

Aluminum-Doping Effects on the Electronic States of Graphene Nanoflake: Diffusion and Hydrogen Storage Mechanism

Abstract
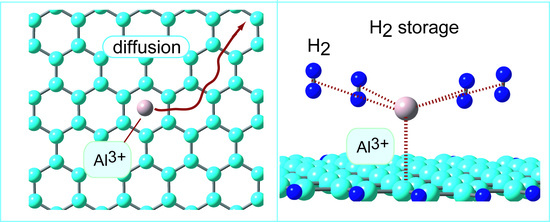
:
1. Introduction
2. Method of Calculation
3. Results
3.1. Structure of GR-Al
3.2. Effects of Al Doping on Excitation Energies of Graphene
3.3. Diffusion of Al on Graphene Surface
3.4. Structures of Molecular Hydrogen Bound to GR-Al
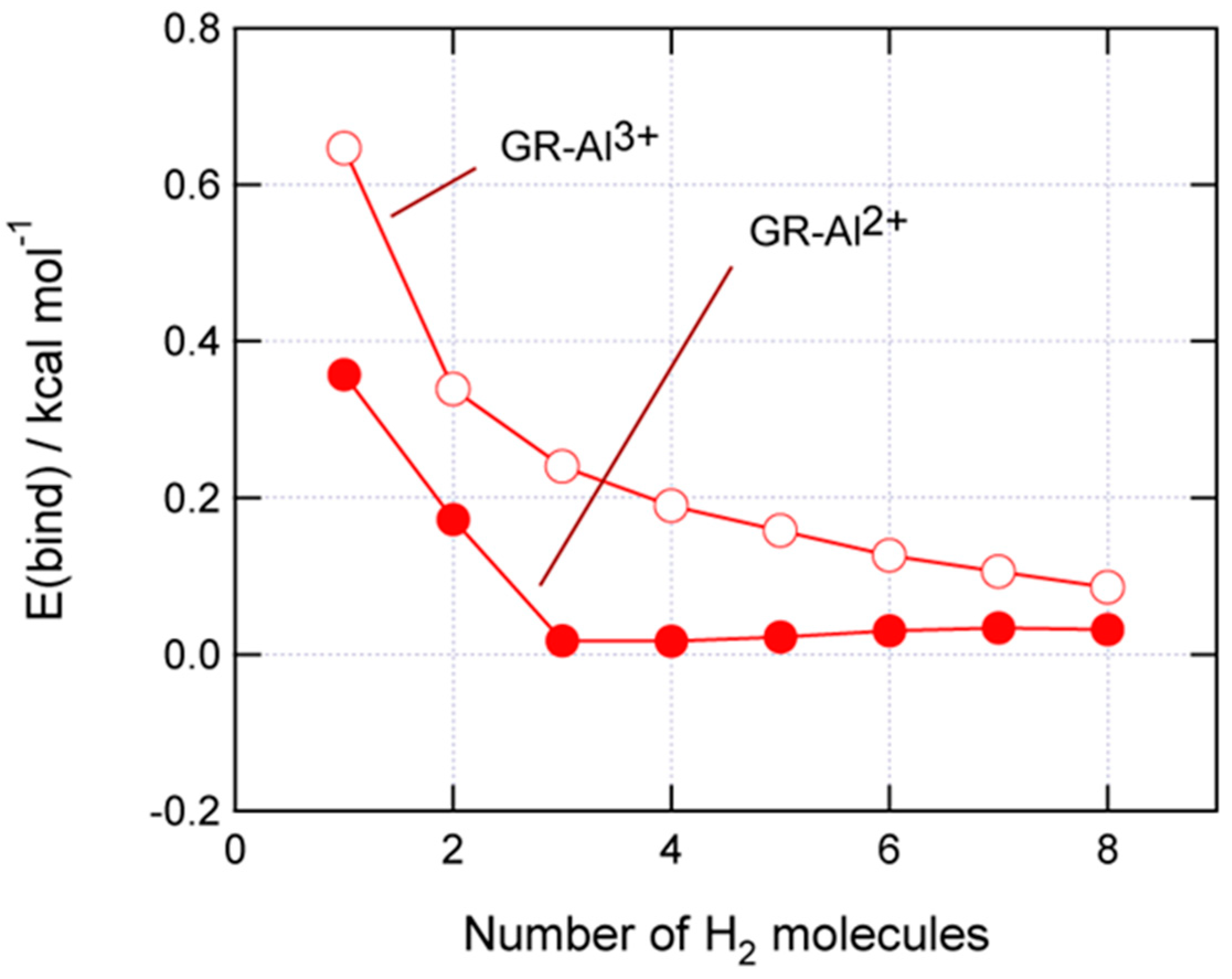
3.5. H2-Binding Energies to GR-Al
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liu, H.; Wang, Z.; Pan, B.; Li, M.; Huang, S.; Lee, J.H.; Kim, N.H. Carbon felt/nitrogen-doped graphene/paraffin wax 3D skeleton regulated tribological performances of the MCPA6 composites: A novel strategy of oil embedding and transporting. Tribol. Int. 2023, 184, 108440. [Google Scholar] [CrossRef]
- Devynck, M.; Rostirolla, B.; Watson, C.P.; Taylor, D.M. Photo-Response of a P3HT:PCBM Blend in Metal-Insulator-Semiconductor Capacitors. Appl. Phys. Lett. 2014, 105, 183301. [Google Scholar] [CrossRef]
- Gajdos, F.; Oberhofer, H.; Dupuis, M.; Blumberger, J. On the Inapplicability of Electron-hopping Models for the Organic Semiconductor Phenyl-C61-butyric Acid Methyl Ester (PCBM). J. Phys. Chem. Lett. 2013, 4, 1012–1017. [Google Scholar] [CrossRef] [PubMed]
- Borges, I., Jr.; Aquino, A.J.A.; Köhn, A.; Nieman, R.; Hase, W.L.; Chen, L.X.; Lischka, H. Ab Initio Modeling of Excitonic and Charge-Transfer States in Organic Semiconductors: The PTB1/PCBM Low Band Gap System. J. Am. Chem. Soc. 2013, 135, 18252–18255. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, O.; Lungenschmied, C.; Bauer, S. Investigation of Trap States and Mobility in Organic Semiconductor Devices by Dielectric Spectroscopy: Oxygen-doped P3HT:PCBM Solar Cells. Phys. Rev. B 2012, 86, 235201. [Google Scholar] [CrossRef]
- Armbruster, O.; Lungenschmied, C.; Bauer, S. Dielectric Response of Doped Organic Semiconductor Devices: P3HT:PCBM Solar Cells. Phys. Rev. B 2011, 84, 085208. [Google Scholar] [CrossRef]
- Watanabe, S.; Tanaka, H.; Ito, H.; Marumoto, K.; Kuroda, S. ESR Studies of Ambipolar Charge Carriers in Metal-Insulator-Semiconductor Diodes of Regioregular Poly(3-hexylthiophene)/PCBM Composites. Synth. Metal. 2009, 159, 893–896. [Google Scholar] [CrossRef]
- Narayanan, B.; Zhao, Y.; Ciobanu, C.V. Migration Mechanism for Atomic Hydrogen in Porous Carbon Materials. Appl. Phys. Lett. 2012, 100, 203901. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wei, J.; Vajtai, R.; Ajayan, P.M.; Barrera, E.V. Iodine Doped Carbon Nanotube Cables Exceeding Specific Electrical Conductivity of Metals. Sci. Rep. 2011, 1, 83. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Fang, L.; Zhang, B.; Gong, J.R. Graphene Doping: A Review. Insci. J. 2011, 1, 80–89. [Google Scholar] [CrossRef] [Green Version]
- Hoyt, R.A.; Remillard, E.M.; Cubuk, E.D.; Vecitis, C.D.; Kaxiras, E. Polyiodide-Doped Graphene. J. Phys. Chem. C 2017, 121, 609–615. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, W.; Zhao, K.; Guan, F.; Wang, Y. Investigations on the Electronic Structure and Optical Properties of (Ga, N, Ga-N) Doped Graphene by First-Principle Calculations. Int. J. Mod. Phys. B 2021, 35, 2150067. [Google Scholar] [CrossRef]
- Alizadeh, Z.; Mohammadizadeh, M. Predicting Electron-Phonon Coupling Constants of Superconducting Elements by Machine Learning. Phys. C Supercond. Appl. 2018, 558, 7–13. [Google Scholar] [CrossRef]
- Dianat, A.; Zhongquan, L.; Gall, M.; Zhang, T.; Gutierrez, R.; Zschech, E.; Cuniberti, G. Doping of Graphene Induced by Boron/Silicon Substrate. Nanotechnology 2017, 28, 215701. [Google Scholar] [CrossRef]
- Mao, X.; Kour, G.; Yan, C.; Zhu, Z.; Du, A. Single Transition Metal Atom-Doped Graphene Supported on a Nickel Substrate: Enhanced Oxygen Reduction Reactions Modulated by Electron Coupling. J. Phys. Chem. C 2019, 123, 3703–3710. [Google Scholar] [CrossRef]
- Promthong, N.; Tabtimsai, C.; Rakrai, W.; Wanno, B. Transition Metal-doped Graphene Nanoflakes for CO and CO2 Storage and Sensing Applications: A DFT Study. Struct. Chem. 2020, 31, 2237–2247. [Google Scholar] [CrossRef]
- Tang, H.; Xiang, Y.; Zhan, H.; Kang, J.; Zhou, Y. DFT Investigation of Transition Metal-Doped Graphene for the Adsorption of HCl Gas. Diam. Relat. Mater. 2023, 136, 109995. [Google Scholar] [CrossRef]
- Lim, D.-H.; Negreira, A.S.; Wilcox, J. DFT Studies on the Interaction of Defective Graphene-Supported Fe and Al Nanoparticles. J. Phys. Chem. C 2011, 115, 18. [Google Scholar] [CrossRef]
- Zhou, X.; Zhao, C.; Wu, G.; Chen, J.; Li, Y. DFT Study on the Electronic Structure and Optical Properties of N, Al, and N-Al Doped Graphene. Appl. Surf. Sci. 2018, 459, 354–362. [Google Scholar] [CrossRef]
- Tachikawa, H.; Iyama, T. Mechanism of Hydrogen Storage in the Graphene Nanoflake-Lithium-H2 System. J. Phys. Chem. C 2019, 123, 8709–8716. [Google Scholar] [CrossRef]
- Tachikawa, H.; Yi, H.; Iyama, T.; Yamasaki, S.; Azumi, K. Hydrogen Storage Mechanism in Sodium-Based Graphene Nanoflakes: A Density Functional Theory Study. Hydrogen 2022, 3, 43–52. [Google Scholar] [CrossRef]
- Tachikawa, H.; Izumi, Y.; Iyama, T.; Azumi, K. Molecular Design of a Reversible Hydrogen Storage Device Composed of the Graphene Nanoflake-Magnesium-H2 System. ACS Omega 2021, 6, 7778–7785. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Ab-Initio Calculation Program: Gaussian 09; Revision B.04; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Yanai, T.; Tew, D.; Handy, N. A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian Basis Sets for Molecular Calculations. I. Second Low Atoms, Z = 11 − 18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Foster, J.P.; Weinhold, F. Natural Hybrid Orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Tachikawa, H. Hydrogen Storages Based on Graphene Nano-Flakes: Density Functional Theory Approach. C 2022, 8, 36. [Google Scholar] [CrossRef]
- Tachikawa, H.; Iyama, T. Reactions of Graphene Nano-Flakes in Materials Chemistry and Astrophysics. Physchem 2022, 2, 145–162. [Google Scholar] [CrossRef]
- Tachikawa, H. Hydrogen Atom Addition to the Surface of Graphene Nanoflakes: A Density Functional Theory Study. Appl. Surf. Sci. 2017, 396, 1335–1342. [Google Scholar] [CrossRef]
- Tachikawa, H. Ionization Dynamics of Water Dimer on Ice Surface. Surf. Sci. 2016, 647, 1–7. [Google Scholar] [CrossRef]
- Tachikawa, H. Mechanism of Li storage on graphene nanoflakes: Density functional theory study. Surf. Sci. 2020, 691, 121489. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Al Species | h | NPA | NPA | E(bind) |
|---|---|---|---|---|
| Al | GR37 | |||
| Al | 2.217 | +0.864 | −0.864 | 1.9 |
| Al+ | 2.273 | +0.931 | +0.069 | 45.3 |
| Al2+ | 2.333 | +0.924 | +1.076 | 297.7 |
| Al3+ | 2.406 | +0.912 | +2.088 | 730.5 |
| Al Species | h | NPA | NPA | E(bind) |
|---|---|---|---|---|
| Al | GR19 | |||
| Al | 2.195 | +0.848 | −0.848 | 0.8 |
| Al+ | 2.240 | +0.861 | +0.139 | 47.8 |
| Al2+ | 2.349 | +0.885 | +1.115 | 280.1 |
| Al3+ | 2.515 | +0.911 | +2.089 | 679.5 |
| Al Species | E(diffuse) | M | E(diffuse) |
|---|---|---|---|
| Al | 2.44 | Li+ | 7.17 |
| Al+ | 2.23 | Na+ | 2.75 |
| Al2+ | 1.53 | - | - |
| Al3+ | 1.67 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tachikawa, H.; Izumi, Y.; Iyama, T.; Abe, S.; Watanabe, I. Aluminum-Doping Effects on the Electronic States of Graphene Nanoflake: Diffusion and Hydrogen Storage Mechanism. Nanomaterials 2023, 13, 2046. https://doi.org/10.3390/nano13142046
Tachikawa H, Izumi Y, Iyama T, Abe S, Watanabe I. Aluminum-Doping Effects on the Electronic States of Graphene Nanoflake: Diffusion and Hydrogen Storage Mechanism. Nanomaterials. 2023; 13(14):2046. https://doi.org/10.3390/nano13142046
Chicago/Turabian StyleTachikawa, Hiroto, Yoshiki Izumi, Tetsuji Iyama, Shigeaki Abe, and Ikuya Watanabe. 2023. "Aluminum-Doping Effects on the Electronic States of Graphene Nanoflake: Diffusion and Hydrogen Storage Mechanism" Nanomaterials 13, no. 14: 2046. https://doi.org/10.3390/nano13142046