Isothermal Amplification Technology for Disease Diagnosis

 ,
,
Abstract
:1. Introduction
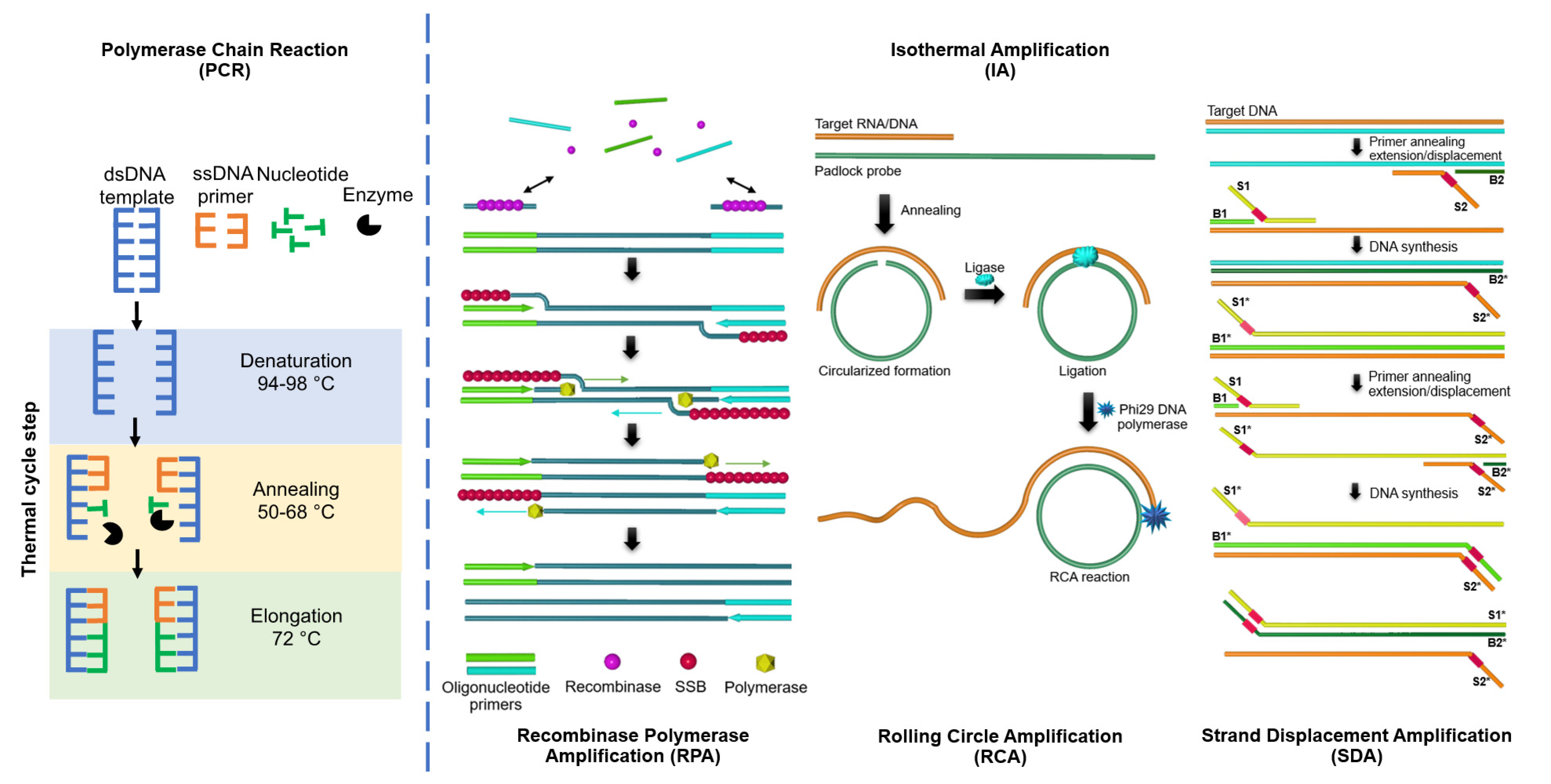
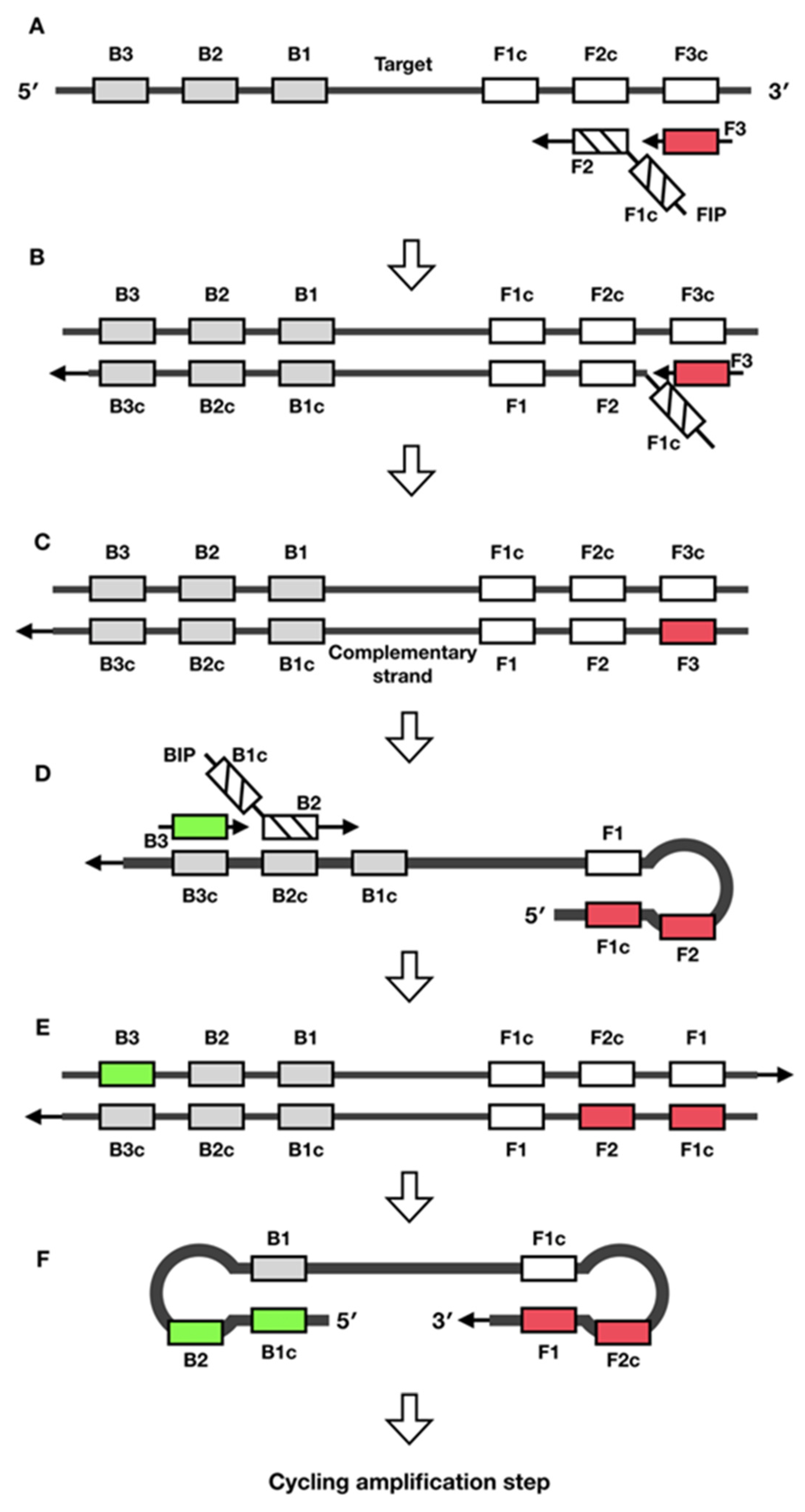
2. Loop-Mediated Isothermal Amplification (LAMP)
3. Rolling Circle Amplification (RCA)
4. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR-Associated Protein (Cas) System
5. Conclusions
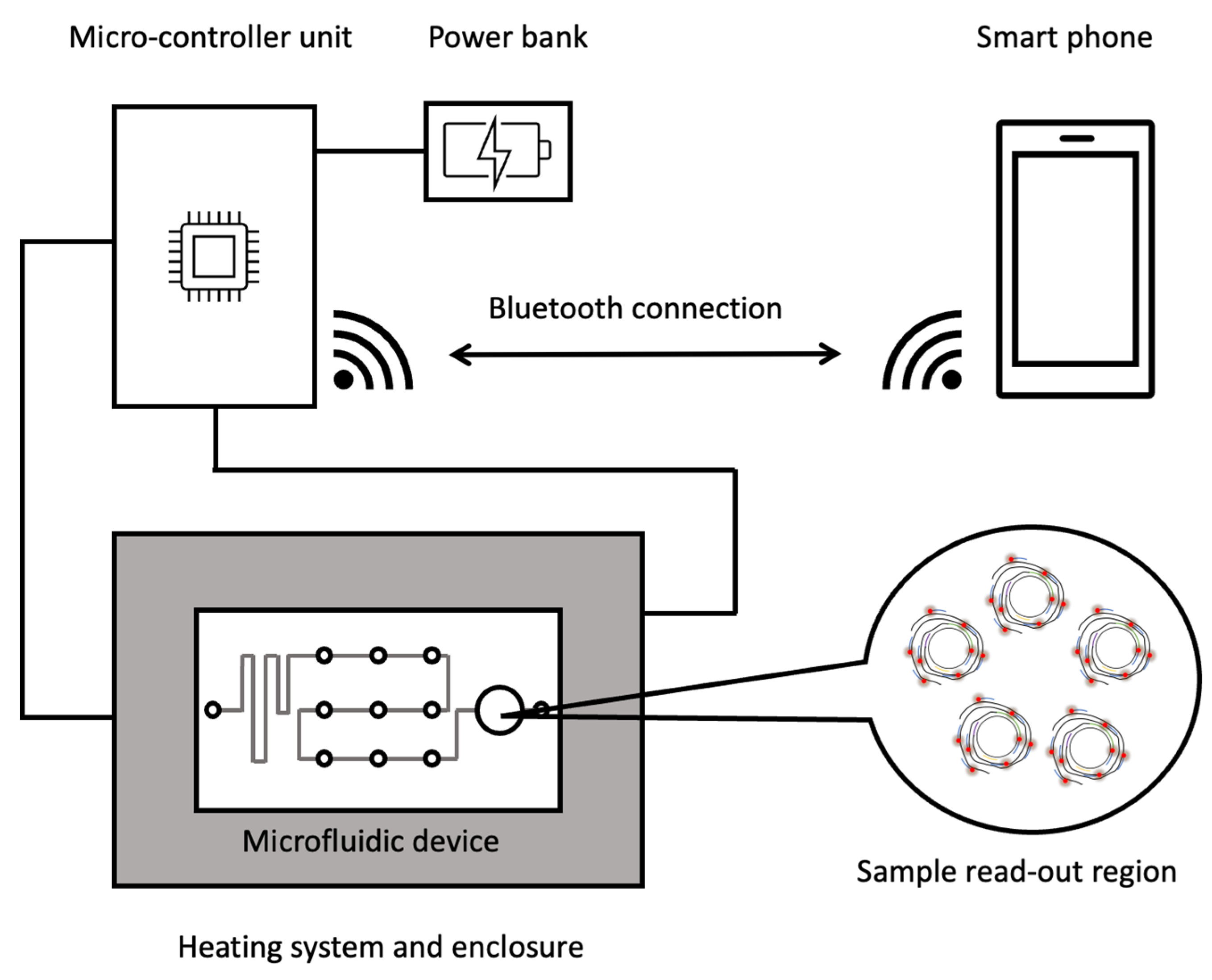
6. Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, E63. [Google Scholar] [CrossRef]
- Mori, Y.; Nagamine, K.; Tomita, N.; Notomi, T. Detection of loop-mediated isothermal amplification reaction by turbidity derived from magnesium pyrophosphate formation. Biochem. Biophys. Res. Commun. 2001, 289, 150–154. [Google Scholar] [CrossRef]
- Soliman, H.; El-Matbouli, M. An inexpensive and rapid diagnostic method of Koi Herpesvirus (KHV) infection by loop-mediated isothermal amplification. Virol. J. 2005, 2, 83. [Google Scholar] [CrossRef]
- Nurul Najian, A.B.; Foo, P.C.; Ismail, N.; Kim-Fatt, L.; Yean, C.Y. Probe-specific loop-mediated isothermal amplification magnetogenosensor assay for rapid and specific detection of pathogenic Leptospira. Mol. Cell Probes 2019, 44, 63–68. [Google Scholar] [CrossRef]
- Iwamoto, T.; Sonobe, T.; Hayashi, K. Loop-mediated isothermal amplification for direct detection of Mycobacterium tuberculosis complex, M. avium, and M. intracellulare in sputum samples. J. Clin. Microbiol. 2003, 41, 2616–2622. [Google Scholar] [CrossRef]
- Lee, S.H.; Baek, Y.H.; Kim, Y.H.; Choi, Y.K.; Song, M.S.; Ahn, J.Y. One-Pot reverse transcriptional loop-mediated isothermal amplification (RT-LAMP) for detecting MERS-CoV. Front. Microbiol. 2016, 7, 2166. [Google Scholar] [CrossRef]
- Shirato, K.; Yano, T.; Senba, S.; Akachi, S.; Kobayashi, T.; Nishinaka, T.; Matsuyama, S. Detection of Middle East respiratory syndrome coronavirus using reverse transcription loop-mediated isothermal amplification (RT-LAMP). Virol. J. 2014, 11, 139. [Google Scholar] [CrossRef]
- Kim, J.H.; Kang, M.; Park, E.; Chung, D.R.; Kim, J.; Hwang, E.S. A simple and multiplex loop-mediated isothermal amplification (LAMP) assay for rapid detection of SARS-CoV. Biochip. J. 2019, 13, 341–351. [Google Scholar] [CrossRef]
- Lamb, L.E.; Bartolone, S.N.; Ward, E.; Chancellor, M.B. Rapid detection of novel coronavirus/Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) by reverse transcription-loop-mediated isothermal amplification. PLoS ONE 2020, 15, e0234682. [Google Scholar]
- Hsieh, K.; Mage, P.L.; Csordas, A.T.; Eisenstein, M.; Soh, H.T. Simultaneous elimination of carryover contamination and detection of DNA with uracil-DNA-glycosylase-supplemented loop-mediated isothermal amplification (UDG-LAMP). Chem Commun. 2014, 50, 3747–3749. [Google Scholar] [CrossRef]
- Watts, M.R.; James, G.; Sultana, Y.; Ginn, A.N.; Outhred, A.C.; Kong, F.; Verweij, J.J.; Iredell, J.R.; Chen, S.C.; Lee, R. A loop-mediated isothermal amplification (LAMP) assay for Strongyloides stercoralis in stool that uses a visual detection method with SYTO-82 fluorescent dye. Am. J. Trop. Med. Hyg. 2014, 90, 306–311. [Google Scholar] [CrossRef] [Green Version]
- Fire, A.; Xu, S.Q. Rolling replication of short DNA circles. Proc. Natl. Acad. Sci. USA 1995, 92, 4641–4645. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.; Li, F.; Zhang, Z.Q.; Zhang, K.X.; Kang, D.K.; Ankrum, J.A.; Le, X.C.; Zhao, W. Rolling circle amplification: A versatile tool for chemical biology, materials science and medicine. Chem. Soc. Rev. 2014, 43, 3324–3341. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, H.; Cao, H.; Jiang, Y.; Mao, K.; Yang, Z. Rolling circle amplification as an efficient analytical tool for rapid detection of contaminants in aqueous environments. Biosensors 2021, 11, 352. [Google Scholar] [CrossRef]
- Zhao, W.; Ali, M.M.; Brook, M.A.; Li, Y. Rolling circle amplification: Applications in nanotechnology and biodetection with functional nucleic acids. Angew. Chem. Int. Ed. Engl. 2008, 47, 6330–6337. [Google Scholar] [CrossRef]
- Zhao, Y.; Chen, F.; Li, Q.; Wang, L.; Fan, C. Isothermal amplification of nucleic acids. Chem. Rev. 2015, 115, 12491–12545. [Google Scholar] [CrossRef]
- Liu, D.; Daubendiek, S.L.; Zillman, M.A.; Ryan, K.; Kool, E.T. Rolling circle DNA synthesis: Small circular oligonucleotides as efficient templates for DNA polymerases. J. Am. Chem. Soc. 1996, 118, 1587–1594. [Google Scholar] [CrossRef]
- Lizardi, P.M.; Huang, X.; Zhu, Z.; Bray-Ward, P.; Thomas, D.C.; Ward, D.C. Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat. Genet. 1998, 19, 225–232. [Google Scholar] [CrossRef]
- Murakami, T.; Sumaoka, J.; Komiyama, M. Sensitive isothermal detection of nucleic-acid sequence by primer generation-rolling circle amplification. Nucleic Acids Res. 2009, 37, e19. [Google Scholar] [CrossRef]
- Mohsen, M.G.; Kool, E.T. The discovery of rolling circle amplification and rolling Ccrcle transcription. Acc. Chem. Res. 2016, 49, 2540–2550. [Google Scholar] [CrossRef]
- Sun, J.; de Hoog, S. Hyperbranching rolling circle amplification, an improved protocol for discriminating between closely related fungal species. Methods Mol. Biol. 2013, 968, 167–175. [Google Scholar]
- Gao, Z.; Wu, C.; Lv, S.; Wang, C.; Zhang, N.; Xiao, S.; Han, Y.; Xu, H.; Zhang, Y.; Li, F.; et al. Nicking-enhanced rolling circle amplification for sensitive fluorescent detection of cancer-related microRNAs. Anal. Bioanal. Chem. 2018, 410, 6819–6826. [Google Scholar] [CrossRef]
- Ge, J.; Hu, Y.; Deng, R.; Li, Z.; Zhang, K.; Shi, M.; Yang, D.; Cai, R.; Tan, W. Highly sensitive microRNA detection by coupling nicking-enhanced rolling circle amplification with MoS2 quantum dots. Anal. Chem. 2020, 92, 13588–13594. [Google Scholar] [CrossRef]
- Dahl, F.; Baner, J.; Gullberg, M.; Mendel-Hartvig, M.; Landegren, U.; Nilsson, M. Circle-to-circle amplification for precise and sensitive DNA analysis. Proc. Natl. Acad. Sci. USA 2004, 101, 4548–4553. [Google Scholar] [CrossRef]
- Xu, S.; Lou, Z. Ultrasensitive detection of nasopharyngeal carcinoma-related miRNA through garland rolling circle amplification integrated catalytic hairpin assembly. ACS Omega 2021, 6, 6460–6465. [Google Scholar] [CrossRef]
- Bialy, R.M.; Ali, M.M.; Li, Y.; Brennan, J.D. Protein-mediated suppression of rolling circle amplification for biosensing with an aptamer-containing DNA primer. Chemistry 2020, 26, 5085–5092. [Google Scholar] [CrossRef]
- Qiu, Z.; Shu, J.; He, Y.; Lin, Z.; Zhang, K.; Lv, S.; Tang, D. CdTe/CdSe quantum dot-based fluorescent aptasensor with hemin/G-quadruplex DNzyme for sensitive detection of lysozyme using rolling circle amplification and strand hybridization. Biosens. Bioelectron. 2017, 87, 18–24. [Google Scholar] [CrossRef]
- Li, J.; Deng, T.; Chu, X.; Yang, R.; Jiang, J.; Shen, G.; Yu, R. Rolling circle amplification combined with gold nanoparticle aggregates for highly sensitive identification of single-nucleotide polymorphisms. Anal. Chem. 2010, 82, 2811–2816. [Google Scholar] [CrossRef]
- Sanchez Martin, D.; Oropesa-Nunez, R.; Zardan Gomez de la Torre, T. Formation of visible aggregates between rolling circle amplification products and magnetic nanoparticles as a strategy for point-of-care diagnostics. ACS Omega 2021, 6, 32970–32976. [Google Scholar] [CrossRef]
- Fan, T.; Mao, Y.; Liu, F.; Zhang, W.; Lin, J.S.; Yin, J.; Tan, Y.; Huang, X.; Jiang, Y. Label-free fluorescence detection of circulating microRNAs based on duplex-specific nuclease-assisted target recycling coupled with rolling circle amplification. Talanta 2019, 200, 480–486. [Google Scholar] [CrossRef]
- Wen, Y.; Xu, Y.; Mao, X.; Wei, Y.; Song, H.; Chen, N.; Huang, Q.; Fan, C.; Li, D. DNAzyme-based rolling-circle amplification DNA machine for ultrasensitive analysis of microRNA in Drosophila larva. Anal. Chem. 2012, 84, 7664–7669. [Google Scholar] [CrossRef]
- Schopf, E.; Liu, Y.; Deng, J.C.; Yang, S.; Cheng, G.; Chen, Y. Mycobacterium tuberculosis detection via rolling circle amplification. Anal. Methods 2011, 3, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, S.V.; Ghourchian, H.; Tavoosidana, G. Real-time detection of H5N1 influenza virus through hyperbranched rolling circle amplification. Analyst 2015, 140, 1502–1509. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Potter, S.J.; Lin, Y.; Cunningham, A.L.; Dwyer, D.E.; Su, Y.; Ma, X.; Hou, Y.; Saksena, N.K. Rapid and sensitive detection of severe acute respiratory syndrome coronavirus by rolling circle amplification. J. Clin. Microbiol. 2005, 43, 2339–2344. [Google Scholar] [CrossRef]
- Hong, C.; Baek, A.; Hah, S.S.; Jung, W.; Kim, D.E. Fluorometric detection of microRNA using isothermal gene amplification and graphene oxide. Anal. Chem. 2016, 88, 2999–3003. [Google Scholar] [CrossRef] [PubMed]
- Khoothiam, K.; Treerattrakoon, K.; Iempridee, T.; Luksirikul, P.; Dharakul, T.; Japrung, D. Ultrasensitive detection of lung cancer-associated miRNAs by multiple primer-mediated rolling circle amplification coupled with a graphene oxide fluorescence-based (MPRCA-GO) sensor. Analyst 2019, 144, 4180–4187. [Google Scholar] [CrossRef]
- Treerattrakoon, K.; Jiemsakul, T.; Tansarawiput, C.; Pinpradup, P.; Iempridee, T.; Luksirikul, P.; Khoothiam, K.; Dharakul, T.; Japrung, D. Rolling circle amplification and graphene-based sensor-on-a-chip for sensitive detection of serum circulating miRNAs. Anal Biochem. 2019, 577, 89–97. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Sashital, D.G. Pathogen detection in the CRISPR-Cas era. Genome Med. 2018, 10, 32. [Google Scholar] [CrossRef]
- Li, Y.; Li, S.; Wang, J.; Liu, G. CRISPR/Cas Systems towards Next-Generation Biosensing. Trends Biotechnol. 2019, 37, 730–743. [Google Scholar] [CrossRef]
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed]
- Adli, M. The CRISPR tool kit for genome editing and beyond. Nat. Commun. 2018, 9, 1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Hu, L.; Ying, L.; Zhao, Z.; Chu, P.K.; Yu, X.F. A CRISPR-Cas9-triggered strand displacement amplification method for ultrasensitive DNA detection. Nat. Commun. 2018, 9, 5012. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.Y.; Zhu, L.Y.; Zhu, C.S.; Ma, J.X.; Hou, T.; Wu, X.M.; Xie, S.-S.; Min, L.; Tan, D.-A.; Zhang, D.-Y.; et al. Highly effective and low-cost microRNA detection with CRISPR-Cas9. ACS Synth. Biol. 2018, 7, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.; Smargon, A.; Scott, D.; Cox, D.; Pyzocha, N.; Yan, W.; Abudayyeh, O.O.; Gootenberg, J.S.; Makarova, K.S.; Wolf, Y.I.; et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol. 2017, 15, 169–182. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef]
- Kellner, M.J.; Koob, J.G.; Gootenberg, J.S.; Abudayyeh, O.O.; Zhang, F. SHERLOCK: Nucleic acid detection with CRISPR nucleases. Nat. Protoc. 2019, 14, 2986–3012. [Google Scholar] [CrossRef]
- Gootenberg, J.S.; Abudayyeh, O.O.; Kellner, M.J.; Joung, J.; Collins, J.J.; Zhang, F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018, 360, 439–444. [Google Scholar] [CrossRef]
- Zhang, F. Development of CRISPR-Cas systems for genome editing and beyond. Q. Rev. Biophys. 2019, 52, E6. [Google Scholar] [CrossRef]
- Li, S.Y.; Cheng, Q.X.; Liu, J.K.; Nie, X.Q.; Zhao, G.P.; Wang, J. CRISPR-Cas12a has both cis- and trans-cleavage activities on single-stranded DNA. Cell Res. 2018, 28, 491–493. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef]
- Tsou, J.H.; Leng, Q.; Jiang, F. A CRISPR test for detection of circulating nuclei acids. Transl Oncol. 2019, 12, 1566–1573. [Google Scholar] [CrossRef] [PubMed]
- Broughton, J.P.; Deng, X.; Yu, G.; Fasching, C.L.; Servellita, V.; Singh, J.; Miao, X.; Streithorst, J.A.; Granados, A.; Sotomayor-Gonzalez, A.; et al. CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 2020, 38, 870–874. [Google Scholar] [CrossRef]
- Ding, X.; Yin, K.; Li, Z.; Lalla, R.V.; Ballesteros, E.; Sfeir, M.M.; Liu, C. Ultrasensitive and visual detection of SARS-CoV-2 using all-in-one dual CRISPR-Cas12a assay. Nat Commun. 2020, 11, 4711. [Google Scholar] [CrossRef]
- Mukama, O.; Wu, J.; Li, Z.; Liang, Q.; Yi, Z.; Lu, X.; Liu, Y.; Liu, Y.; Hussain, M.; Makafe, G.G.; et al. An ultrasensitive and specific point-of-care CRISPR/Cas12 based lateral flow biosensor for the rapid detection of nucleic acids. Biosens. Bioelectron. 2020, 159, 112143. [Google Scholar] [CrossRef]
- Dai, Y.; Somoza, R.A.; Wang, L.; Welter, J.F.; Li, Y.; Caplan, A.I.; Liu, C.C. Exploring the trans-cleavage activity of CRISPR-Cas12a (cpf1) for the development of a universal electrochemical biosensor. Angew. Chem. Int. Ed. Engl. 2019, 58, 17399–17405. [Google Scholar] [CrossRef]
- Li, F.; Ye, Q.; Chen, M.; Zhou, B.; Zhang, J.; Pang, R.; Xue, L.; Wang, J.; Zeng, H.; Wu, S.; et al. An ultrasensitive CRISPR/Cas12a based electrochemical biosensor for Listeria monocytogenes detection. Biosens. Bioelectron. 2021, 179, 113073. [Google Scholar] [CrossRef]
- Qing, M.; Chen, S.L.; Sun, Z.; Fan, Y.; Luo, H.Q.; Li, N.B. Universal and programmable rolling circle amplification-CRISPR/Cas12a-mediated immobilization-free electrochemical biosensor. Anal. Chem. 2021, 93, 7499–7507. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isothermal Amplification Technique | Application | Target | Detection | References |
|---|---|---|---|---|
| Loop-mediated isothermal amplification (LAMP) | Detection of animal pathogen DNA | Koi Herpesvirus (KHV) DNA | Fluorescent dyes | [3] |
| Detection of human pathogen DNA | Leptospira DNA | Fluorescent labeled probes | [4] | |
| Detection of human pathogen DNA | Mycobacterium spp DNA | Fluorescent dyes | [5] | |
| Detection of viral RNA | Middle East Respiratory Syndrome-Coronavirus (MERS-CoV) | Fluorescent dyes | [6,7] | |
| Detection of viral RNA | Severe Acute Respiratory Syndrome-related coronavirus (SARS-CoV) | Fluorescent dyes | [7] | |
| Detection of viral RNA | Novel coronavirus (COVID-19) | Fluorescent dyes | [8,9] | |
| Rolling circle amplification (RCA) | Detection of food-borne pathogens | Listeria monocytogenase DNA | Fluorescent dyes | [19] |
| Detection of pathogen DNA | Mycobacterium spp. genomic DNA | Fluorescent labeled probes | [32] | |
| Detection of viral RNA | Influenza virus (H5N1) | Fluorescent dyes | [33] | |
| Detection of viral RNA | Severe Acute Respiratory Syndrome-related coronavirus (SARS-CoV) | Fluorescent dyes | [34] | |
| Detection of miRNAs | miRNA in serum | Fluorescent labeled probes | [35] | |
| Detection of cancer biomarker | miRNA in serum | Fluorescent labeled probes | [36] | |
| CRISPR-Cas system | Detection of DNA | Human cell lines (HEK293) and human cancer cell lines (MCF-7) | Fluorescent labeled probes | [44] |
| Detection of miRNAs | miRNA in serum from cancer patient | Colorimetric | [45] | |
| Detection of viral RNA | Zika virus (ZIKV) and Dengue virus (DENV) | Fluorescent labeled probes | [48] | |
| Detection of viral DNA | Human papillomavirus (HPV) | Fluorescent labeled probes | [53] | |
| Detection of viral RNA | Novel coronavirus (COVID-19) | CRISPR/Cas-LAMP lateral flow | [55] | |
| Detection of bacterial DNA | Pseudomonas aeruginosa | CRISPR/Cas-LAMP | [57] | |
| Detection of bacterial DNA | Listeria monocytogenes | Electrochemical biosensor | [58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boonbanjong, P.; Treerattrakoon, K.; Waiwinya, W.; Pitikultham, P.; Japrung, D. Isothermal Amplification Technology for Disease Diagnosis. Biosensors 2022, 12, 677. https://doi.org/10.3390/bios12090677
Boonbanjong P, Treerattrakoon K, Waiwinya W, Pitikultham P, Japrung D. Isothermal Amplification Technology for Disease Diagnosis. Biosensors. 2022; 12(9):677. https://doi.org/10.3390/bios12090677
Chicago/Turabian StyleBoonbanjong, Poramin, Kiatnida Treerattrakoon, Wassa Waiwinya, Piyawat Pitikultham, and Deanpen Japrung. 2022. "Isothermal Amplification Technology for Disease Diagnosis" Biosensors 12, no. 9: 677. https://doi.org/10.3390/bios12090677