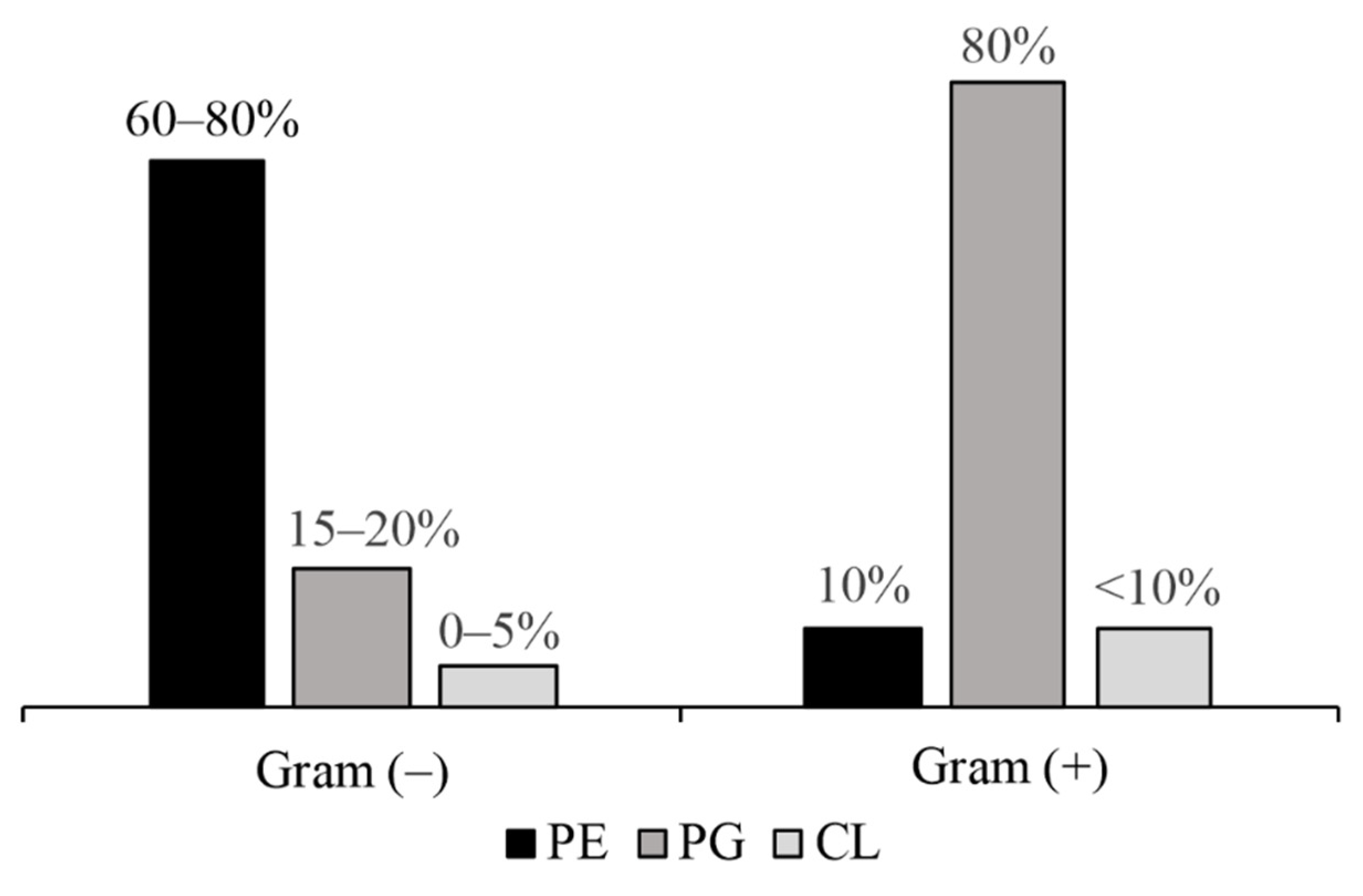
3. Cell Envelope: Gram-Negative Bacteria’s Lipid Rampart
3.1. Outer Membrane (OM)
3.2. Inner Membrane

4. Engineering AMPs against Gram-Negative Bacteria
5. Challenges and Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Global Health Estimates 2019: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2019; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- WHO. Global Priority List of Antibiotic-Resistant Bacteria to Guide Research, Discovery, and Development of New Antibiotics; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Fernandes, P.; Martens, E. Antibiotics in late clinical development. Biochem. Pharmacol. 2017, 133, 152–163. [Google Scholar] [CrossRef] [Green Version]
- Theuretzbacher, U.; Outterson, K.; Engel, A.; Karlén, A. The global preclinical antibacterial pipeline. Nat. Rev. Genet. 2019, 18, 275–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boparai, J.K.; Sharma, P.K. Mini Review on Antimicrobial Peptides, Sources, Mechanism and Recent Applications. Protein Pept. Lett. 2020, 27, 4–16. [Google Scholar] [CrossRef]
- Ghai, I.; Ghai, S. Understanding antibiotic resistance via outer membrane permeability. Infect. Drug Resist. 2018, 11, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Greber, K.E.; Dawgul, M. Antimicrobial Peptides Under Clinical Trials. Curr. Top. Med. Chem. 2016, 17, 620–628. [Google Scholar] [CrossRef]
- Mazer-Amirshahi, M.; Pourmand, A.; May, L. Newly approved antibiotics and antibiotics reserved for resistant infections: Implications for emergency medicine. Am. J. Emerg. Med. 2017, 35, 154–158. [Google Scholar] [CrossRef]
- Rodríguez-Rojas, A.; Moreno-Morales, J.; Mason, J.; Rolff, J. Cationic antimicrobial peptides do not change recombination frequency in Escherichia coli. Biol. Lett. 2018, 14, 20180006. [Google Scholar] [CrossRef] [Green Version]
- Bechinger, B.; Gorr, S.-U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2016, 96, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.-H.; Hall, K.N.; Aguilar, M.-I. Antimicrobial Peptide Structure and Mechanism of Action: A Focus on the Role of Membrane Structure. Curr. Top. Med. Chem. 2016, 16, 25–39. [Google Scholar] [CrossRef]
- Cardoso, P.; Glossop, H.; Meikle, T.G.; Aburto-Medina, A.; Conn, C.E.; Sarojini, V.; Valery, C. Molecular engineering of antimicrobial peptides: Microbial targets, peptide motifs and translation opportunities. Biophys. Rev. 2021, 13, 35–69. [Google Scholar] [CrossRef]
- Giamarellou, H. Multidrug-resistant Gram-negative bacteria: How to treat and for how long. Int. J. Antimicrob. Agents 2010, 36 (Suppl. 2), S50–S54. [Google Scholar] [CrossRef] [Green Version]
- Barreto-Santamaría, A.; Patarroyo, M.E.; Curtidor, H. Designing and optimizing new antimicrobial peptides: All targets are not the same. Crit. Rev. Clin. Lab. Sci. 2019, 56, 351–373. [Google Scholar] [CrossRef]
- León-Buitimea, A.; Garza-Cárdenas, C.R.; Garza-Cervantes, J.A.; Lerma-Escalera, J.A.; Morones-Ramírez, J.R. The Demand for New Antibiotics: Antimicrobial Peptides, Nanoparticles, and Combinatorial Therapies as Future Strategies in Antibacterial Agent Design. Front. Microbiol. 2020, 11, 1669. [Google Scholar] [CrossRef] [PubMed]
- Browne, K.; Chakraborty, S.; Chen, R.; Willcox, M.D.; Black, D.S.; Walsh, W.R.; Kumar, N. A New Era of Antibiotics: The Clinical Potential of Antimicrobial Peptides. Int. J. Mol. Sci. 2020, 21, 7047. [Google Scholar] [CrossRef] [PubMed]
- Huynh, L.; Velásquez, J.; Rabara, R.; Basu, S.; Nguyen, H.B.; Gupta, G. Rational design of antimicrobial peptides targeting Gram-negative bacteria. Comput. Biol. Chem. 2021, 92, 107475. [Google Scholar] [CrossRef]
- Juretić, D.; Simunić, J. Design of α-helical antimicrobial peptides with a high selectivity index. Expert Opin. Drug Discov. 2019, 14, 1053–1063. [Google Scholar] [CrossRef]
- Gottler, L.M.; Ramamoorthy, A. Structure, membrane orientation, mechanism, and function of pexiganan—A highly potent antimicrobial peptide designed from magainin. Biochim. Biophys. Acta (BBA)-Biomembr. 2009, 1788, 1680–1686. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.H.; Lu, T.K. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics 2020, 9, 24. [Google Scholar] [CrossRef] [Green Version]
- Patel, J.B.; Cockerill, F.; Bradford, P.A. Performance standards for antimicrobial susceptibility testing: Twenty-fifth informational supplement. Clin. Lab. Stand. Inst. 2015, 35, 29–50. [Google Scholar]
- Walkty, A.; DeCorby, M.; Nichol, K.; Karlowsky, J.A.; Hoban, D.J.; Zhanel, G.G. In Vitro Activity of Colistin (Polymyxin E) against 3480 Isolates of Gram-Negative Bacilli Obtained from Patients in Canadian Hospitals in the CANWARD Study, 2007–2008. Antimicrob. Agents Chemother. 2009, 53, 4924–4926. [Google Scholar] [CrossRef] [Green Version]
- Naghmouchi, K.; Baah, J.; Hober, D.; Jouy, E.; Rubrecht, C.; Sane, F.; Drider, D. Synergistic Effect between Colistin and Bacteriocins in Controlling Gram-Negative Pathogens and Their Potential to Reduce Antibiotic Toxicity in Mammalian Epithelial Cells. Antimicrob. Agents Chemother. 2013, 57, 2719–2725. [Google Scholar] [CrossRef] [Green Version]
- Florio, W.; Rizzato, C.; Becherini, S.; Guazzelli, L.; D’Andrea, F.; Lupetti, A. Synergistic activity between colistin and the ionic liquids 1-methyl-3-dodecylimidazolium bromide, 1-dodecyl-1-methylpyrrolidinium bromide, or 1-dodecyl-1-methylpiperidinium bromide against Gram-negative bacteria. J. Glob. Antimicrob. Resist. 2020, 21, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Flamm, R.K.; Rhomberg, P.R.; Farrell, D.J.; Jones, R.N. In vitro spectrum of pexiganan activity; bactericidal action and resistance selection tested against pathogens with elevated MIC values to topical agents. Diagn. Microbiol. Infect. Dis. 2016, 86, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Flamm, R.K.; Rhomberg, P.; Simpson, K.; Farrell, D.J.; Sader, H.; Jones, R.N. In VitroSpectrum of Pexiganan Activity When Tested against Pathogens from Diabetic Foot Infections and with Selected Resistance Mechanisms. Antimicrob. Agents Chemother. 2015, 59, 1751–1754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Feng, S.; Qie, J.; Wei, X.; Yan, H.; Liu, K. Polyion complexes of a cationic antimicrobial peptide as a potential systemically administered antibiotic. Int. J. Pharm. 2019, 554, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, D.; Jaśkiewicz, M.; Migoń, D.; Bauer, M.; Sikora, K.; Sikorska, E.; Kamysz, E.; Kamysz, W. Retro analog concept: Comparative study on physico-chemical and biological properties of selected antimicrobial peptides. Amino Acids 2017, 49, 1755–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabisiak, A.; Murawska, N.; Fichna, J. LL-37: Cathelicidin-related antimicrobial peptide with pleiotropic activity. Pharmacol. Rep. 2016, 68, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Dürr, U.H.; Sudheendra, U.; Ramamoorthy, A. LL-37, the only human member of the cathelicidin family of antimicrobial peptides. Biochim. Biophys. Acta (BBA)-Biomembr. 2006, 1758, 1408–1425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grönberg, A.; Mahlapuu, M.; Ståhle, M.; Whately-Smith, C.; Rollman, O. Treatment with LL-37 is safe and effective in enhancing healing of hard-to-heal venous leg ulcers: A randomized, placebo-controlled clinical trial. Wound Repair Regen. 2014, 22, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.; Kaempfer, R.; Chung, C.-S.; Shirvan, A.; Chahin, A.B.; Palardy, J.E.; Parejo, N.A.; Chen, Y.; Whitford, M.; Arad, G.; et al. CD28 Homodimer Interface Mimetic Peptide Acts as a Preventive and Therapeutic Agent in Models of Severe Bacterial Sepsis and Gram-Negative Bacterial Peritonitis. J. Infect. Dis. 2015, 211, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- North, J.R.; Takenaka, S.; Rozek, A.; Kielczewska, A.; Opal, S.; Morici, L.A.; Finlay, B.B.; Schaber, C.J.; Straube, R.; Donini, O. A novel approach for emerging and antibiotic resistant infections: Innate defense regulators as an agnostic therapy. J. Biotechnol. 2016, 226, 24–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.A.; Vierboom, M.P.M.; van Holten-Neelen, C.; Breedveld, E.; Zuiderwijk-Sick, E.; Khan, A.; Kondova, I.; Braskamp, G.; Savelkoul, H.F.J.; Dik, W.A.; et al. Mitigation of septic shock in mice and rhesus monkeys by human chorionic gonadotrophin-related oligopeptides. Clin. Exp. Immunol. 2010, 160, 466–478. [Google Scholar] [CrossRef] [PubMed]
- van Groenendael, R.; Beunders, R.; Kox, M.; van Eijk, L.T.; Pickkers, P. The Human Chorionic Gonadotropin Derivate EA-230 Modulates the Immune Response and Exerts Renal Protective Properties: Therapeutic Potential in Humans. Semin. Nephrol. 2019, 39, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, I.; Hilpert, K.; Hancock, R.E.W. Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc. 2008, 3, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Mercer, D.K.; Torres, M.; Duay, S.S.; Lovie, E.; Simpson, L.; Von Köckritz-Blickwede, M.; De La Fuente-Nunez, C.; O’Neil, D.A.; Angeles-Boza, A.M. Antimicrobial Susceptibility Testing of Antimicrobial Peptides to Better Predict Efficacy. Front. Cell. Infect. Microbiol. 2020, 10, 326. [Google Scholar] [CrossRef]
- Loffredo, M.R.; Savini, F.; Bobone, S.; Casciaro, B.; Franzyk, H.; Mangoni, M.L.; Stella, L. Inoculum effect of antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2021, 118, e2014364118. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 30 November 2021).
- Falagas, M.E.; Kasiakou, S.K.; Saravolatz, L.D. Colistin: The Revival of Polymyxins for the Management of Multidrug-Resistant Gram-Negative Bacterial Infections. Clin. Infect. Dis. 2005, 40, 1333–1341. [Google Scholar] [CrossRef] [Green Version]
- Falagas, M.E.; Grammatikos, A.P.; Michalopoulos, A. Potential of old-generation antibiotics to address current need for new antibiotics. Expert Rev. Anti-Infect. Ther. 2008, 6, 593–600. [Google Scholar] [CrossRef]
- WH0. World Health Organization Model List of Essential Medicines: 21st List 2019; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Lamb, H.M.; Wiseman, L.R. Pexiganan Acetate. Drugs 1998, 56, 1047–1052, discussion 1053–1044. [Google Scholar] [CrossRef]
- Scott, A.; Weldon, S.; Buchanan, P.J.; Schock, B.; Ernst, R.; McAuley, D.; Tunney, M.; Irwin, C.; Elborn, J.; Taggart, C. Evaluation of the Ability of LL-37 to Neutralise LPS In Vitro and Ex Vivo. PLoS ONE 2011, 6, e26525. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Narayana, J.L.; Mishra, B.; Zhang, Y.; Wang, F.; Wang, C.; Zarena, D.; Lushnikova, T.; Wang, X. Design of Antimicrobial Peptides: Progress Made with Human Cathelicidin LL-37. Adv. Exp. Med. Biol. 2019, 1117, 215–240. [Google Scholar] [CrossRef] [PubMed]
- FDA The Drug Development Process. Available online: https://www.fda.gov/patients/learn-about-drug-and-device-approvals/drug-development-process (accessed on 30 November 2021).
- Vincent, J.-L.; Marshall, J.C.; Dellinger, R.P.; Simonson, S.S.; Guntupalli, K.K.; Levy, M.M.; Singer, M.; Malik, R.R. Oral tAlactoferrin in Severe sepsIS Study Investigators. Talactoferrin in Severe Sepsis. Crit. Care Med. 2015, 43, 1832–1838. [Google Scholar] [CrossRef]
- Kollef, M.; Pittet, D.; García, M.S.; Chastre, J.; Fagon, J.-Y.; Bonten, M.; Hyzy, R.; Fleming, T.R.; Fuchs, H.; Bellm, L.; et al. A Randomized Double-Blind Trial of Iseganan in Prevention of Ventilator-associated Pneumonia. Am. J. Respir. Crit. Care Med. 2006, 173, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Giles, F.J.; Rodriguez, R.; Weisdorf, D.; Wingard, J.R.; Martin, P.J.; Fleming, T.R.; Goldberg, S.L.; Anaissie, E.J.; Bolwell, B.J.; Chao, N.J.; et al. A phase III, randomized, double-blind, placebo-controlled, study of iseganan for the reduction of stomatitis in patients receiving stomatotoxic chemotherapy. Leuk. Res. 2004, 28, 559–565. [Google Scholar] [CrossRef]
- Romano, K.P.; Warrier, T.; Poulsen, B.E.; Nguyen, P.H.; Loftis, A.R.; Saebi, A.; Pentelute, B.L.; Hung, D.T. Mutations in pmrB Confer Cross-Resistance between the LptD Inhibitor POL7080 and Colistin in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63, e00511-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, B.; Park, I.-S.; Bang, J.-K.; Shin, S.Y. Short KR-12 analogs designed from human cathelicidin LL-37 possessing both antimicrobial and antiendotoxic activities without mammalian cell toxicity. J. Pept. Sci. 2013, 19, 700–707. [Google Scholar] [CrossRef]
- Wang, C.; Zhao, G.; Wang, S.; Chen, Y.; Gong, Y.; Chen, S.; Xu, Y.; Hu, M.; Wang, X.; Zeng, H.; et al. A Simplified Derivative of Human Defensin 5 with Potent and Efficient Activity against Multidrug-Resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 2018, 62, e01504–e01517. [Google Scholar] [CrossRef] [Green Version]
- Cirioni, O.; Giacometti, A.; Ghiselli, R.; Orlando, F.; Kamysz, W.; D’Amato, G.; Mocchegiani, F.; Lukasiak, J.; Silvestri, C.; Saba, V.; et al. Potential Therapeutic Role of Histatin Derivative P-113din Experimental Rat Models ofPseudomonas aeruginosaSepsis. J. Infect. Dis. 2004, 190, 356–364. [Google Scholar] [CrossRef] [Green Version]
- Hirano, M.; Saito, C.; Yokoo, H.; Goto, C.; Kawano, R.; Misawa, T.; Demizu, Y. Development of Antimicrobial Stapled Peptides Based on Magainin 2 Sequence. Molecules 2021, 26, 444. [Google Scholar] [CrossRef]
- Srinivas, N.; Jetter, P.; Ueberbacher, B.J.; Werneburg, M.; Zerbe, K.; Steinmann, J.; Van der Meijden, B.; Bernardini, F.; Lederer, A.; Dias, R.L.A.; et al. Peptidomimetic Antibiotics Target Outer-Membrane Biogenesis in Pseudomonas aeruginosa. Science 2010, 327, 1010–1013. [Google Scholar] [CrossRef] [Green Version]
- Simons, A.; Alhanout, K.; Duval, R.E. Bacteriocins, Antimicrobial Peptides from Bacterial Origin: Overview of Their Biology and Their Impact against Multidrug-Resistant Bacteria. Microorganisms 2020, 8, 639. [Google Scholar] [CrossRef]
- Duwadi, D.; Shrestha, A.; Yilma, B.; Kozlovski, I.; Sa-Eed, M.; Dahal, N.; Jukosky, J. Identification and screening of potent antimicrobial peptides in arthropod genomes. Peptides 2018, 103, 26–30. [Google Scholar] [CrossRef]
- Münzker, L.; Oddo, A.; Hansen, P.R. Chemical Synthesis of Antimicrobial Peptides. Adv. Struct. Saf. Stud. 2017, 1548, 35–49. [Google Scholar] [CrossRef]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta (BBA)-Biomembr. 2009, 1788, 1687–1692. [Google Scholar] [CrossRef] [Green Version]
- Zgurskaya, H.I.; López, C.A.; Gnanakaran, S. Permeability Barrier of Gram-Negative Cell Envelopes and Approaches to Bypass It. ACS Infect. Dis. 2015, 1, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Benz, R.; Bauer, K. Permeation of hydrophilic molecules through the outer membrane of gram-negativ bacteria. Review of becterial porins. JBIC J. Biol. Inorg. Chem. 1988, 176, 1–19. [Google Scholar] [CrossRef]
- Arcidiacono, S.; Soares, J.W.; Meehan, A.M.; Marek, P.; Kirby, R. Membrane permeability and antimicrobial kinetics of cecropin P1 against Escherichia coli. J. Pept. Sci. 2009, 15, 398–403. [Google Scholar] [CrossRef]
- Florin, T.; Maracci, C.; Graf, M.; Karki, P.; Klepacki, D.; Berninghausen, O.; Beckmann, R.; Vázquez-Laslop, N.; Wilson, D.; Rodnina, M.V.; et al. An antimicrobial peptide that inhibits translation by trapping release factors on the ribosome. Nat. Struct. Mol. Biol. 2017, 24, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Gutsmann, T.; Hagge, S.O.; David, A.; Roes, S.; Böhling, A.; Hammer, M.U.; Seydel, U. Lipid-mediated resistance of Gram-negative bacteria against various pore-forming antimicrobial peptides. J. Endotoxin Res. 2005, 11, 167–173. [Google Scholar] [CrossRef]
- Dalebroux, Z.D.; Miller, S.I. Salmonellae PhoPQ regulation of the outer membrane to resist innate immunity. Curr. Opin. Microbiol. 2014, 17, 106–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Needham, B.; Trent, M.S. Fortifying the barrier: The impact of lipid A remodelling on bacterial pathogenesis. Nat. Rev. Genet. 2013, 11, 467–481. [Google Scholar] [CrossRef]
- Nelson, N.; Opene, B.; Ernst, R.K.; Schwartz, D.K. Antimicrobial peptide activity is anticorrelated with lipid a leaflet affinity. PLoS ONE 2020, 15, e0242907. [Google Scholar] [CrossRef]
- Nummila, K.; Kilpeläinen, I.; Zähringer, U.; Vaara, M.; Helander, I.M. Lipopolysaccharides of polymyxin B-resistant mutants of Escherichia coii are extensively substituted by 2-aminoethyl pyrophosphate and contain aminoarabinose in lipid A. Mol. Microbiol. 1995, 16, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.C.S.; Santos, I.; Campos, C.C.; Rezende, I.N.; Ferreira, Y.M.; Chaves, C.E.V.; Rocha, C.; Carvalho-Assef, A.P.D.; Chang, M.R. Non-clonal occurrence of pmrB mutations associated with polymyxin resistance in carbapenem-resistant Klebsiella pneumoniae in Brazil. Memórias Inst. Oswaldo Cruz 2019, 114, e180555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quaglio, D.; Mangoni, M.L.; Stefanelli, R.; Corradi, S.; Casciaro, B.; Vergine, V.; Lucantoni, F.; Cavinato, L.; Cammarone, S.; Loffredo, M.R.; et al. ent-Beyerane Diterpenes as a Key Platform for the Development of ArnT-Mediated Colistin Resistance Inhibitors. J. Org. Chem. 2020, 85, 10891–10901. [Google Scholar] [CrossRef]
- Liu, Y.; Li, R.; Xiao, X.; Wang, Z. Antibiotic adjuvants: An alternative approach to overcome multi-drug resistant Gram-negative bacteria. Crit. Rev. Microbiol. 2019, 45, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Barker, W.T.; Jania, L.A.; Melander, R.J.; Koller, B.H.; Melander, C. Eukaryotic phosphatase inhibitors enhance colistin efficacy in gram-negative bacteria. Chem. Biol. Drug Des. 2020, 96, 1180–1186. [Google Scholar] [CrossRef]
- Lu, H.-F.; Wu, B.-K.; Huang, Y.-W.; Lee, M.-Z.; Li, M.-F.; Ho, H.-J.; Yang, H.-C.; Yang, T.-C. PhoPQ two-component regulatory system plays a global regulatory role in antibiotic susceptibility, physiology, stress adaptation, and virulence in Stenotrophomonas maltophilia. BMC Microbiol. 2020, 20, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, K.; Schromm, A.B.; Weindl, G.; Heinbockel, L.; Correa, W.; Mauss, K.; de Tejada, G.M.; Garidel, P. An update on endotoxin neutralization strategies in Gram-negative bacterial infections. Expert Rev. Anti-Infect. Ther. 2021, 19, 495–517. [Google Scholar] [CrossRef]
- Chou, H.-T.; Wen, H.-W.; Kuo, T.-Y.; Lin, C.-C.; Chen, W.-J. Interaction of cationic antimicrobial peptides with phospholipid vesicles and their antibacterial activity. Peptides 2010, 31, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, V.; Feio, M.; Bastos, M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51, 149–177. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.F.; Savage, P.B.; Epand, R. Bacterial lipid composition and the antimicrobial efficacy of cationic steroid compounds (Ceragenins). Biochim. Biophys. Acta (BBA)-Biomembr. 2007, 1768, 2500–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arouri, A.; Dathe, M.; Blume, A. Peptide induced demixing in PG/PE lipid mixtures: A mechanism for the specificity of antimicrobial peptides towards bacterial membranes? Biochim. Biophys. Acta (BBA)-Biomembr. 2009, 1788, 650–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreto-Santamaría, A.; Curtidor, H.; Arévalo-Pinzón, G.; Herrera, C.; Suárez, D.; Pérez, W.H.; Patarroyo, M.-E. A New Synthetic Peptide Having Two Target of Antibacterial Action in E. coli ML35. Front. Microbiol. 2016, 7, 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epand, R.F.; Schmitt, M.A.; Gellman, S.H.; Epand, R. Role of membrane lipids in the mechanism of bacterial species selective toxicity by two α/β-antimicrobial peptides. Biochim. Biophys. Acta (BBA)-Biomembr. 2006, 1758, 1343–1350. [Google Scholar] [CrossRef] [Green Version]
- Daleke, D.L. Regulation of phospholipid asymmetry in the erythrocyte membrane. Curr. Opin. Hematol. 2008, 15, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Zawadzka, K.; Bernat, P.; Felczak, A.; Różalska, S.; Lisowska, K. Antibacterial activity of high concentrations of carvedilol against Gram-positive and Gram-negative bacteria. Int. J. Antimicrob. Agents 2018, 51, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Zelezetsky, I.; Tossi, A. Alpha-helical antimicrobial peptides—Using a sequence template to guide structure–activity relationship studies. Biochim. Biophys. Acta (BBA)-Biomembr. 2006, 1758, 1436–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogoladze, G.; Grigolava, M.; Vishnepolsky, B.; Chubinidze, M.; Duroux, P.; Lefranc, M.-P.; Pirtskhalava, M. dbaasp: Database of antimicrobial activity and structure of peptides. FEMS Microbiol. Lett. 2014, 357, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Pirtskhalava, M.; Gabrielian, A.; Cruz, P.; Griggs, H.L.; Squires, R.B.; Hurt, D.E.; Grigolava, M.; Chubinidze, M.; Gogoladze, G.; Vishnepolsky, B.; et al. DBAASP v.2: An enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res. 2016, 44, 6503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, P.; Rosa, R.D.; Destoumieux-Garzón, D. An intimate link between antimicrobial peptide sequence diversity and binding to essential components of bacterial membranes. Biochim. Biophys. Acta (BBA)-Biomembr. 2016, 1858, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Piers, K.L.; Brown, M.; Hancock, R. Improvement of outer membrane-permeabilizing and lipopolysaccharide-binding activities of an antimicrobial cationic peptide by C-terminal modification. Antimicrob. Agents Chemother. 1994, 38, 2311–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saikia, K.; Chaudhary, N. Interaction of MreB-derived antimicrobial peptides with membranes. Biochem. Biophys. Res. Commun. 2018, 498, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Liscano, Y.; Salamanca, C.H.; Vargas, L.; Cantor, S.; Laverde-Rojas, V.; Oñate-Garzón, J. Increases in Hydrophilicity and Charge on the Polar Face of Alyteserin 1c Helix Change its Selectivity towards Gram-Positive Bacteria. Antibiotics 2019, 8, 238. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Adamiak, J.W.; Bonifay, V.; Mehla, J.; Zgurskaya, H.I.; Tan, D.S. Defining new chemical space for drug penetration into Gram-negative bacteria. Nat. Chem. Biol. 2020, 16, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.Y.; Milton, M.N. The determination and interpretation of the therapeutic index in drug development. Nat. Rev. Drug Discov. 2012, 11, 751–761. [Google Scholar] [CrossRef] [PubMed]
- de la Fuente-Nunez, C.; Torres, M.; Mojica, F.J.; Lu, T.K.; de la Fuente-Nunez, C.; Torres, M.; Mojica, F.J.; Lu, T.K. Next-generation precision antimicrobials: Towards personalized treatment of infectious diseases. Curr. Opin. Microbiol. 2017, 37, 95–102. [Google Scholar] [CrossRef] [Green Version]
- Medhi, B.; Sarma, P.; Mahendiratta, S.; Prakash, A. Specifically targeted antimicrobial peptides: A new and promising avenue in selective antimicrobial therapy. Indian J. Pharmacol. 2018, 50, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Berditsch, M.; Hawecker, J.; Ardakani, M.F.; Gerthsen, D.; Ulrich, A.S. Damage of the Bacterial Cell Envelope by Antimicrobial Peptides Gramicidin S and PGLa as Revealed by Transmission and Scanning Electron Microscopy. Antimicrob. Agents Chemother. 2010, 54, 3132–3142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thennarasu, S.; Lee, D.-K.; Tan, A.; Kari, U.P.; Ramamoorthy, A. Antimicrobial activity and membrane selective interactions of a synthetic lipopeptide MSI-843. Biochim. Biophys. Acta (BBA)-Biomembr. 2005, 1711, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Thennarasu, S.; Huang, R.; Lee, D.-K.; Yang, P.; Maloy, L.; Chen, Z.; Ramamoorthy, A. Limiting an Antimicrobial Peptide to the Lipid−Water Interface Enhances Its Bacterial Membrane Selectivity: A Case Study of MSI-367. Biochemistry 2010, 49, 10595–10605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamysz, E.; Sikorska, E.; Jaśkiewicz, M.; Bauer, M.; Neubauer, D.; Bartoszewska, S.; Barańska-Rybak, W.; Kamysz, W. Lipidated Analogs of the LL-37-Derived Peptide Fragment KR12—Structural Analysis, Surface-Active Properties and Antimicrobial Activity. Int. J. Mol. Sci. 2020, 21, 887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Peyear, T.A.; Bennett, W.F.D.; Andersen, O.S.; Lightstone, F.C.; Ingólfsson, H.I. Molecular Mechanism for Gramicidin Dimerization and Dissociation in Bilayers of Different Thickness. Biophys. J. 2019, 117, 1831–1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, J.; Easton, P.; Hinton, J. Effects of Phenylalanine Substitutions in Gramicidin A on the Kinetics of Channel Formation in Vesicles and Channel Structure in SDS Micelles. Biophys. J. 2005, 88, 224–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agadi, N.; Vasudevan, S.; Kumar, A. Structural insight into the mechanism of action of antimicrobial peptide BMAP-28(1–18) and its analogue mutBMAPJ. Struct. Biol. 2018, 204, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Prada, Y.A.; Guzmán, F.; Rondón, P.; Escobar, P.; Ortíz, C.; Sierra, D.A.; Torres, R.; Mejía-Ospino, E. A New Synthetic Peptide with In vitro Antibacterial Potential Against Escherichia coli O157:H7 and Methicillin-Resistant Staphylococcus aureus (MRSA). Probiotics Antimicrob. Proteins 2016, 8, 134–140. [Google Scholar] [CrossRef]
- Claudia, O.L. Design, synthesis, characterization and in vitro evaluation of antimicrobial peptides against pathogenic bacteria resistant to antibiotics. Rev. Acad. Colomb. Cienc. Exactas Físicas Nat. 2019, 43, 614–627. [Google Scholar] [CrossRef]
- Vishnepolsky, B.; Zaalishvili, G.; Karapetian, M.; Nasrashvili, T.; Kuljanishvili, N.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M.; Grigolava, M.; et al. De Novo Design and In Vitro Testing of Antimicrobial Peptides against Gram-Negative Bacteria. Pharmaceuticals 2019, 12, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M.; Managadze, G.; Grigolava, M.; Makhatadze, G.I.; Pirtskhalava, M. Predictive Model of Linear Antimicrobial Peptides Active against Gram-Negative Bacteria. J. Chem. Inf. Model. 2018, 58, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Rishi, P.; Gautam, A.; Gautam, V.; Tewari, R. In vitro and in silico comparative evaluation of anti-Acinetobacter baumannii peptides. J. Microbiol. Biotechnol. 2015. [Google Scholar] [CrossRef]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef]
- Couet, W. Pharmacokinetics/pharmacodynamics characterization of combined antimicrobial agents: A real challenge and an urgent need. Clin. Microbiol. Infect. 2018, 24, 687–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| AMPs against Gram-Negative Bacteria | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Status/ Clinical Phase | Peptide | Description | Target | In Vitro Gram- Negative MIC | In Vitro Gram- Positive MIC | In Vitro Haemolysis | MoA | Route | Ref. |
| FDA- approved | Colistin or polymyxin E (natural) | Cationic cyclic lipodecapeptide isolated from Paenibacillus polymyxa var colistinus | Gram-negative | ≤2 µg/mL (≤1.7 μM) | - | 0–1.8% at 0.12 µg/mL | Membranolytic | Top, oral, IV | [21,22,23,24,25] |
| Phase III | Pexiganan o MSI-78 (designed) | A 22 amino acid-long magainin cationic analogue | Pathogens associated with diabetic foot infection | 8–16 µg/mL (3.23–6.46 µM) | 8–32 µg/mL (3.23–12.9 µM) | 5–63% at 50–64 µg/mL | Toroidal pore former | Top | [19,26,27,28,29] |
| Phase II | LL-37 | hCAP18 human cathelicidin-derived peptide | Broad bacterial and fungi spectrum | 0.2–72 µg/mL (0.04–16 µM) | 0.7–72 µg/mL (0.16–16 µM) | 1.5–5% in MIC range | Membranolytic, binding to LPS and Immune modulation | Top | [30,31,32] |
| Immunomodulatory Peptides against Gram-Negative Bacteria | |||||||||
| Status/ Clinical Phase | Peptide | Description | Target | In Vitro Gram-Negative MIC | In Vitro Gram- Positive MIC | In Vitro Haemolysis | MoA | Route | Ref. |
| Phase III | AB103 (D-ala-p2TA) (designed) | CD28 homodimer interface mimetic octapeptide (CD288–15) abutted with D-Ala at both termini | Effective in mice challenged with E. coli bacteria | Only acts in vivo | Only acts in vivo | - | Attenuates CD28 T-cell signalling in exacerbated immune responses during infection | IV | [33] |
| Phase III | Dusquetide (IMX942, SGX942) (designed) | Synthetic 5-amino acid-long peptide | Gram-negative (i.e., Burkholderia pseudomallei) and Gram-positive (i.e., S. aureus) bacterial infections | Only acts in vivo | Only acts in vivo | - | Innate defence regulator (IDR), complement for antibiotics | IV | [34] |
| Phase IIa | EA-230 (Peptide 46) | Human chorionic gonadotropin derivate tetrapeptide (AQGV) | Effective in cercal ligation and puncture (CLP) mouse model | Only acts in vivo | Only acts in vivo | - | Immune modulation | IV | [35,36] |
| Family | ID | Sequence | Target/MIC | Ref |
|---|---|---|---|---|
| Cathelicidins (LL-37) | KR-12-a5 (α-helix) | KRIVKLILKWLR-NH2 | Both Gram-positive and Gram-negative bacteria/2–8 µM | [52] |
| Defensins | HD5d5 | ARARCRRGRAARRRR LRGVCRIRGRLRRLAAR | A. baumannii/ 40 µg/mL (10.4 µM) | [53] |
| Histatins | P-113D (α-helix) | d-AKRHHGYKRKFH-NH2 | Both Gram-positive, Gram-negative and Candida/ P. aeruginosa: 2 µg/mL (1.28 µM) | [54] |
| Magainins | Mag2 peptide 2 | H-S5IKKS5LKSAKKFVKAFK-NH2 | Both Gram-positive and Gram-negative bacteria/Gram-negative: 1.56–3.1 µM | [55] |
| Protegrins | L27-11 (β-sheet) | TWLKKRRWKKAK | Pseudomonas spp./ 0.004–0.01 µg/mL (0.0026–0.0064 µM) | [56] |
| Bacteriocins | Microcin J25 (MccJ25) Recombinant (β-sheet) | GGAGHVPEYFVGIGTPISFYG | Gram-negative/3.2–10.6 µg/mL (1.5–5 µM) | [57] |
| Cecropins | DAN2 (α-helix) | RWKFLKKIEKVGRKVRDGVIKAGPAVGVVGQATSIYK-NH2 | Gram-negative/2–16 µg/mL (0.49–3.92 µM) | [58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barreto-Santamaría, A.; Arévalo-Pinzón, G.; Patarroyo, M.A.; Patarroyo, M.E. How to Combat Gram-Negative Bacteria Using Antimicrobial Peptides: A Challenge or an Unattainable Goal? Antibiotics 2021, 10, 1499. https://doi.org/10.3390/antibiotics10121499
Barreto-Santamaría A, Arévalo-Pinzón G, Patarroyo MA, Patarroyo ME. How to Combat Gram-Negative Bacteria Using Antimicrobial Peptides: A Challenge or an Unattainable Goal? Antibiotics. 2021; 10(12):1499. https://doi.org/10.3390/antibiotics10121499
Chicago/Turabian StyleBarreto-Santamaría, Adriana, Gabriela Arévalo-Pinzón, Manuel A. Patarroyo, and Manuel E. Patarroyo. 2021. "How to Combat Gram-Negative Bacteria Using Antimicrobial Peptides: A Challenge or an Unattainable Goal?" Antibiotics 10, no. 12: 1499. https://doi.org/10.3390/antibiotics10121499