
Fabrication of Glutaraldehyde Vapor Treated PVA/SA/GO/ZnO Electrospun Nanofibers with High Liquid Absorbability for Antimicrobial of Staphylococcus aureus
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
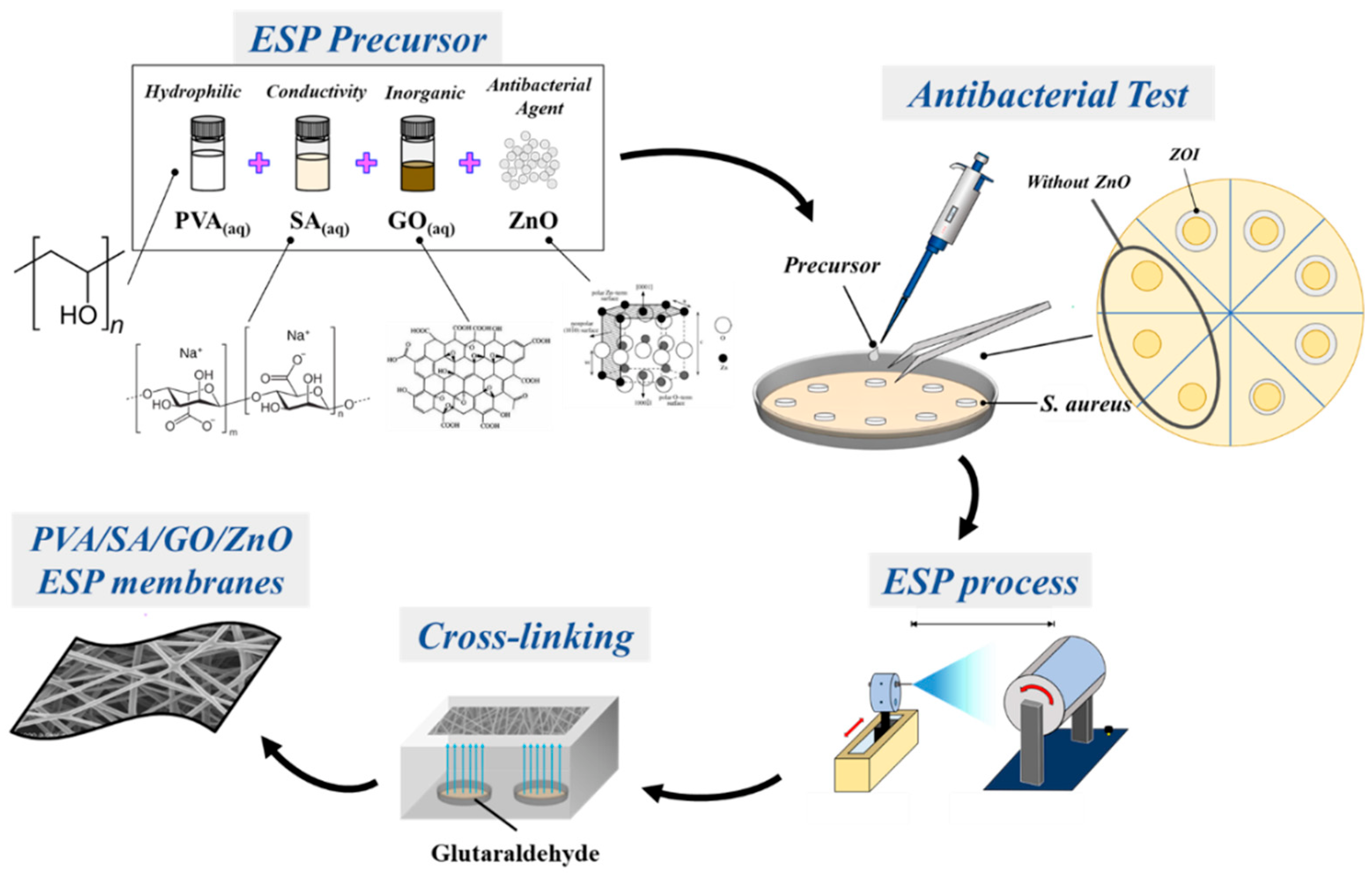
2. Materials and Methods
2.1. Materials
2.2. Synthesis of PVA/SA Nanofiber
2.3. Synthesis of Graphene Oxide (GO)
2.4. Synthesis of PVA/SA/GO Nanofiber
2.5. Synthesis of ZnO Powder by Precipitation Method
2.6. PVA/SA/GO/ZnO Antibacterial Solution Test
2.7. PVA/SA/GO/ZnO Vapor Crosslinking with Glutaraldehyde (GA)

2.8. Swelling Percentage and Water Retention
2.9. Characterization
3. Results and Discussion
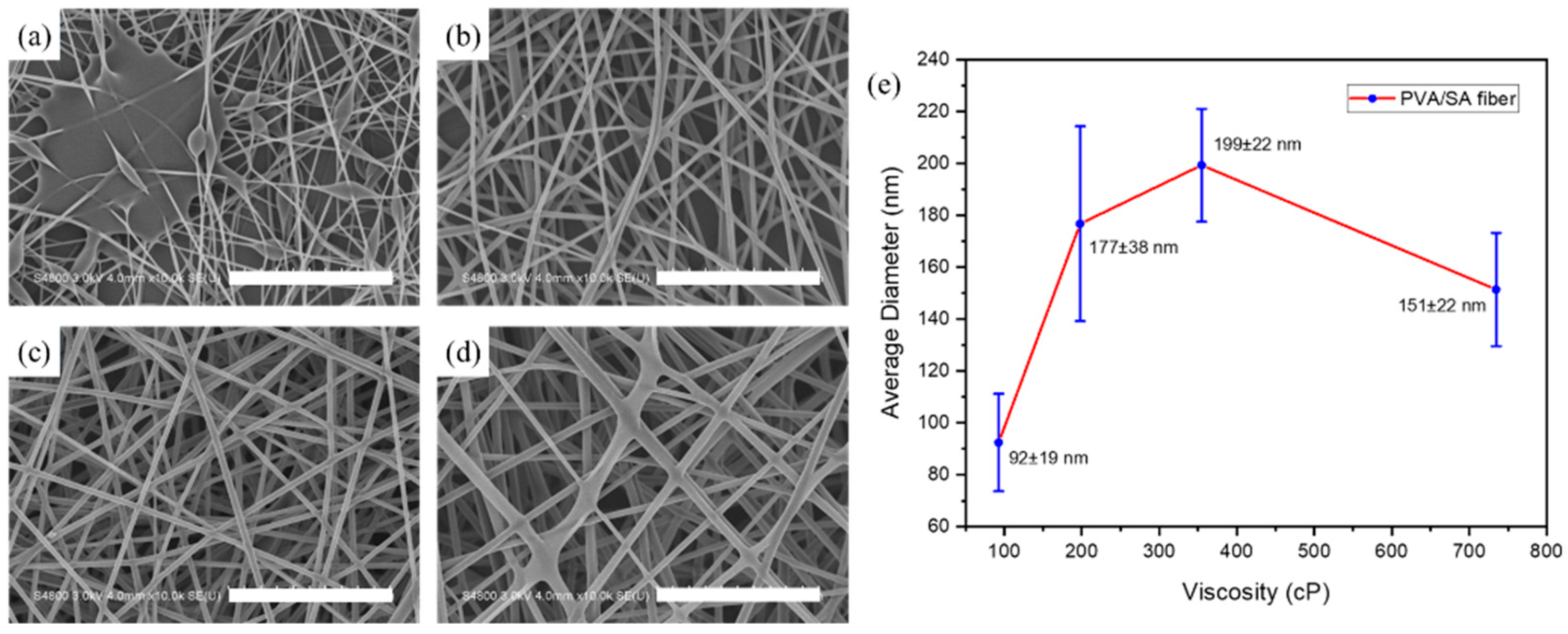
3.1. PVA/SA Electrospun Nanofibers
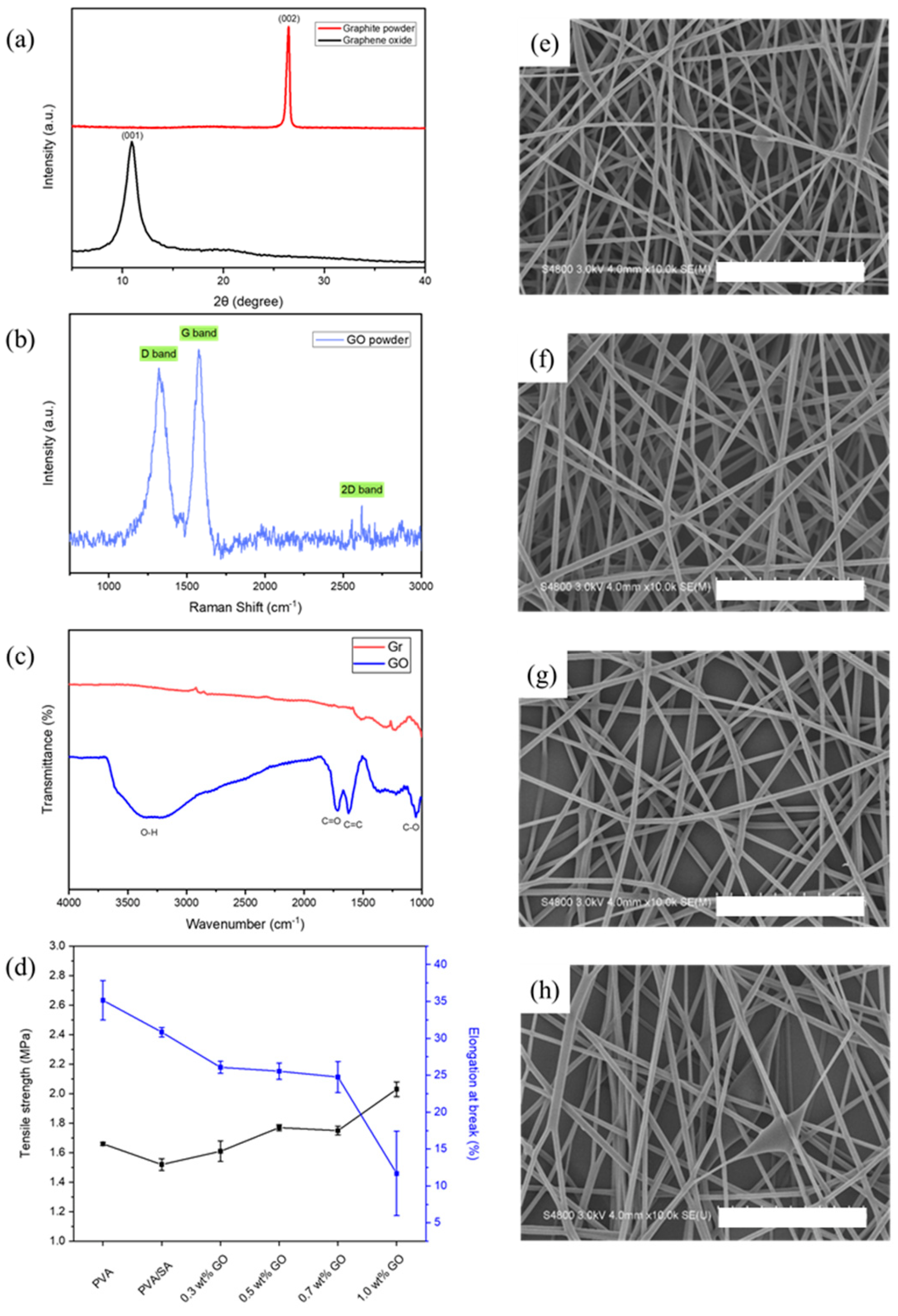
3.2. PVA/SA/GO Electrospun Nanofibers
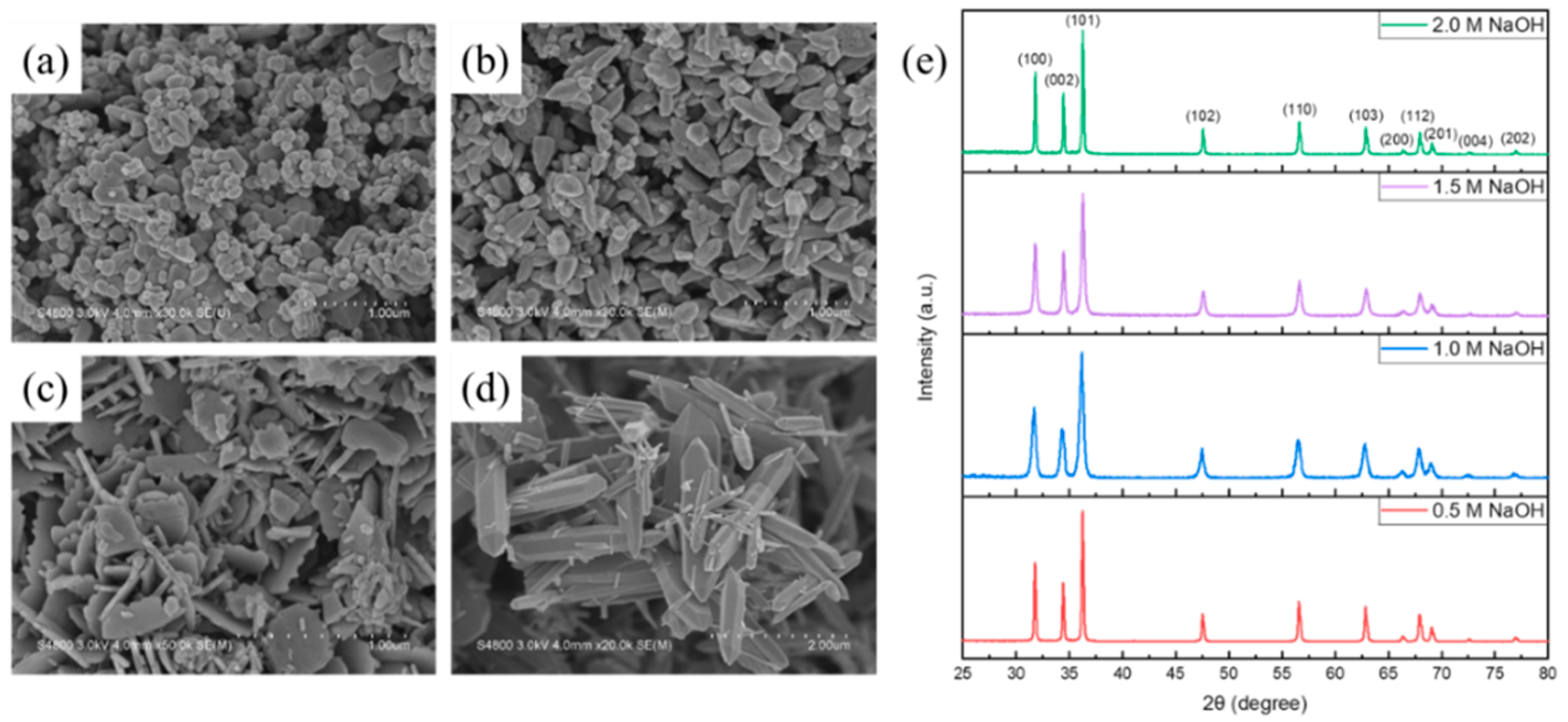
3.3. ZnO Nanomaterials
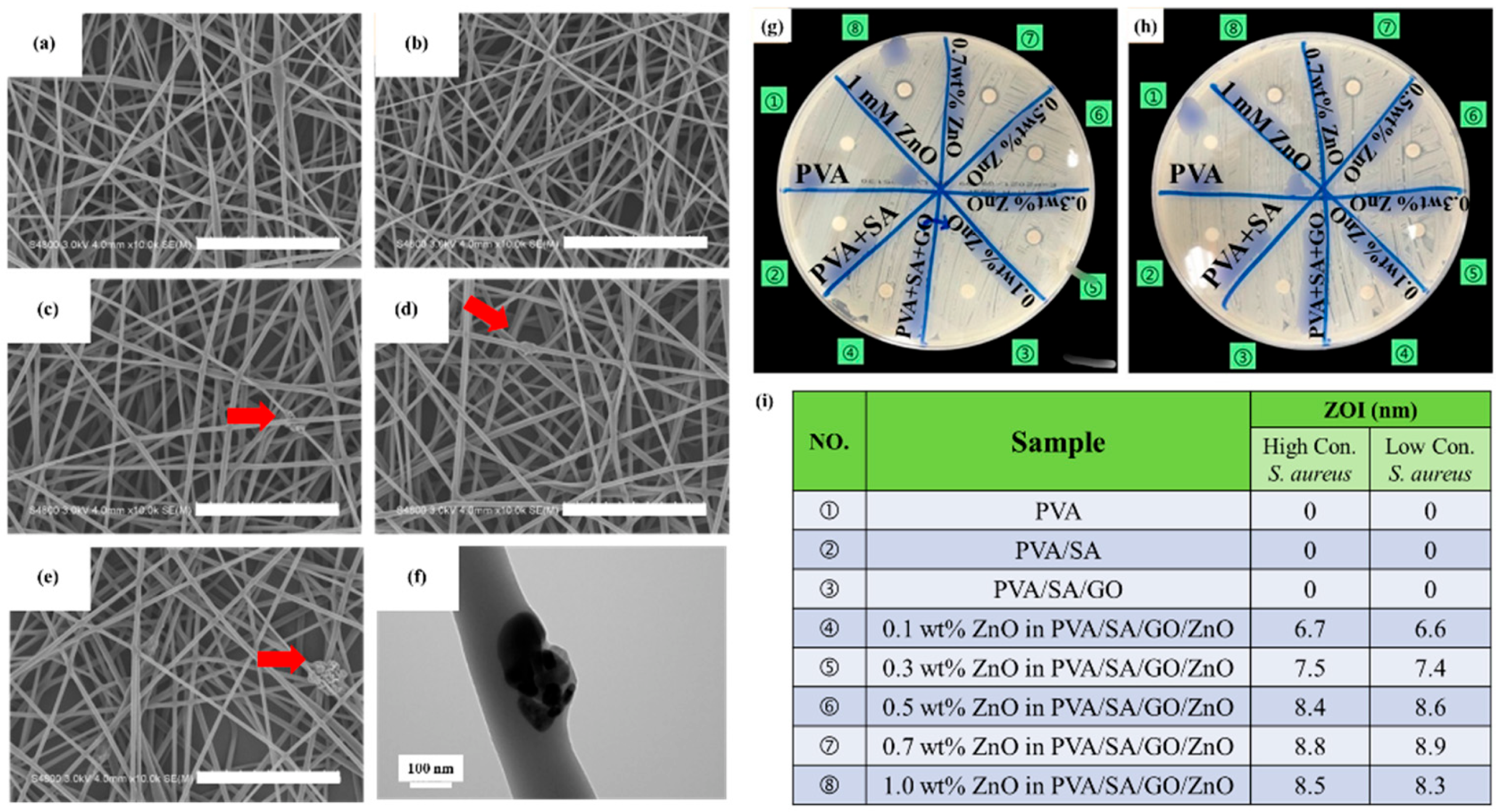
3.4. PVA/SA/GO/ZnO Electrospun Nanofibers and the Antibacterial Test
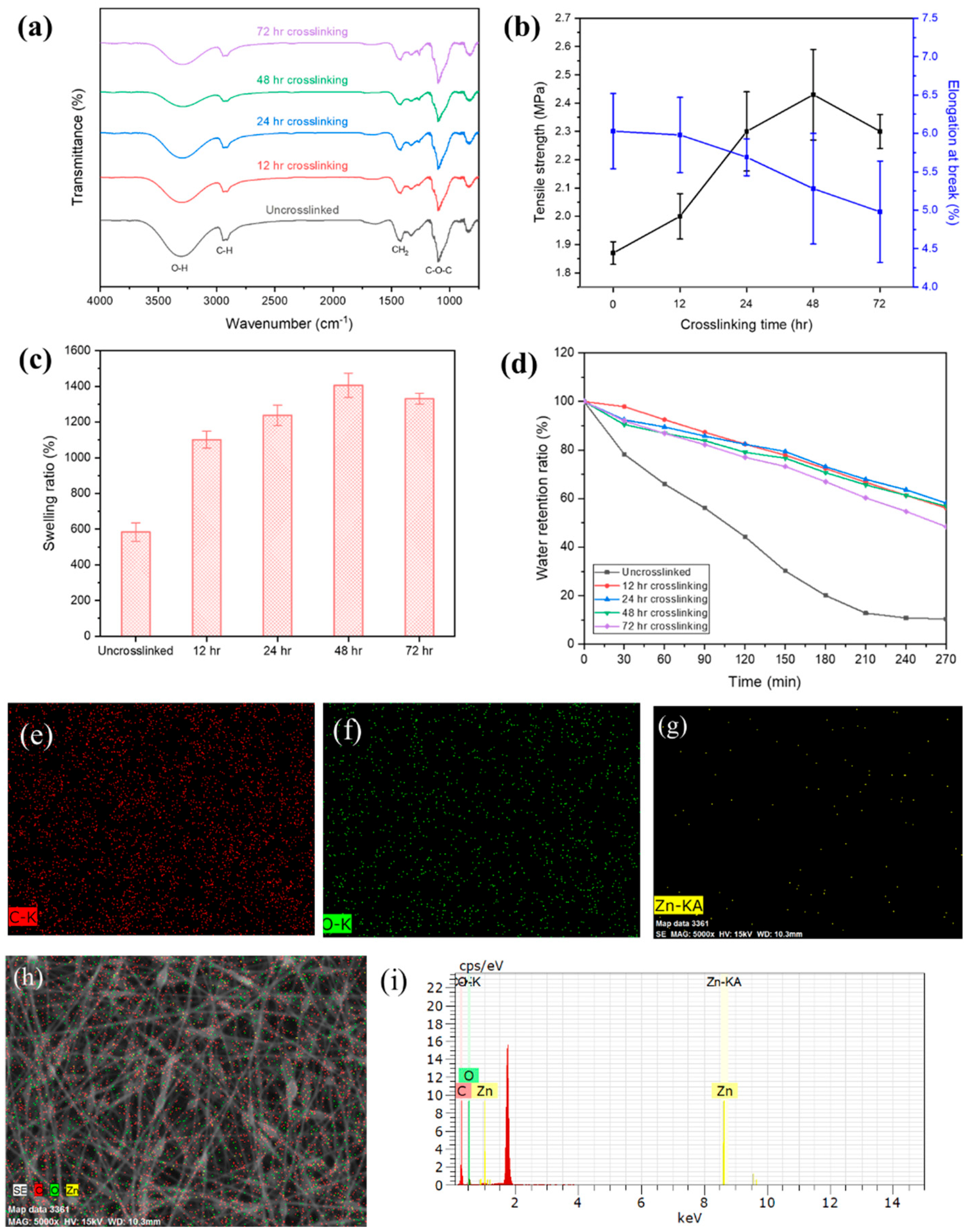
3.5. Glutaraldehyde Vapor Treated PVA/SA/GO/ZnO Nanofibers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yu, B.; He, C.; Wang, W.; Ren, Y.; Yang, J.; Guo, S.; Zheng, Y.; Shi, X. Asymmetric wettable composite wound dressing prepared by electrospinning with bioinspired micropatterning enhances diabetic wound healing. ACS Appl. Bio Mater. 2020, 3, 5383–5394. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; He, J.; Guo, B. Functional hydrogels as wound dressing to enhance wound healing. ACS Nano 2021, 15, 12687–12722. [Google Scholar] [CrossRef]
- Luraghi, A.; Peri, F.; Moroni, L. Electrospinning for drug delivery applications: A review. J. Control. Release 2021, 334, 463–484. [Google Scholar] [CrossRef]
- Cacciotti, I.; Pallotto, F.; Scognamiglio, V.; Moscone, D.; Arduini, F. Reusable optical multi-plate sensing system for pesticide detection by using electrospun membranes as smart support for acetylcholinesterase immobilisation. Mater. Sci. Eng. C 2020, 111, 110744. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Shi, S.; Hu, J. Evaluation of high fluoride contaminated fractured rock aquifer in South Africa–Geochemical and chemometric approaches. Nano Today 2023, 48, 101723. [Google Scholar]
- Kabir, S.; Van Cleve, T.; Khandavalli, S.; Medina, S.; Pylypenko, S.; Mauger, S.; Ulsh, M.; Neyerlin, K.C. Toward Optimizing Electrospun Nanofiber Fuel Cell Catalyst Layers: Microstructure and Pt Accessibility. ACS Appl. Energy Mater. 2021, 4, 3341–3351. [Google Scholar] [CrossRef]
- Lee, C.H.; Tiwari, B.; Zhang, D.; Yap, Y.K. Water purification: Oil–water separation by nanotechnology and environmental concerns. Environ. Sci. Nano 2017, 4, 514–525. [Google Scholar] [CrossRef]
- Yang, X.; Pu, Y.; Li, S.; Liu, X.; Wang, Z.; Yuan, D.; Ning, X. Electrospun polymer composite membrane with superior thermal stability and excellent chemical resistance for high-efficiency PM2. 5 capture. ACS Appl. Mater. Interfaces 2019, 11, 43188–43199. [Google Scholar] [CrossRef]
- Reneker, D.H.; Chun, I. Nanometre diameter fibres of polymer, produced by electrospinning. Nanotechnology 1996, 7, 216. [Google Scholar] [CrossRef] [Green Version]
- Zong, X.; Kim, K.; Fang, D.; Ran, S.; Hsiao, B.S.; Chu, B. Structure and process relationship of electrospun bioabsorbable nanofiber membranes. Polymer 2002, 43, 4403–4412. [Google Scholar] [CrossRef]
- Kade, J.C.; Dalton, P. Polymers for Melt Electrowriting. Adv. Healthc. Mater. 2020, 10, 2001232. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.J.; Pérez-Nava, A.; Ali, S.C.; González-Campos, J.B.; Holloway, J.L.; Cosgriff-Hernandez, E.M. Comparative analysis of fiber alignmentmethods in electrospinning. Matter 2021, 4, 821–844. [Google Scholar] [CrossRef] [PubMed]
- Yen, S.-C.; Liu, Z.-W.; Juang, R.-S.; Sahoo, S.; Huang, C.-H.; Chen, P.; Hsiao, Y.-S.; Fang, J.-T. Carbon nanotube/conducting polymer hybrid nanofibers as novel organic bioelectronic interfaces for efficient removal of protein-bound uremic toxins. ACS Appl. Mater. Interfaces 2019, 11, 43843–43856. [Google Scholar] [CrossRef] [PubMed]
- Pan, F.; Amarjargal, A.; Altenried, S.; Liu, M.; Zuber, F.; Zeng, Z.; Rossi, R.M.; Maniura-Weber, K.; Ren, Q. Bioresponsive hybrid nanofibers enable controlled drug delivery through glass transition switching at physiological temperature. ACS Appl. Bio Mater. 2021, 4, 4271–4279. [Google Scholar] [CrossRef]
- Ramírez-Agudelo, R.; Scheuermann, K.; Gala-García, A.; Monteiro, A.P.F.; Pinzón-García, A.D.; Cortés, M.E.; Sinisterra, R.D. Hybrid nanofibers based on poly-caprolactone/gelatin/hydroxyapatite nanoparticles-loaded Doxycycline: Effective anti-tumoral and antibacterial activity. Mater. Sci. Eng. C 2018, 83, 25–34. [Google Scholar] [CrossRef]
- Wang, X.; Fang, D.; Yoon, K.; Hsiao, B.S.; Chu, B. High performance ultrafiltration composite membranes based on poly (vinyl alcohol) hydrogel coating on crosslinked nanofibrous poly (vinyl alcohol) scaffold. J. Membr. Sci. 2006, 278, 261–268. [Google Scholar] [CrossRef]
- Liu, H.; Gough, C.; Deng, Q.; Gu, Z.; Wang, F.; Hu, X. Recent Advances in Electrospun SustainableComposites for Biomedical, Environmental, Energy,and Packaging Applications. Int. J. Mol. Sci. 2020, 21, 4019. [Google Scholar]
- Adeli-Sardou, M.; Yaghoobi, M.M.; Torkzadeh-Mahani, M.; Dodel, M. Controlled release of lawsone from polycaprolactone/gelatin electrospun nano fibers for skin tissue regeneration. Int. J. Biol. Macromol. 2019, 124, 478–491. [Google Scholar] [CrossRef]
- Asadi, N.; Alizadeh, E.; Salehi, R.; Khalandi, B.; Davaran, S.; Akbarzadeh, A. Nanocomposite hydrogels for cartilage tissue engineering: A review. Artif. Cells Nanomed. Biotechnol. 2018, 46, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Zare, P.; Aleemardani, M.; Seifalian, A.; Bagher, Z.; Seifalian, A.M. Graphene oxide: Opportunities and challenges in biomedicine. Nanomaterials 2021, 11, 1083. [Google Scholar] [CrossRef]
- Rijal, N.P.; Adhikari, U.; Khanal, S.; Pai, D.; Sankar, J.; Bhattarai, N. Magnesium oxide-poly (ε-caprolactone)-chitosan-based composite nanofiber for tissue engineering applications. Mater. Sci. Eng. B 2018, 228, 18–27. [Google Scholar] [CrossRef]
- Sumitha, M.; Shalumon, K.; Sreeja, V.; Jayakumar, R.; Nair, S.V.; Menon, D. Biocompatible and antibacterial nanofibrous poly (ϵ-caprolactone)-nanosilver composite scaffolds for tissue engineering applications. J. Macromol. Sci. Part A 2012, 49, 131–138. [Google Scholar] [CrossRef]
- Lee, Y.J.; Shin, D.S.; Kwon, O.W.; Park, W.H.; Choi, H.G.; Lee, Y.R.; Han, S.S.; Noh, S.K.; Lyoo, W.S. Preparation of atactic poly (vinyl alcohol)/sodium alginate blend nanowebs by electrospinning. J. Appl. Polym. Sci. 2007, 106, 1337–1342. [Google Scholar] [CrossRef]
- Zhu, B.; Li, W.; Chi, N.; Lewis, R.V.; Osamor, J.; Wang, R. Optimization of glutaraldehyde vapor treatment for electrospun collagen/silk tissue engineering scaffolds. ACS Omega 2017, 2, 2439–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulupi, M.; Haryadi, H. Synthesis and characterization of electrospinning PVA nanofiber-crosslinked by glutaraldehyde. Mater. Today Proc. 2019, 13, 199–204. [Google Scholar] [CrossRef]
- You, X.L.; He, J.X.; Nan, N.; Sun, X.Q.; Qi, K.; Zhou, Y.M.; Shao, W.L.; Liu, F.; Cui, S.Z. Stretchable capacitive fabric electronic skin woven by electrospun nanofiber coated yarns for detecting tactile and multimodal mechanical stimuli. J. Mater. Chem. C 2018, 6, 12981–12991. [Google Scholar] [CrossRef]
- Zaaba, N.I.; Foo, K.L.; Hashim, U.; Tan, S.J.; Liu, W.W.; Voon, C.H. Synthesis of Graphene Oxide using Modified Hummers Method: Solvent Influence. Procedia Eng. 2017, 184, 469–477. [Google Scholar] [CrossRef]
- Gatou, M.A.; Lagopati, N.; Vagena, I.A.; Gazouli, M.; Pavlatou, E.A. ZnO Nanoparticles from Different Precursors and Their Photocatalytic Potential for Biomedical Use. Nanomaterials 2023, 13, 122. [Google Scholar] [CrossRef]
- Aladejana, J.T.; Wu, Z.; Li, D.; Guelifack, K.; Wei, W.; Wang, X.; Xie, Y. Facile Approach for Glutaraldehyde Cross-Linking of PVA/Aluminophosphate Adhesives for Wood-Based Panels. ACS Sustain. Chem. Eng. 2019, 7, 18524–18533. [Google Scholar] [CrossRef]
- Premanathan, M.; Karthikeyan, K.; Jeyasubramanian, K.; Manivannan, G. Selective toxicity of ZnO nanoparticles toward Gram-positive bacteria and cancer cells by apoptosis through lipid peroxidation. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 184–192. [Google Scholar] [CrossRef]
- Moezzi, A.; Cortie, M.; McDonagh, A. Aqueous pathways for the formation of zinc oxide nanoparticles. Dalton Trans. 2011, 40, 4871–4878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palanikumar, L.; Ramasamy, S.N.; Balachandran, C. Size-dependent antimicrobial response of zinc oxide nanoparticles. IET Nanobiotechnol. 2014, 8, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Mirhosseini, F.; Amiri, M.; Daneshkazemi, A.; Zandi, H.; Javadi, Z.S. Antimicrobial effect of different sizes of nano zinc oxide on oral microorganisms. Front. Dent. 2019, 16, 105. [Google Scholar] [CrossRef] [PubMed]
- Bursali, E.A.; Coskun, S.; Kizil, M.; Yurdakoc, M. Synthesis, characterization and in vitro antimicrobial activities of boron/starch/polyvinyl alcohol hydrogels. Carbohydr. Polym. 2011, 83, 1377–1383. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chien, Y.-H.; Ho, M.-T.; Feng, C.-H.; Yen, J.-H.; Chang, Y.-C.; Lai, C.-S.; Louh, R.-F. Fabrication of Glutaraldehyde Vapor Treated PVA/SA/GO/ZnO Electrospun Nanofibers with High Liquid Absorbability for Antimicrobial of Staphylococcus aureus. Nanomaterials 2023, 13, 932. https://doi.org/10.3390/nano13050932
Chien Y-H, Ho M-T, Feng C-H, Yen J-H, Chang Y-C, Lai C-S, Louh R-F. Fabrication of Glutaraldehyde Vapor Treated PVA/SA/GO/ZnO Electrospun Nanofibers with High Liquid Absorbability for Antimicrobial of Staphylococcus aureus. Nanomaterials. 2023; 13(5):932. https://doi.org/10.3390/nano13050932
Chicago/Turabian StyleChien, Yi-Hsin, Meng-Tzu Ho, Chin-Hsign Feng, Jung-Hsign Yen, Yi-Chan Chang, Chih-Sheng Lai, and Rong-Fuh Louh. 2023. "Fabrication of Glutaraldehyde Vapor Treated PVA/SA/GO/ZnO Electrospun Nanofibers with High Liquid Absorbability for Antimicrobial of Staphylococcus aureus" Nanomaterials 13, no. 5: 932. https://doi.org/10.3390/nano13050932