
Redox Chemistry of the Subphases of α-CsPbI2Br and β-CsPbI2Br: Theory Reveals New Potential for Photostability

 , , ,
, , ,  , and
, and
Abstract
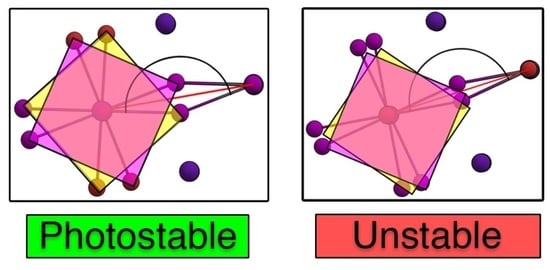
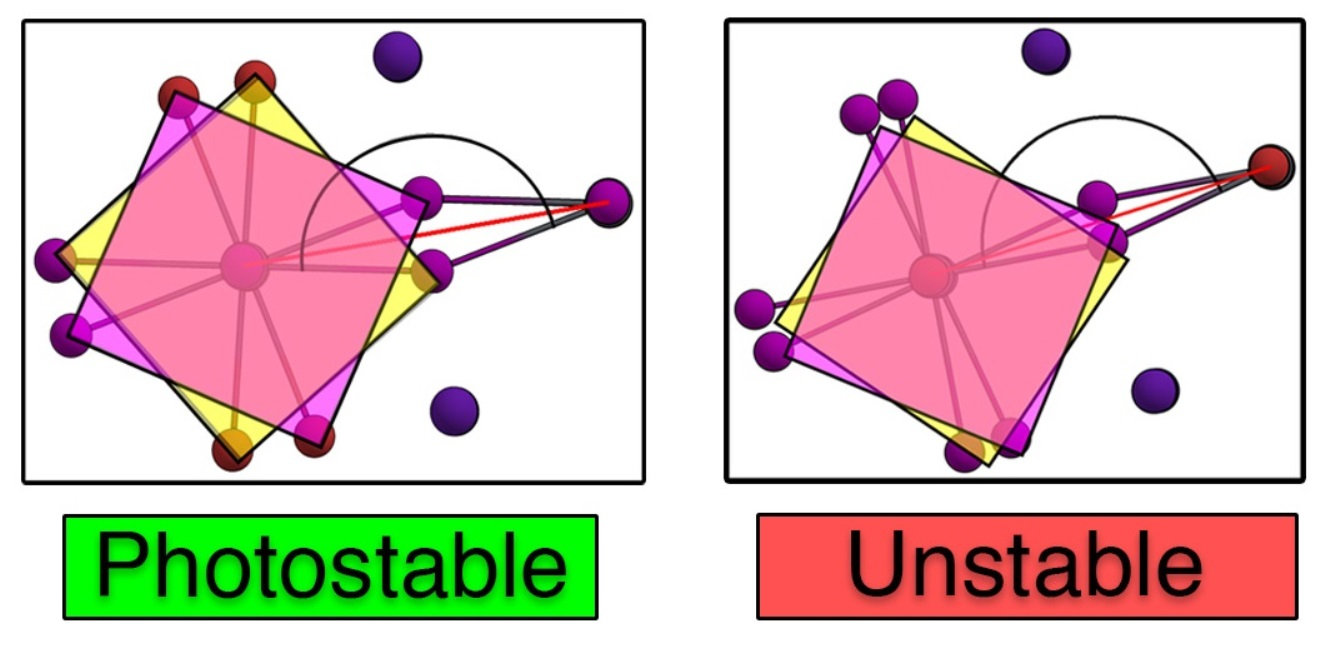
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
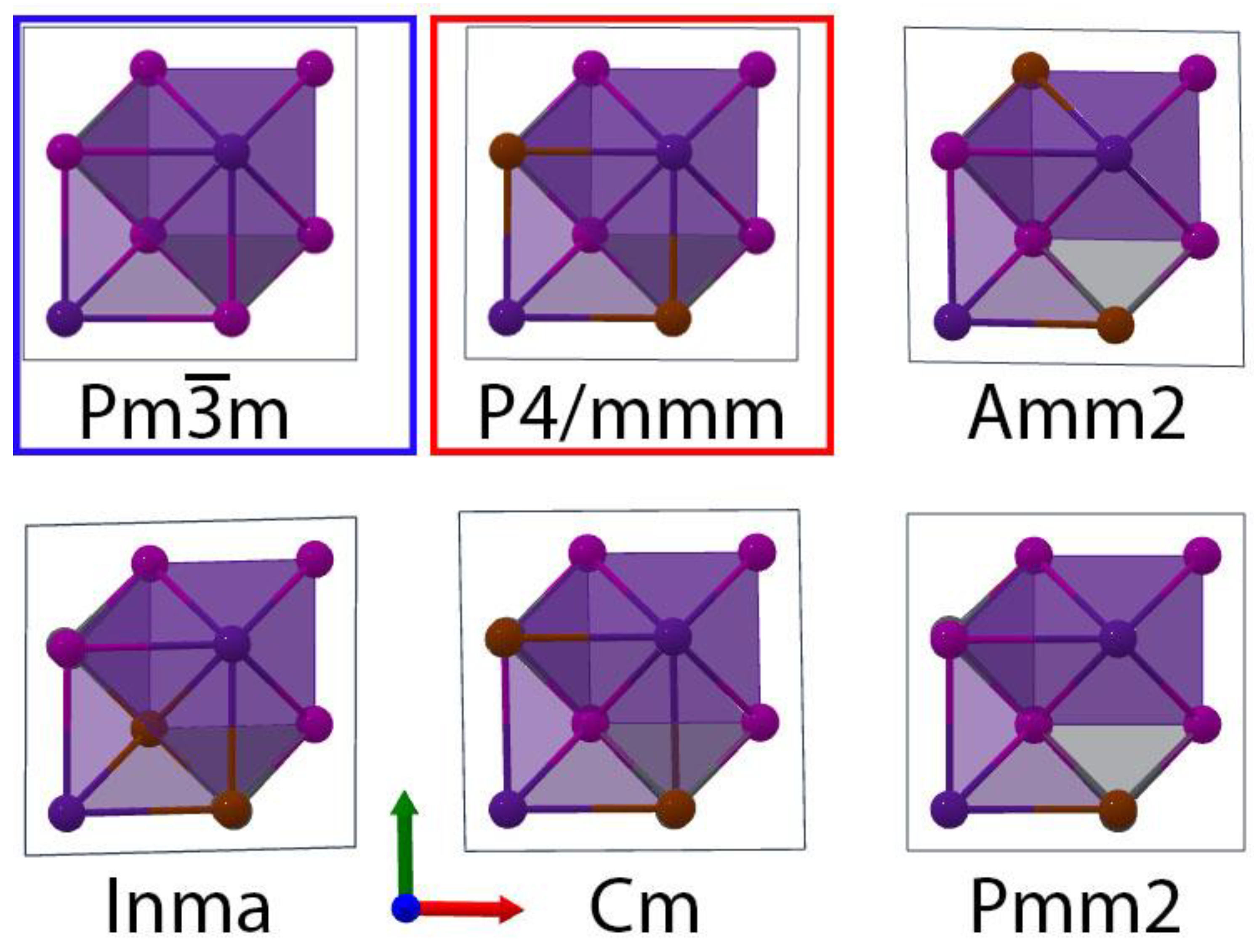
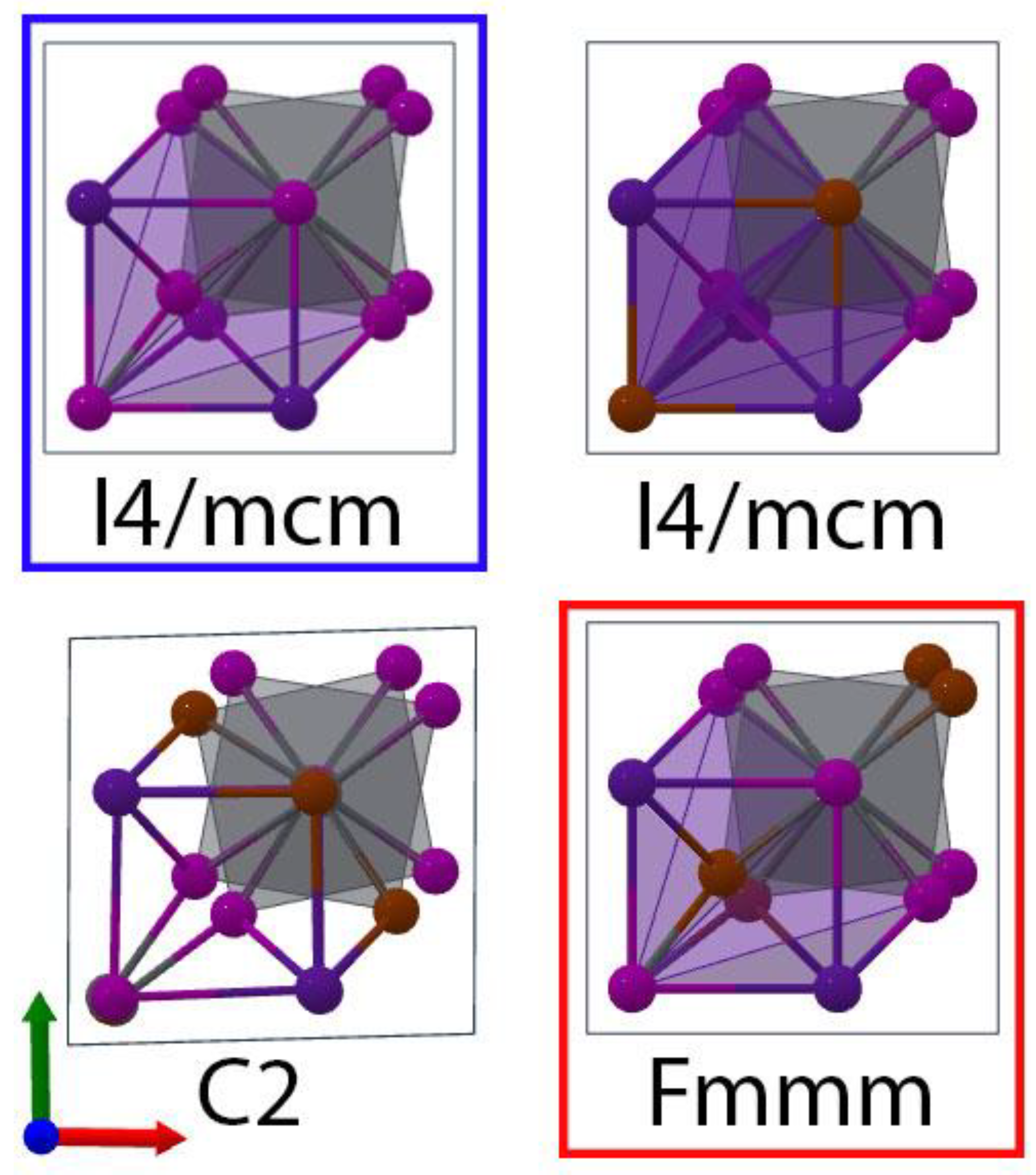
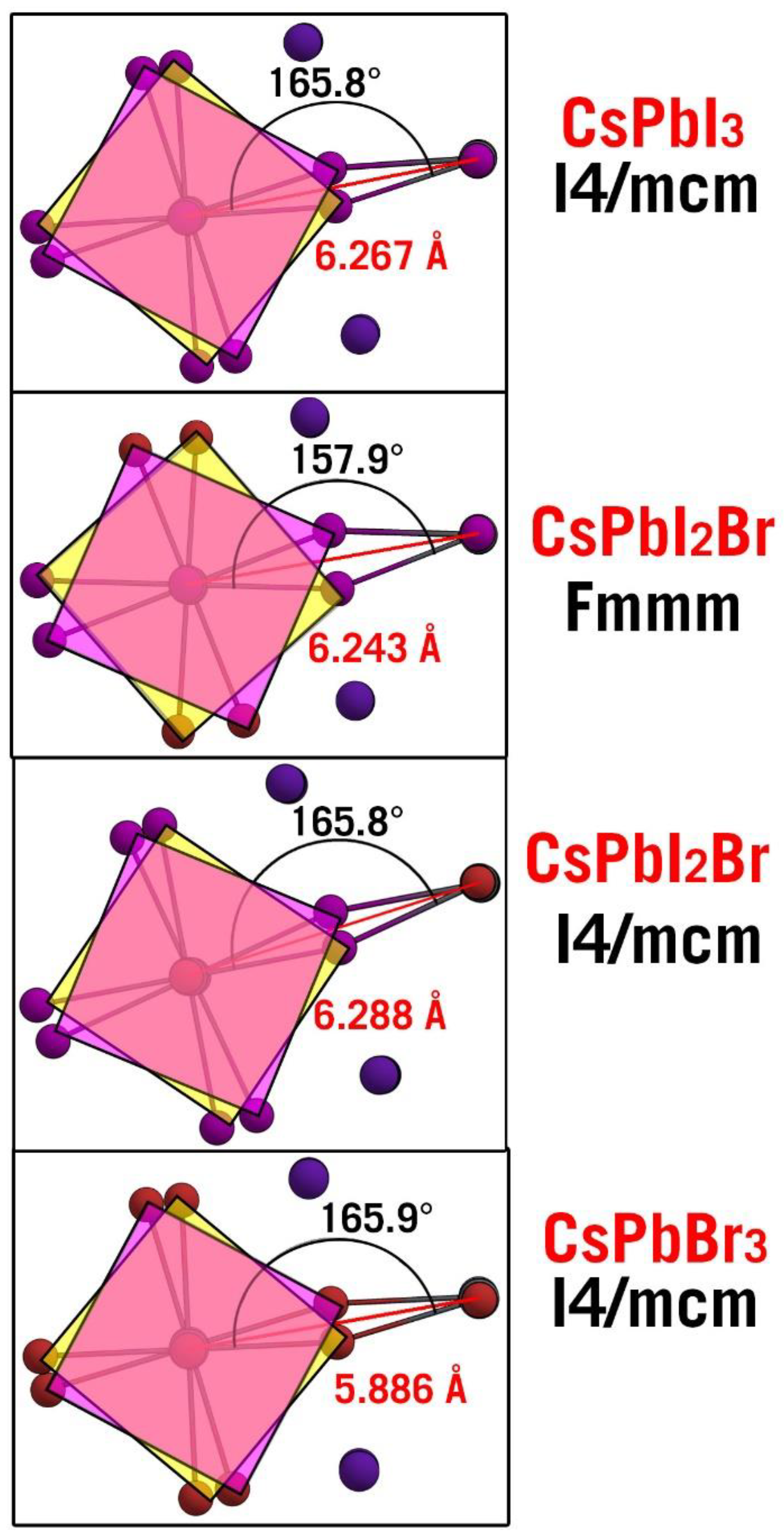
3.1. Lattice Constants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Lattice Parameter | Energy/f.u. (eV/cell) | ||
| a | ||||
| Exp | α-CsPbI3 | 6.29 [31], 6.297 [52] | - | |
| DFT | α-CsPbI3 | 6.18 [52], 6.26 [56], 6.27 (6.30), 6.41 [57] | −15.28 | |
| Exp | α-CsPbI2Br | 6.138 [30] | − | |
| DFT | α-CsPbI2Br P4/mmm | 6.18 (6.17) 0.22, 6.26 [29] | −15.84 | |
| α-CsPbI2Br C1 | 6.190.02 | −15.82 | ||
| α-CsPbI2Br Inma | 6.180.05 | −15.84 | ||
| α-CsPbI2Br Cm | 6.180.13 | −15.83 | ||
| α-CsPbI2Br Pmm2 | 6.190.04 | −15.82 | ||
| α-CsPbI2Br Amm2 | 6.190.05 | −15.80 | ||
| Phase | Lattice Parameters | Energy/f.u. (eV/cell) | ||
| a | b | |||
| Exp | β-CsPbI3 | 6.241 [52] | 6.299 [52] | - |
| DFT | β-CsPbI3 I4/mcm | 5.97 [52], 6.28 | 6.273 [52], 6.33 | −15.30 |
| Exp | β-CsPbI2Br | 6.130 [30] | 6.088 [30] | - |
| DFT | β-CsPbI2Br I4/mcm | 6.33 [29], 6.29 | 6.03 [29], 5.96 | −15.84 |
| β-CsPbI2Br Fmmm | 6.08 [55], 6.01 (6.03) | 6.45 [55], 6.38 (6.37) | −15.92 | |
| β-CsPbI2Br C2 | 6.14 | 6.19 | −15.87 | |
3.2. Mechanical Properties
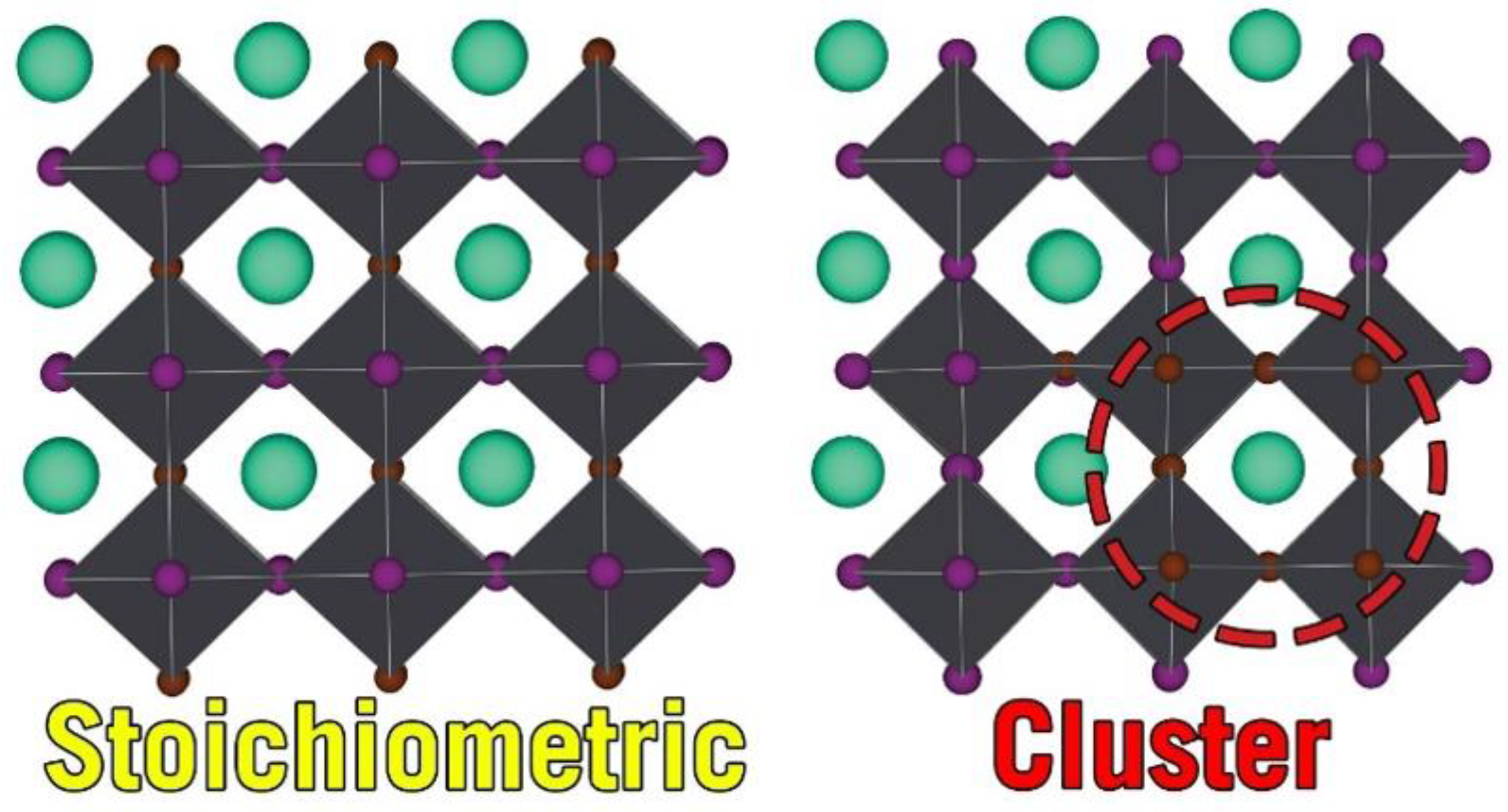
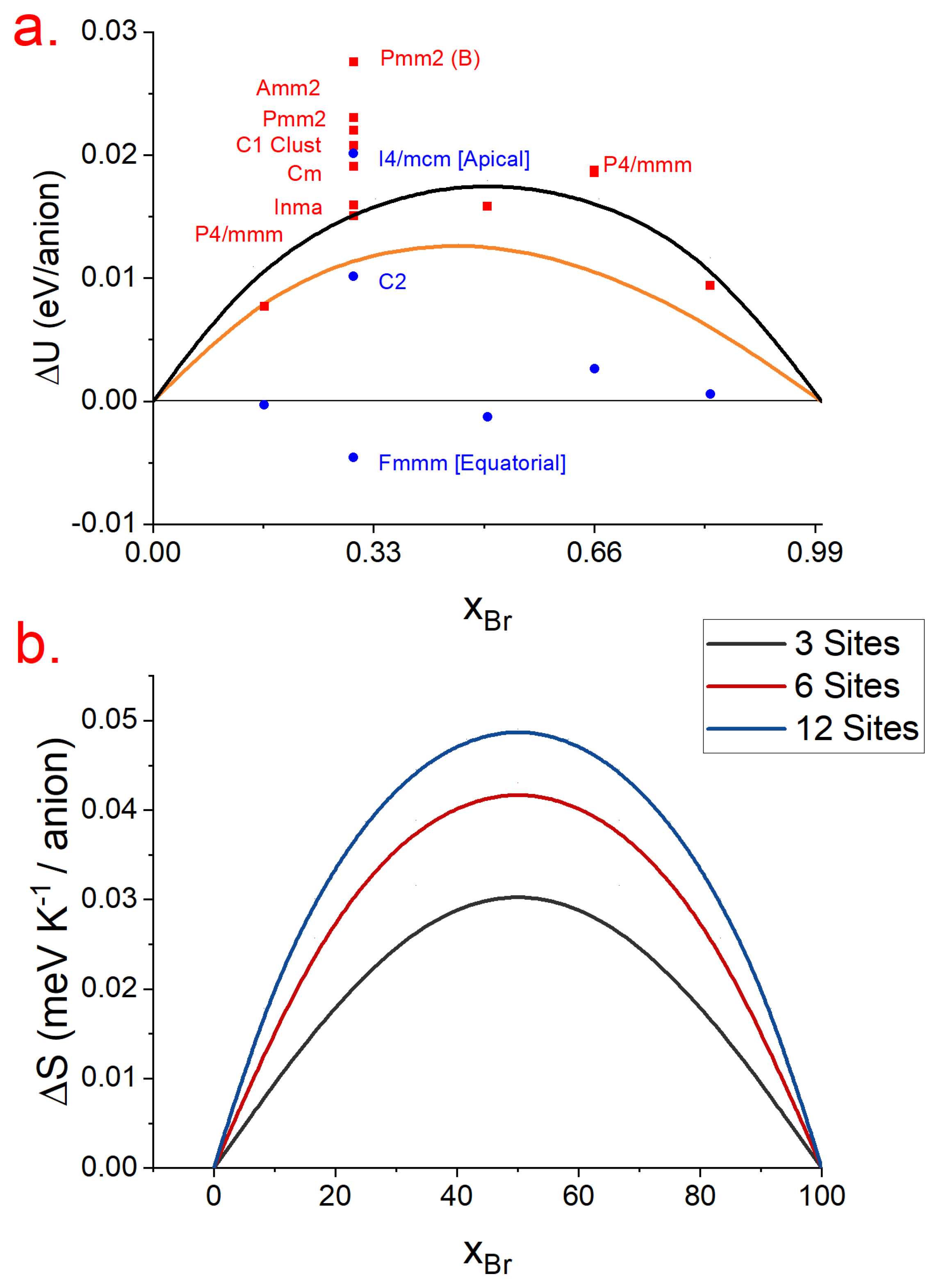
3.3. Mixing Energy
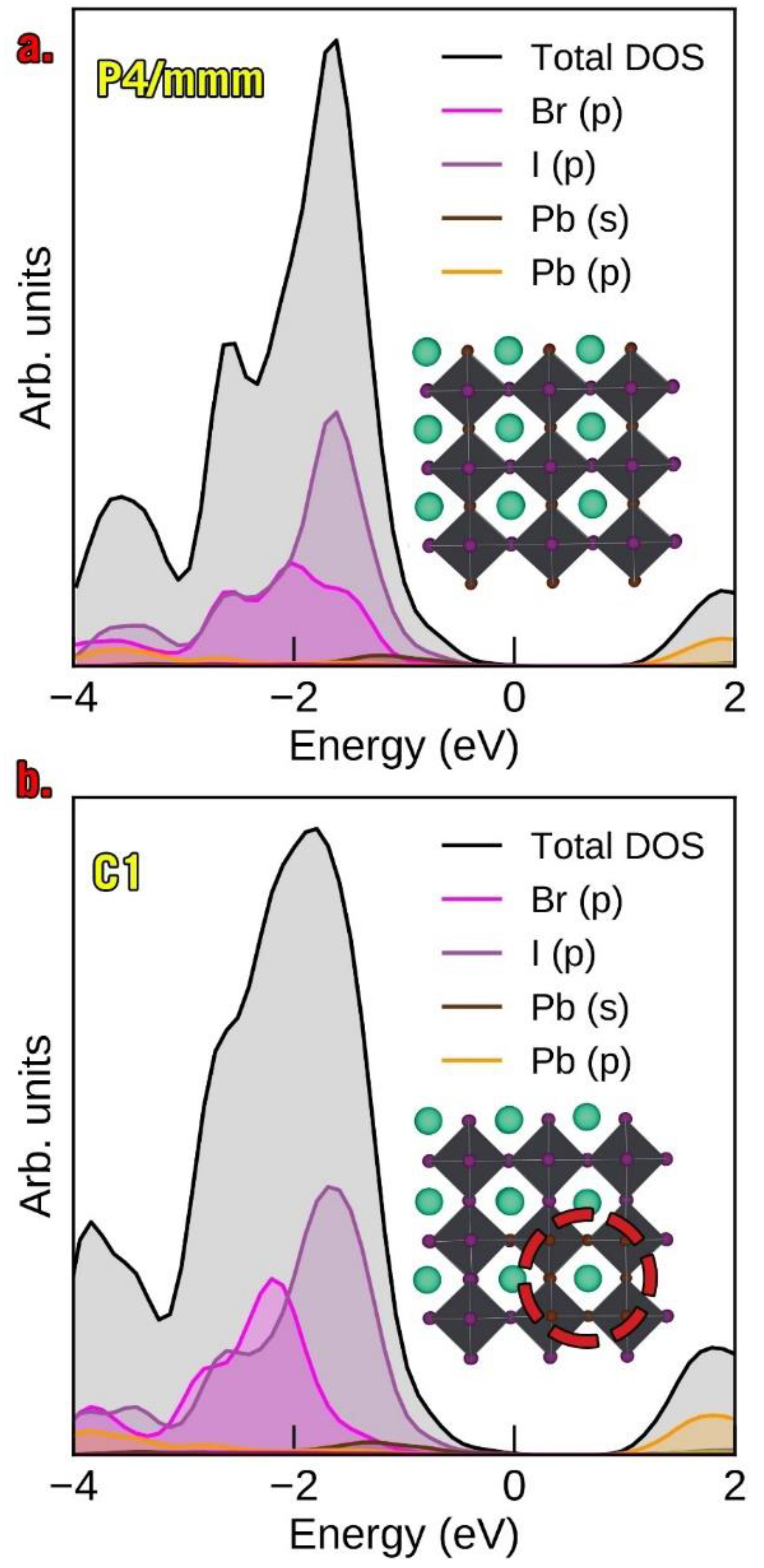
3.4. Redox Properties of α-CsPbI2Br and β-CsPbI2Br
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density functional theory |
| GGA | Generalized gradient approximation |
| MGGA | Metageneralized gradient approximation |
| VASP | Vienna Ab initio Simulation Package |
| PAW | Projector augmented-wave |
| PV | Photovoltaic |
| PCE | Photoconversion efficiency |
| CFD | Charged Frenkel defect |
| SOC | Spin–orbit coupling |
| EOS | Equation of state |
| DOS | Density of states |
| pDOS | Partial density of states |
| EV | Energy versus volume |
References
- Jena, A.K.; Kulkarni, A.; Miyasaka, T. Halide Perovskite Photovoltaics: Background, Status, and Future Prospects. Chem. Rev. 2019, 119, 3036–3103. [Google Scholar] [CrossRef] [PubMed]
- NREL. Best Research-Cell Efficiency Chart. Available online: https://www.nrel.gov/pv/cell-efficiency.html (accessed on 22 November 2020).
- Davies, C.L.; Filip, M.R.; Patel, J.B.; Crothers, T.W.; Verdi, C.; Wright, A.D.; Milot, R.L.; Giustino, F.; Johnston, M.B.; Herz, L.M. Bimolecular Recombination in Methylammonium Lead Triiodide Perovskite Is an Inverse Absorption Process. Nat. Commun. 2018, 9, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R.; Mujahid, M.; Duan, Y.; Wang, Z.; Xue, J.; Yang, Y. A Review of Perovskites Solar Cell Stability. Adv. Funct. Mater. 2019, 29, 1808843. [Google Scholar] [CrossRef]
- Wang, D.; Wright, M.; Elumalai, N.K.; Uddin, A. Stability of Perovskite Solar Cells. Sol. Energy Mater. Sol. Cells 2016, 147, 255–275. [Google Scholar] [CrossRef]
- Asghar, M.I.; Zhang, J.; Wang, H.; Lund, P.D. Device Stability of Perovskite Solar Cells—A Review. Renew. Sustain. Energy Rev. 2017, 77, 131–146. [Google Scholar] [CrossRef] [Green Version]
- Walsh, A.; Scanlon, D.O.; Chen, S.; Gong, X.G.; Wei, S.-H.H. Self-Regulation Mechanism for Charged Point Defects in Hybrid Halide Perovskites. Angew. Chemie Int. Ed. 2015, 54, 1791–1794. [Google Scholar] [CrossRef] [Green Version]
- Brommer, P.; Quigley, D. Automated Effective Band Structures for Defective and Mismatched Supercells. J. Phys. Condens. Matter 2014, 26, 48. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Tan, L.Z.; Shen, X.; Hu, T.; Miyata, K.; Tuan Trinh, M.; Li, R.; Coffee, R.; Liu, S.; Egger, D.A.; et al. Light-Induced Picosecond Rotational Disordering of the Inorganic Sublattice in Hybrid Perovskites. Sci. Adv. 2017, 3, e1602388. [Google Scholar] [CrossRef] [Green Version]
- Neukirch, A.J.; Nie, W.; Blancon, J.C.; Appavoo, K.; Tsai, H.; Sfeir, M.Y.; Katan, C.; Pedesseau, L.; Even, J.; Crochet, J.J.; et al. Polaron Stabilization by Cooperative Lattice Distortion and Cation Rotations in Hybrid Perovskite Materials. Nano Lett. 2016, 16, 3809–3816. [Google Scholar] [CrossRef] [Green Version]
- Hajjiah, A.; Gamal, M.; Kandas, I.; Gorji, N.E.; Shehata, N. DFT and AMPS-1D Simulation Analysis of All-Perovskite Solar Cells Based on CsPbI3/FAPbI3 Bilayer Structure. Sol. Energy Mater. Sol. Cells 2022, 248, 112026. [Google Scholar] [CrossRef]
- Lv, Y.; Zhang, H.; Liu, R.; Sun, Y.; Huang, W. Composite Encapsulation Enabled Superior Comprehensive Stability of Perovskite Solar Cells. ACS Appl. Mater. Interfaces 2020, 12, 27277–27285. [Google Scholar] [CrossRef]
- Jung, M.; Shin, T.J.; Seo, J.; Kim, G.; Seok, S. Il Structural Features and Their Functions in Surfactant-Armoured Methylammonium Lead Iodide Perovskites for Highly Efficient and Stable Solar Cells. Energy Environ. Sci. 2018, 11, 2188–2197. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, Y.; Zhang, X.; Zhao, J.; Zhao, Z.; Su, X.; Hua, Z.; Zhang, J.; Cao, J.; Feng, J.; et al. General Decomposition Pathway of Organic–Inorganic Hybrid Perovskites through an Intermediate Superstructure and Its Suppression Mechanism. Adv. Mater. 2020, 32, 2001107. [Google Scholar] [CrossRef]
- Kim, G.Y.; Senocrate, A.; Yang, T.Y.; Gregori, G.; Grätzel, M.; Maier, J. Large Tunable Photoeffect on Ion Conduction in Halide Perovskites and Implications for Photodecomposition. Nat. Mater. 2018, 17, 445–449. [Google Scholar] [CrossRef]
- Zhao, Y.C.; Zhou, W.K.; Zhou, X.; Liu, K.H.; Yu, D.P.; Zhao, Q. Quantification of Light-Enhanced Ionic Transport in Lead Iodide Perovskite Thin Films and Its Solar Cell Applications. Light Sci. Appl. 2017, 6, e16243. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, E.; Meggiolaro, D.; Snaith, H.J.; Stranks, S.D.; De Angelis, F. Light-Induced Annihilation of Frenkel Defects in Organo-Lead Halide Perovskites. Energy Environ. Sci. 2016, 9, 3180–3187. [Google Scholar] [CrossRef]
- Popov, A.I.; Kotomin, E.A.; Maier, J. Analysis of Self-Trapped Hole Mobility in Alkali Halides and Metal Halides. Solid State Ion. 2017, 302, 3–6. [Google Scholar] [CrossRef]
- Peng, C.; Wang, J.; Wang, H.; Hu, P. Unique Trapped Dimer State of the Photogenerated Hole in Hybrid Orthorhombic CH3NH3PbI3 Perovskite: Identification, Origin, and Implications. Nano Lett. 2017, 17, 7724–7730. [Google Scholar] [CrossRef] [Green Version]
- Kang, B.; Biswas, K. Shallow Trapping vs. Deep Polarons in a Hybrid Lead Halide Perovskite, CH3NH3PbI3. Phys. Chem. Chem. Phys. 2017, 19, 27184–27190. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.H.; Lee, J.H.; Hong, K.H. The Role of Intrinsic Defects in Methylammonium Lead Iodide Perovskite. J. Phys. Chem. Lett. 2014, 5, 1312–1317. [Google Scholar] [CrossRef]
- Schulz, P.; Cahen, D.; Kahn, A. Halide Perovskites: Is It All about the Interfaces? Chem. Rev. 2019, 119, 3349–3417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frolova, L.A.; Luchkin, S.Y.; Lekina, Y.; Gutsev, L.G.; Tsarev, S.A.; Zhidkov, I.S.; Kurmaev, E.Z.; Shen, Z.X.; Stevenson, K.J.; Aldoshin, S.M.; et al. Reversible Pb2+/Pb0 and I−/I3− Redox Chemistry Drives the Light-Induced Phase Segregation in All-Inorganic Mixed Halide Perovskites. Adv. Energy Mater. 2021, 11, 2002934. [Google Scholar] [CrossRef]
- Taylor, T.R.; Asmis, K.R.; Zanni, M.T.; Neumark, D.M. Characterization of the I3 Radical by Anion Photoelectron Spectroscopy. J. Chem. Phys. 1999, 110, 7607–7609. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.S.P.; Visscher, L.; Bolvin, H.; Saue, T.; Knecht, S.; Fleig, T.; Eliav, E. The Electronic Structure of the Triiodide Ion from Relativistic Correlated Calculations: A Comparison of Different Methodologies. J. Chem. Phys. 2010, 133, 064305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoke, E.T.; Slotcavage, D.J.; Dohner, E.R.; Bowring, A.R.; Karunadasa, H.I.; McGehee, M.D. Reversible Photo-Induced Trap Formation in Mixed-Halide Hybrid Perovskites for Photovoltaics. Chem. Sci. 2015, 6, 613–617. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Brocks, G.; Tao, S.; Bobbert, P.A. Unified Theory for Light-Induced Halide Segregation in Mixed Halide Perovskites. Nat. Commun. 2021, 12, 2687. [Google Scholar] [CrossRef]
- Suchan, K.; Just, J.; Becker, P.; Rehermann, C.; Merdasa, A.; Suchan, K.; Scheblykin, I.G.; Unger, E. Multi-Stage Phase-Segregation of Mixed Halide Perovskites under Illumination: A Quantitative Comparison of Experimental Observations and Thermodynamic Models. arXiv, 2022; in preprint. arXiv:2205.10867,v1. [Google Scholar] [CrossRef]
- Chen, Y.; Shi, T.; Liu, P.; Xie, W.; Chen, K.; Xu, X.; Shui, L.; Shang, C.; Chen, Z.; Yip, H.L.; et al. The Distinctive Phase Stability and Defect Physics in CsPbI2Br Perovskite. J. Mater. Chem. A 2019, 7, 20201–20207. [Google Scholar] [CrossRef]
- Breniaux, E.; Dufour, P.; Guillemet-Fritsch, S.; Tenailleau, C. Unraveling All-Inorganic CsPbI3 and CsPbI2Br Perovskite Thin Films Formation—Black Phase Stabilization by Cs2PbCl2I2 Addition and Flash-Annealing. Eur. J. Inorg. Chem. 2021, 2021, 3059–3073. [Google Scholar] [CrossRef]
- Trots, D.M.; Myagkota, S.V. High-Temperature Structural Evolution of Caesium and Rubidium Triiodoplumbates. J. Phys. Chem. Solids 2008, 69, 2520–2526. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Y.; Ding, C.; Kobayashi, S.; Izuishi, T.; Nakazawa, N.; Toyoda, T.; Ohta, T.; Hayase, S.; Minemoto, T.; et al. Highly Luminescent Phase-Stable CsPbI3 Perovskite Quantum Dots Achieving Near 100% Absolute Photoluminescence Quantum Yield. ACS Nano 2017, 11, 10373–10383. [Google Scholar] [CrossRef]
- Nations, S.; Gutsev, L.; Ramachandran, B.; Aldoshin, S.; Duan, Y.; Wang, S. First-Principles Study of the Defect-Activity and Optical Properties of FAPbCl3. Mater. Adv. 2022, 3, 3897–3905. [Google Scholar] [CrossRef]
- Datta, K.; van Gorkom, B.T.; Chen, Z.; Dyson, M.J.; van der Pol, T.P.A.; Meskers, S.C.J.; Tao, S.; Bobbert, P.A.; Wienk, M.M.; Janssen, R.A.J. Effect of Light-Induced Halide Segregation on the Performance of Mixed-Halide Perovskite Solar Cells. ACS Appl. Energy Mater. 2021, 4, 6650–6658. [Google Scholar] [CrossRef]
- Kuznetsov, M.K.; Emelianov, N.A.; Korchagin, D.V.; Shilov, G.V.; Aldoshin, S.M.; Troshin, P.A.; Frolova, L.A. Enhanced Photostability of CsPbI2Br-Based Perovskite Solar Cells through Suppression of Phase Segregation Using a Zwitterionic Additive. Sustain. Energy Fuels 2022, 6, 3536–3541. [Google Scholar] [CrossRef]
- Ozturk, T.; Akman, E.; Shalan, A.E.; Akin, S. Composition Engineering of Operationally Stable CsPbI2Br Perovskite Solar Cells with a Record Efficiency over 17%. Nano Energy 2021, 87, 106157. [Google Scholar] [CrossRef]
- Dong, Y.; Guo, Y.; Wang, M.; Zhu, R.; Ma, D.; Jia, Y. Designing Multifunctional Donor–Acceptor-Type Molecules to Passivate Surface Defects Efficiently and Enhance Charge Transfer of CsPbI2Br Perovskite for High Power Conversion Efficiency. Inorg. Chem. 2022, 61, 9469–9479. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab Initio Molecular-Dynamics Simulation of the Liquid-Metalamorphous- Semiconductor Transition in Germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Jong, U.G.; Yu, C.J.; Kim, Y.S.; Kye, Y.H.; Kim, C.H. First-Principles Study on the Material Properties of the Inorganic Perovskite Rb1-XCsxPbI3 for Solar Cell Applications. Phys. Rev. B 2018, 98, 32–39. [Google Scholar] [CrossRef] [Green Version]
- Płowaś-Korus, I.; Kaczkowski, J. Comparative Density Functional Studies of BiMO3 Polymorphs (M = Al, Ga, In) Based on LDA, GGA, and Meta-GGA Functionals. New J. Chem. 2022, 46, 15381–15391. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid Functionals Based on a Screened Coulomb Potential. J. Chem. Phys. 2003, 118, 8207. [Google Scholar] [CrossRef] [Green Version]
- Xue, H.; Brocks, G.; Tao, S. First-Principles Calculations of Defects in Metal Halide Perovskites: A Performance Comparison of Density Functionals. Phys. Rev. Mater. 2021, 5, 125408. [Google Scholar] [CrossRef]
- Curtarolo, S.; Setyawan, W.; Hart, G.L.W.; Jahnatek, M.; Chepulskii, R.V.; Taylor, R.H.; Wang, S.; Xue, J.; Yang, K.; Levy, O.; et al. AFLOW: An Automatic Framework for High-Throughput Materials Discovery. Comput. Mater. Sci. 2012, 58, 218–226. [Google Scholar] [CrossRef] [Green Version]
- Meggiolaro, D.; De Angelis, F. First-Principles Modeling of Defects in Lead Halide Perovskites: Best Practices and Open Issues. ACS Energy Lett. 2018, 3, 2206–2222. [Google Scholar] [CrossRef] [Green Version]
- Govinda, S.; Kore, B.P.; Swain, D.; Hossain, A.; De, C.; Guru Row, T.N.; Sarma, D.D. Critical Comparison of FAPbX3 and MAPbX3 (X = Br and Cl): How Do They Differ? J. Phys. Chem. C 2018, 122, 13758–13766. [Google Scholar] [CrossRef]
- Zeng, Q.; Zhang, X.; Liu, C.; Feng, T.; Chen, Z.; Zhang, W.; Zheng, W.; Zhang, H.; Yang, B. Inorganic CsPbI2Br Perovskite Solar Cells: The Progress and Perspective. Sol. RRL 2019, 3, 1800239. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Surrente, A.; Galkowski, K.; Miyata, A.; Portugall, O.; Sutton, R.J.; Haghighirad, A.A.; Snaith, H.J.; Maude, D.K.; Plochocka, P.; et al. Impact of the Halide Cage on the Electronic Properties of Fully Inorganic Cesium Lead Halide Perovskites. ACS Energy Lett. 2017, 2, 1621–1627. [Google Scholar] [CrossRef]
- Marronnier, A.; Roma, G.; Boyer-Richard, S.; Pedesseau, L.; Jancu, J.M.; Bonnassieux, Y.; Katan, C.; Stoumpos, C.C.; Kanatzidis, M.G.; Even, J. Anharmonicity and Disorder in the Black Phases of Cesium Lead Iodide Used for Stable Inorganic Perovskite Solar Cells. ACS Nano 2018, 12, 3477–3486. [Google Scholar] [CrossRef]
- Yu, C.J.; Ko, U.H.; Hwang, S.G.; Kim, Y.S.; Jong, U.G.; Kye, Y.H.; Ri, C.H. First-Principles Study on Material Properties and Stability of Inorganic Halide Perovskite Solid Solutions CsPb(I1-XBrx)3. Phys. Rev. Mater. 2020, 4, 045402. [Google Scholar] [CrossRef]
- Pols, M.; Vicent-Luna, J.M.; Filot, I.; Van Duin, A.C.T.; Tao, S. Atomistic Insights into the Degradation of Inorganic Halide Perovskite CsPbI3: A Reactive Force Field Molecular Dynamics Study. J. Phys. Chem. Lett. 2021, 12, 5519–5525. [Google Scholar] [CrossRef]
- Xu, P. All-Inorganic Perovskite CsPbI2Br as a Promising Photovoltaic Absorber: A First-Principles Study. J. Chem. Sci. 2020, 132, 74. [Google Scholar] [CrossRef]
- Liu, D.; Zha, W.; Guo, Y.; Sa, R. Insight into the Improved Phase Stability of CsPbI3 from First-Principles Calculations. ACS Omega 2020, 5, 893–896. [Google Scholar] [CrossRef] [Green Version]
- Vogel, D.J.; Inerbaev, T.M.; Kilin, D.S. Role of Lead Vacancies for Optoelectronic Properties of Lead-Halide Perovskites. J. Phys. Chem. C 2018, 122, 5216–5226. [Google Scholar] [CrossRef]
- Nations, S.; Jia, T.; Wang, S.; Duan, Y. Electronic and Optical Properties of Orthorhombic (CH3NH3)BX3(B = Sn, Pb; X = F, Cl, Br, I) Perovskites: A First-Principles Investigation. RSC Adv. 2021, 11, 22264–22272. [Google Scholar] [CrossRef]
- Ghaithan, H.M.; Alahmed, Z.A.; Qaid, S.M.H.; Aldwayyan, A.S. Structural, Electronic, and Optical Properties of Cspb(Br1−xclx)3 Perovskite: First-Principles Study with Pbe–Gga and Mbj–Gga Methods. Materials 2020, 13, 4944. [Google Scholar] [CrossRef]
- Ezzeldien, M.; Al-Qaisi, S.; Alrowaili, Z.A.; Alzaid, M.; Maskar, E.; Es-Smairi, A.; Vu, T.V.; Rai, D.P. Electronic and Optical Properties of Bulk and Surface of CsPbBr3 Inorganic Halide Perovskite a First Principles DFT 1/2 Approach. Sci. Rep. 2021, 11, 20622. [Google Scholar] [CrossRef]
- Soni, K.; Lakshmi, N.; Jain, V.; Chandra, A.R.; Jain, R. A Comparative Study of Structural, Electronic and Optical Properties of Cubic CsPbI 3: Bulk and Surface. Bull. Mater. Sci. 2019, 42, 275. [Google Scholar] [CrossRef]
- Brivio, F.; Caetano, C.; Walsh, A. Thermodynamic Origin of Photoinstability in the CH3NH3Pb(I1-XBrx)3 Hybrid Halide Perovskite Alloy. J. Phys. Chem. Lett. 2016, 7, 1083–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.H.; Yin, W.J.; Park, J.S.; Wei, S.H. Self-Regulation of Charged Defect Compensation and Formation Energy Pinning in Semiconductors. Sci. Rep. 2015, 5, 16977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, S.; Schmidt, I.; Brocks, G.; Jiang, J.; Tranca, I.; Meerholz, K.; Olthof, S. Absolute Energy Level Positions in Tin- and Lead-Based Halide Perovskites. Nat. Commun. 2019, 10, 2560. [Google Scholar] [CrossRef] [Green Version]
- Mariotti, S.; Hutter, O.S.; Phillips, L.J.; Yates, P.J.; Kundu, B.; Durose, K. Stability and Performance of CsPbI2Br Thin Films and Solar Cell Devices. ACS Appl. Mater. Interfaces 2018, 10, 3750–3760. [Google Scholar] [CrossRef] [PubMed]
- Beal, R.E.; Slotcavage, D.J.; Leijtens, T.; Bowring, A.R.; Belisle, R.A.; Nguyen, W.H.; Burkhard, G.F.; Hoke, E.T.; McGehee, M.D. Cesium Lead Halide Perovskites with Improved Stability for Tandem Solar Cells. J. Phys. Chem. Lett. 2016, 7, 746–751. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, T.; Xu, F.; Li, Y.; Zhao, Y. A Facile Low Temperature Fabrication of High Performance CsPbI2Br All-Inorganic Perovskite Solar Cells. Sol. RRL 2018, 2, 1700180. [Google Scholar] [CrossRef]
- Frolova, L.A.; Chang, Q.; Luchkin, S.Y.; Zhao, D.; Akbulatov, A.F.; Dremova, N.N.; Ivanov, A.V.; Chia, E.E.M.; Stevenson, K.J.; Troshin, P.A. Efficient and Stable All-Inorganic Perovskite Solar Cells Based on Nonstoichiometric CsxPbI2Brx (x > 1) Alloys. J. Mater. Chem. C 2019, 7, 5314–5323. [Google Scholar] [CrossRef]
- Zheng, K.; Ge, J.; Liu, C.; Lou, Q.; Chen, X.; Meng, Y.; Yin, X.; Bu, S.; Liu, C.; Ge, Z. Improved Phase Stability of CsPbI2Br Perovskite by Released Microstrain toward Highly Efficient and Stable Solar Cells. InfoMat 2021, 3, 1431–1444. [Google Scholar] [CrossRef]







| α-CsPbBr3 | 22.31 | 4.67 | 0.99997 |
| α-CsPbI2Br P4/mmm | 19.20 | 4.85 | 1.00000 |
| α-CsPbI3 | 18.04 | 4.67 | 0.99996 |
| Eox * | Ered * EF = Eg (eV) | Eredox * @ EF = Epin (eV) | Epin * (eV) | EVBM (eV) | Eg (eV) | |
|---|---|---|---|---|---|---|
| -CsPbI3 | 0.25 (0.34) | 0.08 (0.17) | 0.85 (0.94) | 0.60 | 1.765 | 1.362 |
| P4/mmm | 0.25 (0.36) | 0.11 (0.22) | 0.85 (0.99) | 0.63 | 1.550 | 1.402 |
| C1 | 0.26 (0.36) | 0.08 (0.19) | 0.89 (0.93) | 0.57 | 1.657 | 1.311 |
| Fmmm (Equatorial) | 0.17 (0.28) | 0.16 (0.27) | 0.93 (1.04) | 0.75 | 1.440 | 1.526 |
| I4/mcm (Apical) | 0.24 (0.35) | −0.01 (0.09) | 0.85 (0.96) | 0.61 | 1.475 | 1.539 |
| -CsPbBr3 | 0.83 (0.93) | −0.02(0.06) | 1.01 (1.11) | 0.17 | 1.212 | 1.606 |
| Redox Reaction: | (eV) | Redox Reaction | (eV) | |
|---|---|---|---|---|
| 2.84 1.08 | 1.25 0.63 | |||
| α-CsPbI2Br P4/mmm | 2.92 0.97 | 1.21 0.46 | ||
| 1.65 −0.18 | −0.02 −0.01 | |||
| 2.98 1.07 | 1.31 0.56 | |||
| 1.59 −0.27 | −0.12 −0.10 | |||
| α-CsPbI2Br C1 | 1.73 −0.26 | 0.39 0.07 | ||
| 1.93 0.69 | 0.80 0.96 | |||
| 2.29 0.51 | 1.16 0.84 | |||
| 1.37 −0.08 | 0.03 0.19 | |||
| α -CsPbI2Br P4/mmm (rc) | 0.45 −2.06 | −1.24 −1.76 | ||
| 0.54 −1.34 | −1.19 −1.03 | |||
| 1.22 −1.34 | −0.51 −1.03 | |||
| −0.23 −2.07 | −1.92 −1.76 | |||
| β-CsPbI2Br Fmmm(Equatorial) | 3.57 1.85 | 1.73 0.75 | ||
| 3.40 1.51 | 1.58 1.68 | |||
| 4.03 2.33 | 2.21 1.22 | |||
| 2.94 1.04 | 1.11 1.21 | |||
| β-CsPbI2Br I4/mcm(Apical) | 1.25 −1.37 | −1.29 −1.16 | ||
| 0.37 −1.60 | −1.43 −1.34 | |||
| 1.31 −0.58 | −0.49 −0.37 | |||
| 0.31 −2.40 | −2.22 −2.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gutsev, L.G.; Nations, S.; Ramachandran, B.R.; Gutsev, G.L.; Wang, S.; Aldoshin, S.; Duan, Y. Redox Chemistry of the Subphases of α-CsPbI2Br and β-CsPbI2Br: Theory Reveals New Potential for Photostability. Nanomaterials 2023, 13, 276. https://doi.org/10.3390/nano13020276
Gutsev LG, Nations S, Ramachandran BR, Gutsev GL, Wang S, Aldoshin S, Duan Y. Redox Chemistry of the Subphases of α-CsPbI2Br and β-CsPbI2Br: Theory Reveals New Potential for Photostability. Nanomaterials. 2023; 13(2):276. https://doi.org/10.3390/nano13020276
Chicago/Turabian StyleGutsev, Lavrenty Gennady, Sean Nations, Bala Ramu Ramachandran, Gennady Lavrenty Gutsev, Shengnian Wang, Sergei Aldoshin, and Yuhua Duan. 2023. "Redox Chemistry of the Subphases of α-CsPbI2Br and β-CsPbI2Br: Theory Reveals New Potential for Photostability" Nanomaterials 13, no. 2: 276. https://doi.org/10.3390/nano13020276