
First-Principles Calculation of MoO2 and MoO3 Electronic and Optical Properties Compared with Experimental Data

 , , , , and
, , , , and
Abstract
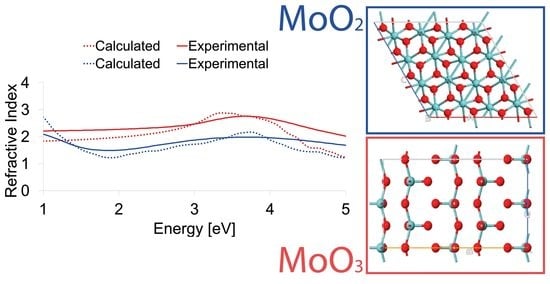
:
1. Introduction
2. Materials and Methods
2.1. Theoretical Modeling and DFT Calculation
2.2. Experimental Section
3. Results and Discussion
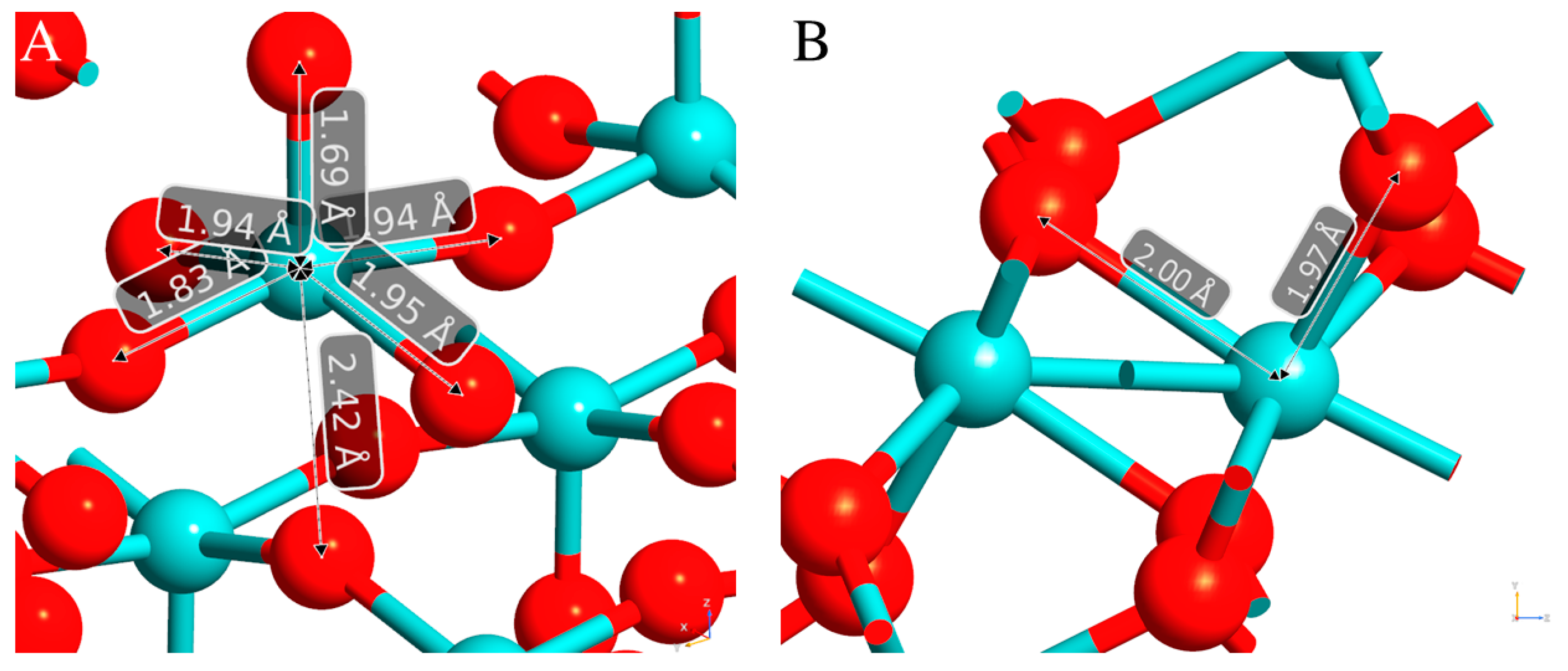
3.1. Geometrical and Lattice Parameters
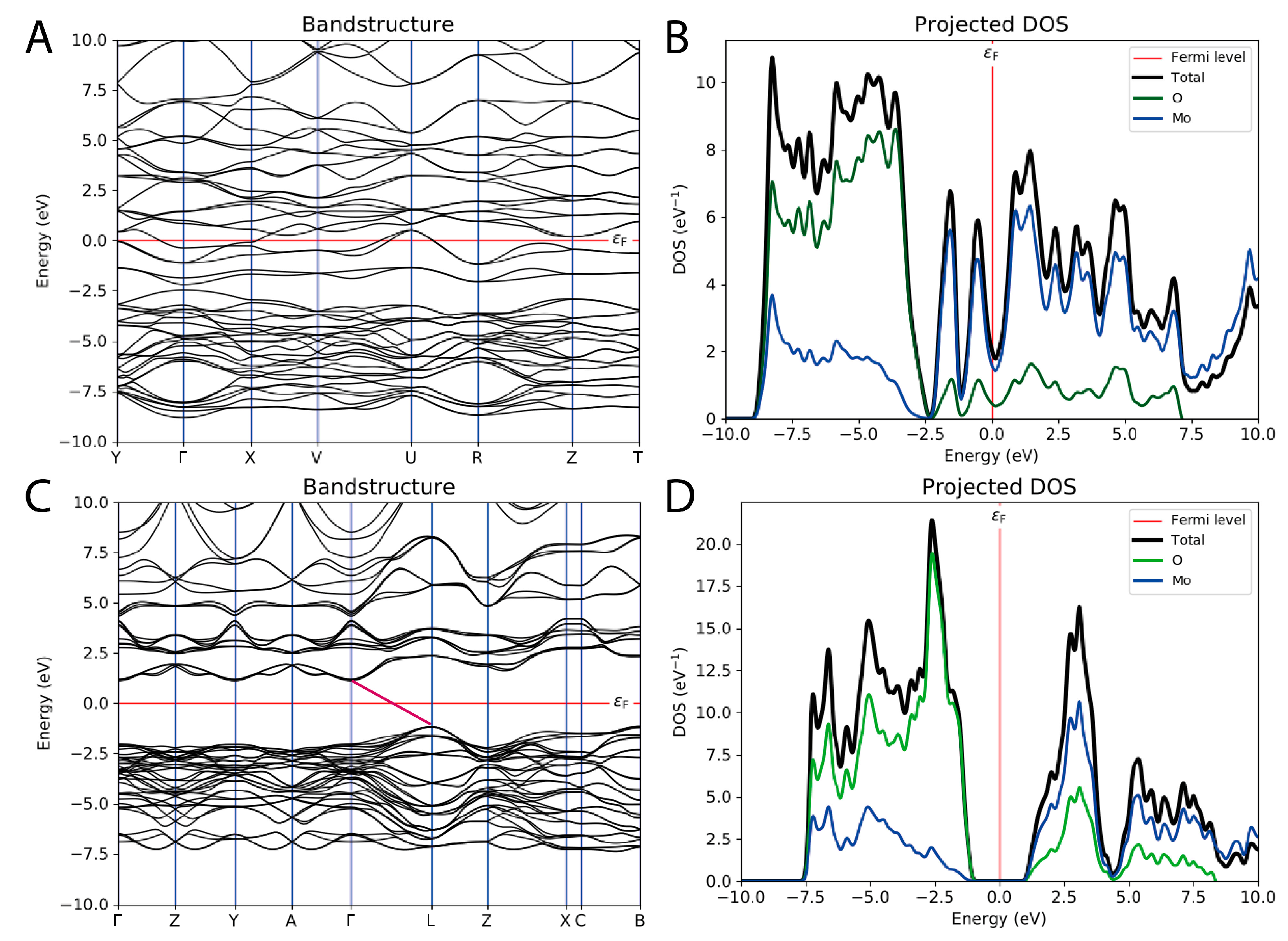
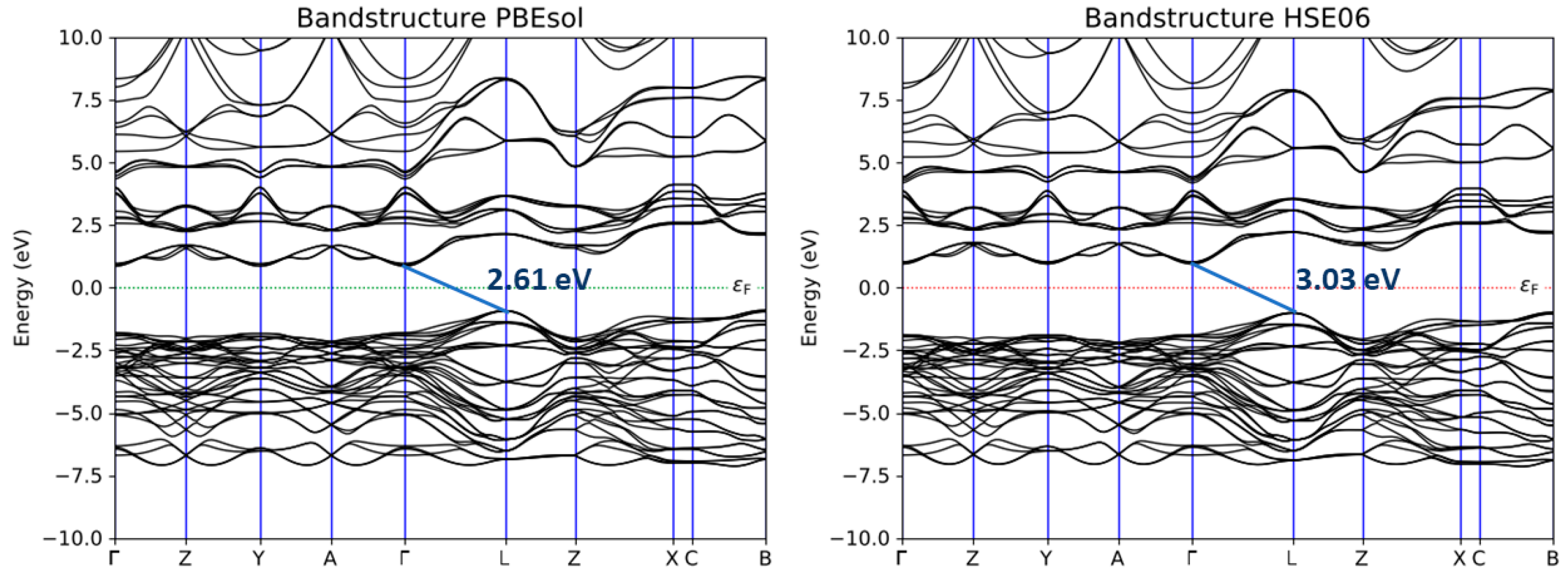
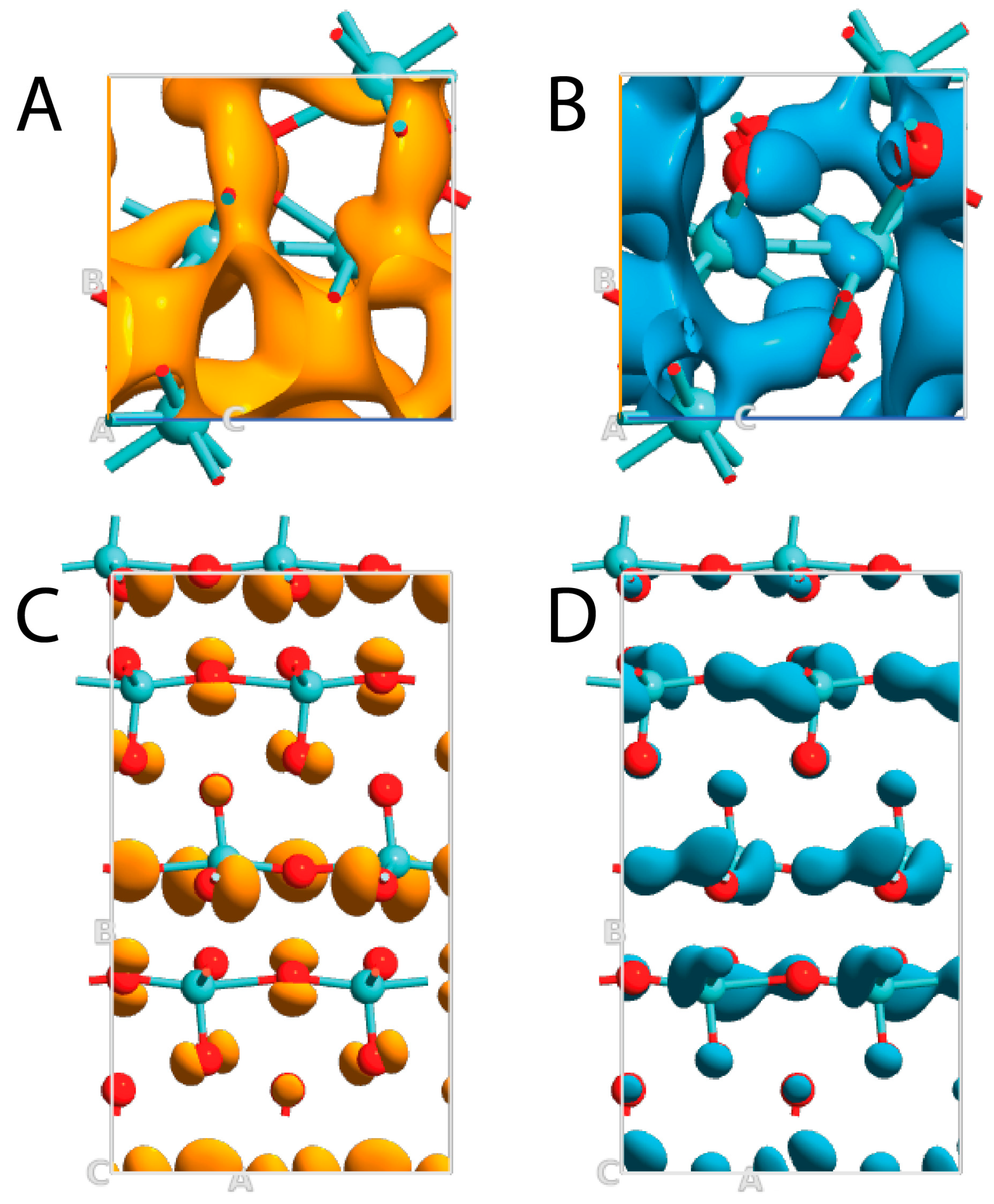
3.2. Band Structure, Band Gap, and Density of the Electronic State
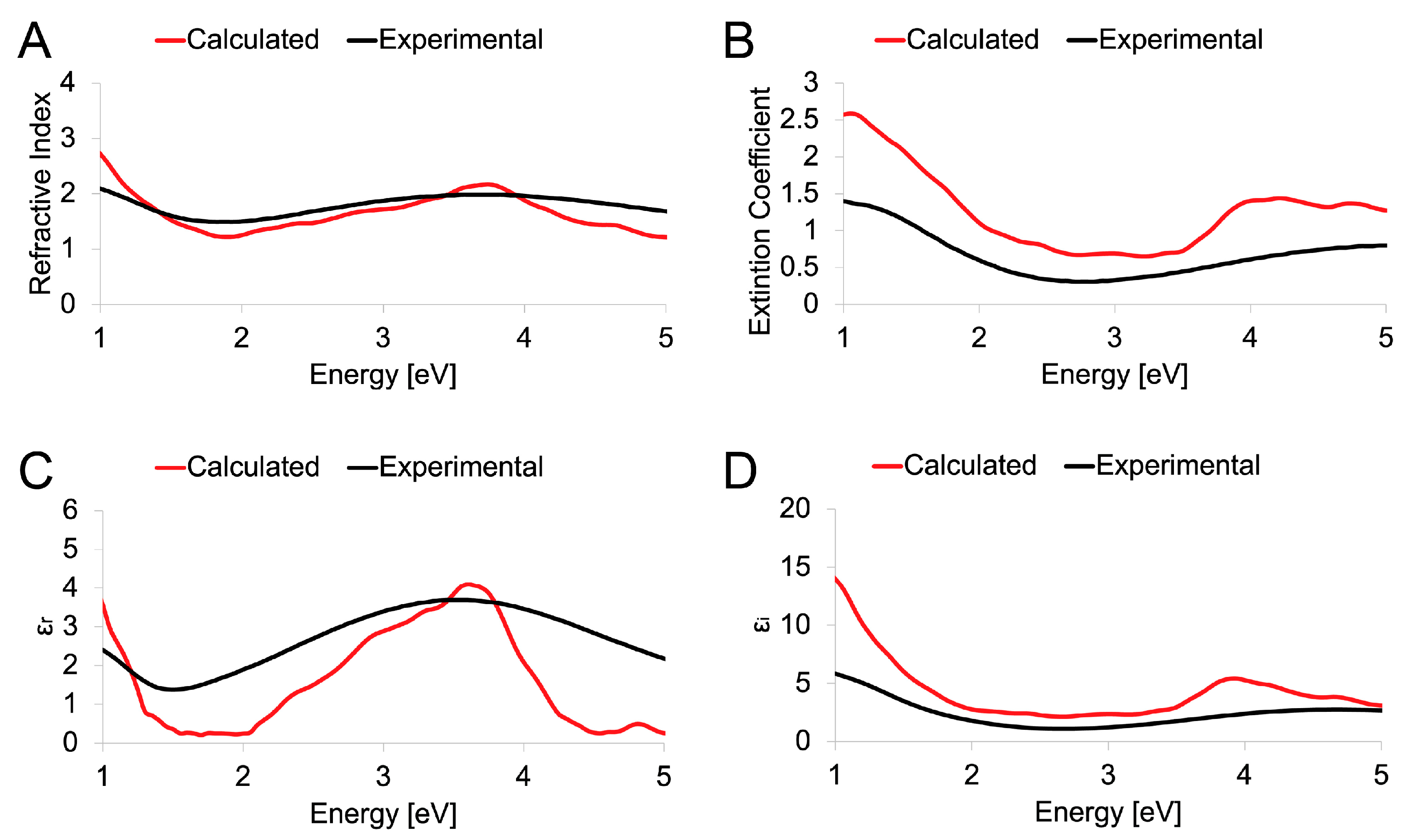
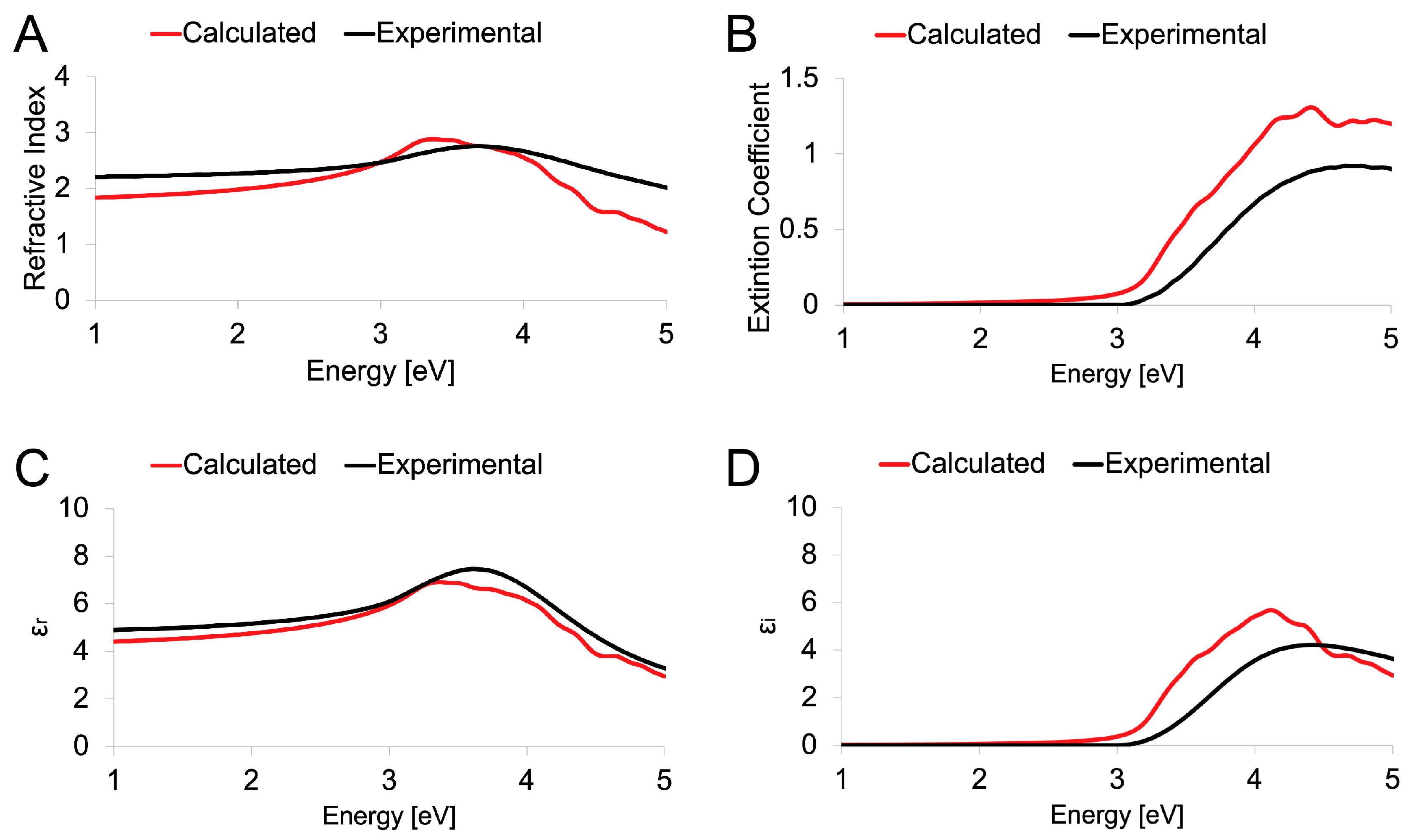
3.3. Experimental and Theoretical Optical Spectra
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Castro, I.A.; Datta, R.S.; Ou, J.Z.; Castellanos-Gomez, A.; Sriram, S.; Daeneke, T.; Kalantar-zadeh, K. Molybdenum Oxides—From Fundamentals to Functionality. Adv. Mater. 2017, 29, 1701619. [Google Scholar] [CrossRef]
- Scanlon, D.O.; Watson, G.W.; Payne, D.J.; Atkinson, G.R.; Egdell, R.G.; Law, D.S.L. Theoretical and Experimental Study of the Electronic Structures of MoO3 and MoO2. J. Phys. Chem. C 2010, 114, 4636–4645. [Google Scholar] [CrossRef]
- Boyadjian, C.; Lefferts, L. Promoting Li/MgO Catalyst with Molybdenum Oxide for Oxidative Conversion of n-Hexane. Catalysts 2020, 10, 354. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.H.; Hargreaves, J.S.J.; Hutchings, G.J.; Joyner, R.W.; Lembacher, C.W. The Partial Oxidation of Methane to Methanol: An Approach to Catalyst Design. Catal. Today 1998, 42, 217–224. [Google Scholar] [CrossRef]
- Balendhran, S.; Walia, S.; Alsaif, M.; Nguyen, E.P.; Ou, J.Z.; Zhuiykov, S.; Sriram, S.; Bhaskaran, M.; Kalantar-Zadeh, K. Field Effect Biosensing Platform Based on 2D α-MoO3. ACS Nano 2013, 7, 9753–9760. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, C.; Yin, X.; Zheng, M.; Sharp, I.D.; Chen, T.; McDonnell, S.; Azcatl, A.; Carraro, C.; Ma, B.; Maboudian, R.; et al. Hole Selective MoOx Contact for Silicon Solar Cells. Nano Lett. 2014, 14, 967–971. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.Y.; Haider, Z.; Van, T.K.; Pawar, A.U.; Kang, M.J.; Kim, C.W.; Kang, Y.S. Tuning of the Crystal Engineering and Photoelectrochemical Properties of Crystalline Tungsten Oxide for Optoelectronic Device Applications. CrystEngComm 2015, 17, 6070–6093. [Google Scholar] [CrossRef]
- Santhosh, S.; Mathankumar, M.; Selva Chandrasekaran, S.; Nanda Kumar, A.K.; Murugan, P.; Subramanian, B. Effect of Ablation Rate on the Microstructure and Electrochromic Properties of Pulsed-Laser-Deposited Molybdenum Oxide Thin Films. Langmuir 2017, 33, 19–33. [Google Scholar] [CrossRef]
- Ranjba, M.; Delalat, F.; Salamati, H. Molybdenum Oxide Nanosheets Prepared by an Anodizing-Exfoliation Process and Observation of Photochromic Properties. Appl. Surf. Sci. 2017, 396, 1752–1759. [Google Scholar] [CrossRef]
- Tomás, S.A.; Arvizu, M.A.; Zelaya-Angel, O.; Rodríguez, P. Effect of ZnSe Doping on the Photochromic and Thermochromic Properties of MoO3 Thin Films. Thin Solid Films 2009, 518, 1332–1336. [Google Scholar] [CrossRef]
- Bandaru, S.; Saranya, G.; English, N.J.; Yam, C.; Chen, M. Tweaking the Electronic and Optical Properties of A-MoO3 by Sulphur and Selenium Doping—A Density Functional Theory Study. Sci. Rep. 2018, 8, 10144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balendhran, S.; Deng, J.; Ou, J.Z.; Walia, S.; Scott, J.; Tang, J.; Wang, K.L.; Field, M.R.; Russo, S.; Zhuiykov, S.; et al. Enhanced Charge Carrier Mobility in Two-Dimensional High Dielectric Molybdenum Oxide. Adv. Mater. 2013, 25, 109–114. [Google Scholar] [CrossRef]
- Mohebbi, E.; Pavoni, E.; Mencarelli, D.; Stipa, P.; Pierantoni, L.; Laudadio, E. Insights into First-Principles Characterization of the Monoclinic VO2(B) Polymorph via DFT + U Calculation: Electronic, Magnetic and Optical Properties. Nanoscale Adv. 2022, 4, 3634–3646. [Google Scholar] [CrossRef]
- Mohebbi, E.; Pavoni, E.; Mencarelli, D.; Stipa, P.; Pierantoni, L.; Laudadio, E. PBEsol/HSE Functional: A Promising Candidate for Vanadium Dioxide (B) Characterization. RSC Adv. 2022, 12, 31255–31263. [Google Scholar] [CrossRef]
- Cueff, S.; John, J.; Zhang, Z.; Parra, J.; Sun, J.; Orobtchouk, R.; Ramanathan, S.; Sanchis, P. VO2 Nanophotonics. APL Photonics 2020, 5, 110901. [Google Scholar] [CrossRef]
- Alves, L.M.S.; Benaion, S.S.; Romanelli, C.M.; dos Santos, C.A.M.; da Luz, M.S.; de Lima, B.S.; Oliveira, F.S.; Machado, A.J.S.; Guedes, E.B.; Abbate, M.; et al. Electrical Resistivity in Non-Stoichiometric MoO2. Braz. J. Phys. 2015, 45, 234–237. [Google Scholar] [CrossRef]
- Boukhachem, A.; Bouzidi, C.; Boughalmi, R.; Ouerteni, R.; Kahlaoui, M.; Ouni, B.; Elhouichet, H.; Amlouk, M. Physical Investigations on MoO3 Sprayed Thin Film for Selective Sensitivity Applications. Ceram. Int. 2014, 40, 13427–13435. [Google Scholar] [CrossRef]
- Pavoni, E.; Mohebbi, E.; Stipa, P.; Mencarelli, D.; Pierantoni, L.; Laudadio, E. The Role of Zr on Monoclinic and Orthorhombic Hfx Zry O2 Systems: A First-Principles Study. Materials 2022, 15, 4175. [Google Scholar] [CrossRef]
- Pavoni, E.; Mohebbi, E.; Mencarelli, D.; Stipa, P.; Laudadio, E.; Pierantoni, L. The Effect of Y Doping on Monoclinic, Orthorhombic, and Cubic Polymorphs of HfO2: A First Principles Study. Nanomaterials 2022, 12, 4324. [Google Scholar] [CrossRef]
- Camacho-López, M.A.; Escobar-Alarcón, L.; Picquart, M.; Arroyo, R.; Córdoba, G.; Haro-Poniatowski, E. Micro-Raman Study of the m-MoO2 to α-MoO3 Transformation Induced by Cw-Laser Irradiation. Opt. Mater. 2011, 33, 480–484. [Google Scholar] [CrossRef]
- Taddei, P.; Ruggiero, A.; Pavoni, E.; Affatato, S. Transfer of Metallic Debris after in Vitro Ceramic-on-Metal Simulation: Wear and Degradation in Biolox® Delta Composite Femoral Heads. Compos. B Eng. 2017, 115, 477–487. [Google Scholar] [CrossRef]
- Feng, Y.; Liu, H.; Liu, Y.; Li, J. Tunable Oxygen Deficient in MoO3-x/MoO2 Heterostructure for Enhanced Lithium Storage Properties. Int. J. Energy Res. 2022, 46, 5789–5799. [Google Scholar] [CrossRef]
- Divigalpitiya, W.M.R.; Frindt, R.F.; Morrison, S.R. Oriented Films of Molybdenum Trioxide. Thin Solid Films 1990, 188, 173–179. [Google Scholar] [CrossRef]
- Balendhran, S.; Walia, S.; Nili, H.; Ou, J.Z.; Zhuiykov, S.; Kaner, R.B.; Sriram, S.; Bhaskaran, M.; Kalantar-Zadeh, K. Two-Dimensional Molybdenum Trioxide and Dichalcogenides. Adv. Funct. Mater. 2013, 23, 3952–3970. [Google Scholar] [CrossRef]
- Pavoni, E.; Bandini, E.; Benaglia, M.; Molloy, J.K.; Bergamini, G.; Ceroni, P.; Armaroli, N. A Tailored RAFT Copolymer for the Dispersion of Single Walled Carbon Nanotubes in Aqueous Media. Polym. Chem. 2014, 5, 6148–6150. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Y.; Yang, Z.; Yan, Y.; Sun, K. Synthesis of MoS2 and MoO2 for Their Applications in H2 Generation and Lithium Ion Batteries: A Review. Sci. Technol. Adv. Mater. 2013, 14, 43501–43513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Li, X.; Ma, Q.; Zhang, Q.; Bai, H.; Yi, W.; Liu, J.; Han, J.; Xi, G. A Metallic Molybdenum Dioxide with High Stability for Surface Enhanced Raman Spectroscopy. Nat. Commun. 2017, 8, 14903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Zhang, W.; Liu, X.; Mei, Y.; Huang, Y. Nanostructured Mo-Based Electrode Materials for Electrochemical Energy Storage. Chem. Soc. Rev. 2015, 44, 2376–2404. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ruzsinszky, A.; Perdew, J.P. Strongly Constrained and Appropriately Normed Semilocal Density Functional. Phys. Rev. Lett. 2015, 115, 36402. [Google Scholar] [CrossRef] [Green Version]
- van Setten, M.J.; Giantomassi, M.; Bousquet, E.; Verstraete, M.J.; Hamann, D.R.; Gonze, X.; Rignanese, G.M. The PSEUDODOJO: Training and Grading a 85 Element Optimized Norm-Conserving Pseudopotential Table. Comput. Phys. Commun. 2018, 226, 39–54. [Google Scholar] [CrossRef] [Green Version]
- Sun, J. (Invited) SCAN Meta-GGA: An Accurate, Efficient, and Physically Sound Density Functional for Materials Discovery and Design. ECS Meet. Abstr. 2019, MA2019-02, 2022. [Google Scholar] [CrossRef]
- Sun, J.; Remsing, R.C.; Zhang, Y.; Sun, Z.; Ruzsinszky, A.; Peng, H.; Yang, Z.; Paul, A.; Waghmare, U.; Wu, X.; et al. Accurate First-Principles Structures and Energies of Diversely Bonded Systems from an Efficient Density Functional. Nat. Chem. 2016, 8, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Waghmare, U.V.; Friend, C.M.; Kaxiras, E. A Density Functional Study of Clean and Hydrogen-Covered α-MoO3 (010): Electronic Structure and Surface Relaxation. J. Chem. Phys. 1998, 109, 6854. [Google Scholar] [CrossRef]
- Rozzi, A.; Manghi, F.; Parmigiani, F. Ab Initio Fermi Surface and Conduction-Band Calculations in Oxygen-Reduced MoO3. Phys. Rev. B 2003, 68, 075110. [Google Scholar] [CrossRef]
- Eyert, V.; Horny, R.; Höck, K.H.; Horn, S. Embedded Peierls instability and the Electronic Structure of MoO2. J. Phys. Condens. Matter 2000, 12, 4923. [Google Scholar] [CrossRef]
- Moosburger-Will, J.; Kündel, J.; Klemm, M.; Horn, S.; Hofmann, P.; Schwingenschlögl, U.; Eyert, V. Fermi Surface of MoO2 Studied by Angle-Resolved Photoemission Spectroscopy, de Haas-van Alphen Measurements, and Electronic Structure Calculations. Phys. Rev. B Condens. Matter Mater. Phys. 2009, 79, 115113. [Google Scholar] [CrossRef] [Green Version]
- Coquet, R.; Willock, D.J. The (010) Surface of α-MoO3, a DFT + U Study. Phys. Chem. Chem. Phys. 2005, 7, 3819–3828. [Google Scholar] [CrossRef]
- Gulomov, J.; Accouche, O.; Al Barakeh, Z.; Aliev, R.; Gulomova, I.; Neji, B. Atom-to-Device Simulation of MoO3/Si Heterojunction Solar Cell. Nanomaterials 2022, 12, 4240. [Google Scholar] [CrossRef]
- Smidstrup, S.; Markussen, T.; Vancraeyveld, P.; Wellendorff, J.; Schneider, J.; Gunst, T.; Verstichel, B.; Stradi, D.; Khomyakov, P.A.; Vej-Hansen, U.G.; et al. QuantumATK: An Integrated Platform of Electronic and Atomic-Scale Modelling Tools. J. Phys. Condens. Matter 2020, 32, 015901. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A Materials Genome Approach to Accelerating Materials Innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef] [Green Version]
- Soler, J.M.; Artacho, E.; Gale, J.D.; García, A.; Junquera, J.; Ordejón, P.; Sánchez-Portal, D. The SIESTA Method for Ab Initio Order-N Materials Simulation. J. Phys. Condens. Matter 2002, 14, 2745. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [PubMed] [Green Version]
- Mortazavi, B.; Shahrokhi, M.; Makaremi, M.; Rabczuk, T. Anisotropic Mechanical and Optical Response and Negative Poisson’s Ratio in Mo2C Nanomembranes Revealed by First-Principles Simulations. Nanotechnology 2017, 28, 115705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiroyuki, F. Spectroscopic Ellipsometry: Principles and Applications; John Wiley & Sons Inc: Hoboken, NJ, USA, 2007. [Google Scholar]
- Jellison, G.E.; Modine, F.A. Parameterization of the Optical Functions of Amorphous Materials in the Interband Region. Appl. Phys. Lett. 1996, 69, 371–373. [Google Scholar] [CrossRef]
- Azzam, R.M.A.; Bashara, N.M. Ellipsometry and Polarized Light, Paperback ed.; North-Holland: Amsterdam, The Netherlands, 1987; ISBN 0444870164. [Google Scholar]
- Brandt, B.; Skapski, A. A Refinement of the Crystal Structure of Molybdenum Dioxide. Acta Chem. Scand. 1967, 21, 661–672. [Google Scholar] [CrossRef]
- Sitepu, H.; O’Connor, B.H.; Li, D. Comparative Evaluation of the March and Generalized Spherical Harmonic Preferred Orientation Models Using X-Ray Diffraction Data for Molybdite and Calcite Powders. J. Appl. Crystallogr. 2005, 38, 158–167. [Google Scholar] [CrossRef]
- Kihlborg, L. Crystal structure of Mo18O52+ existence of homologous series of structures based on MoO3. Ark. Kemi 1964, 21, 443–448. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| This Work | Experimental | This Work | |||
|---|---|---|---|---|---|
| Orthorhombic MoO3 | a | 3.909 Å | 3.962 Å | α | 90° |
| b | 13.855 Å | 13.855 Å | β | 90° | |
| c | 3.681 Å | 3.699 Å | γ | 90° | |
| Monoclinic MoO2 | a | 5.625 Å | 5.611 Å | α | 90° |
| b | 4.872 Å | 4.856 Å | β | 120.5° | |
| c | 5.645 Å | 5.628 Å | γ | 90° |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavoni, E.; Modreanu, M.G.; Mohebbi, E.; Mencarelli, D.; Stipa, P.; Laudadio, E.; Pierantoni, L. First-Principles Calculation of MoO2 and MoO3 Electronic and Optical Properties Compared with Experimental Data. Nanomaterials 2023, 13, 1319. https://doi.org/10.3390/nano13081319
Pavoni E, Modreanu MG, Mohebbi E, Mencarelli D, Stipa P, Laudadio E, Pierantoni L. First-Principles Calculation of MoO2 and MoO3 Electronic and Optical Properties Compared with Experimental Data. Nanomaterials. 2023; 13(8):1319. https://doi.org/10.3390/nano13081319
Chicago/Turabian StylePavoni, Eleonora, Mircea Gabriel Modreanu, Elaheh Mohebbi, Davide Mencarelli, Pierluigi Stipa, Emiliano Laudadio, and Luca Pierantoni. 2023. "First-Principles Calculation of MoO2 and MoO3 Electronic and Optical Properties Compared with Experimental Data" Nanomaterials 13, no. 8: 1319. https://doi.org/10.3390/nano13081319