
Ab Initio Study of Octane Moiety Adsorption on H- and Cl-Functionalized Silicon Nanowires


 , , and
, , and
Abstract
:1. Introduction
2. Methods
3. Results and Discussion
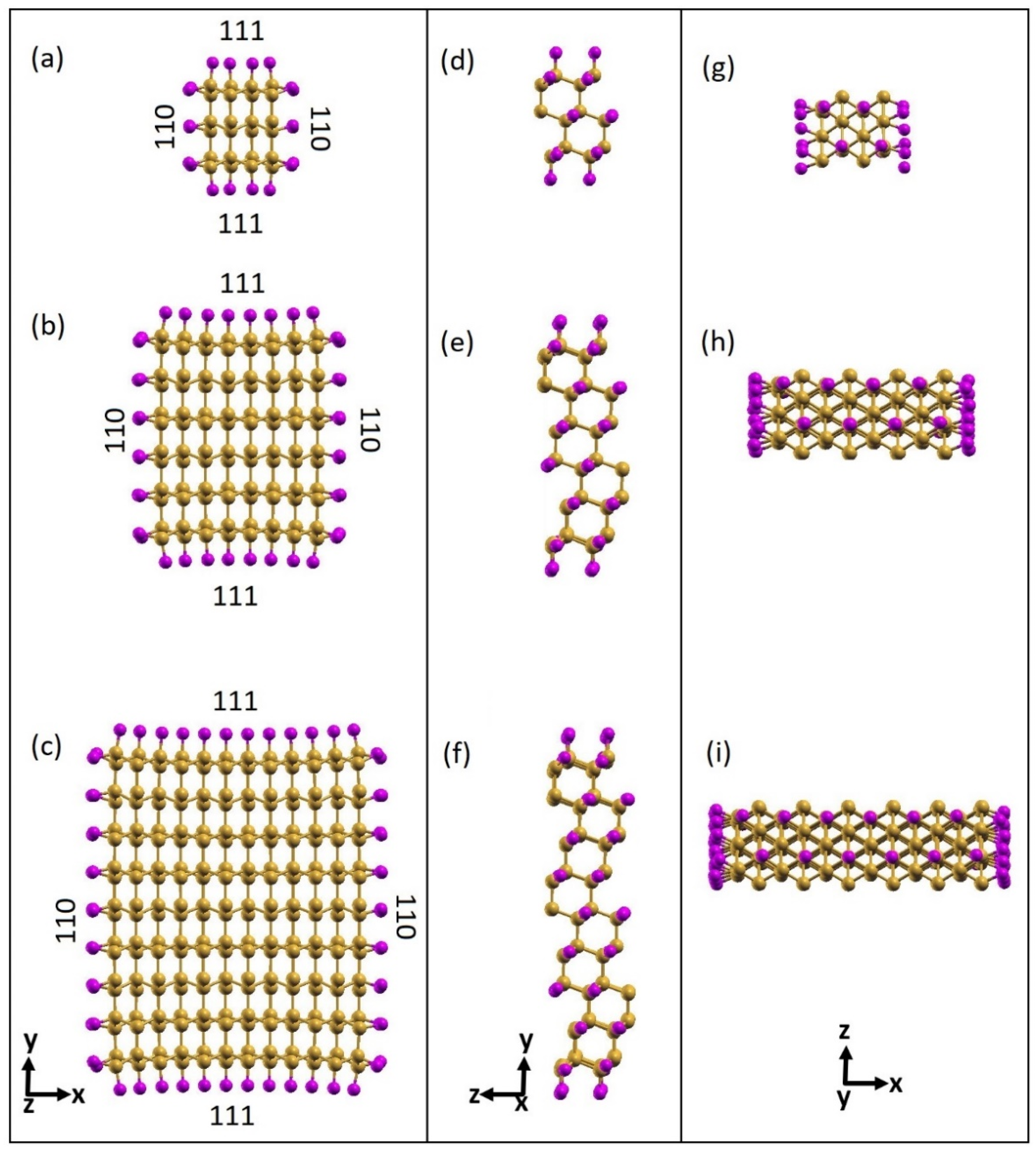
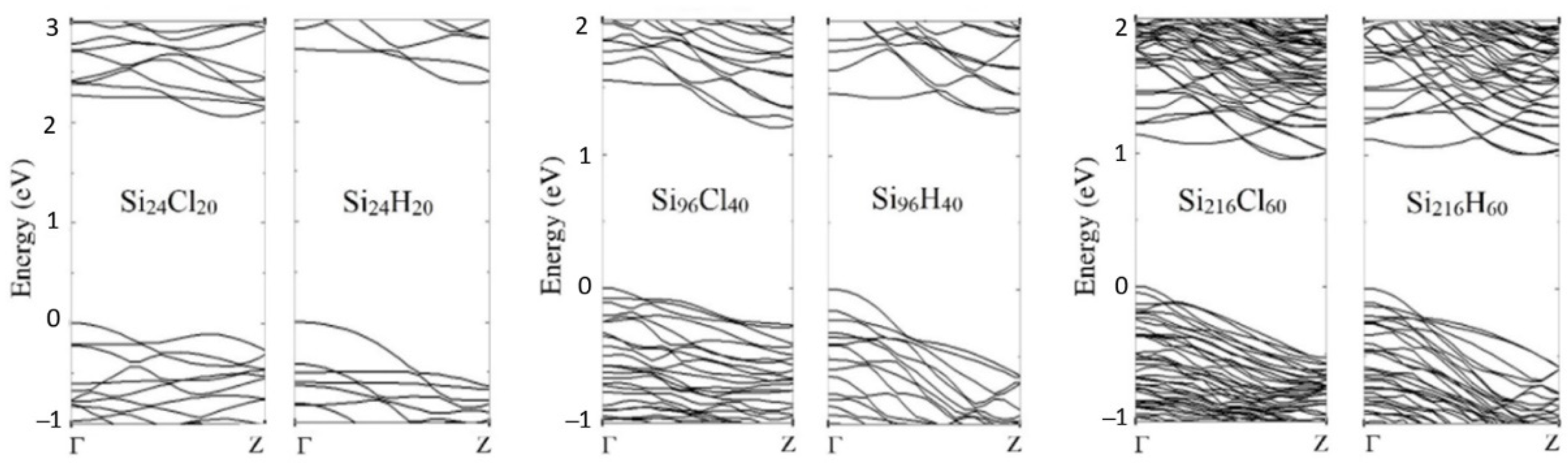
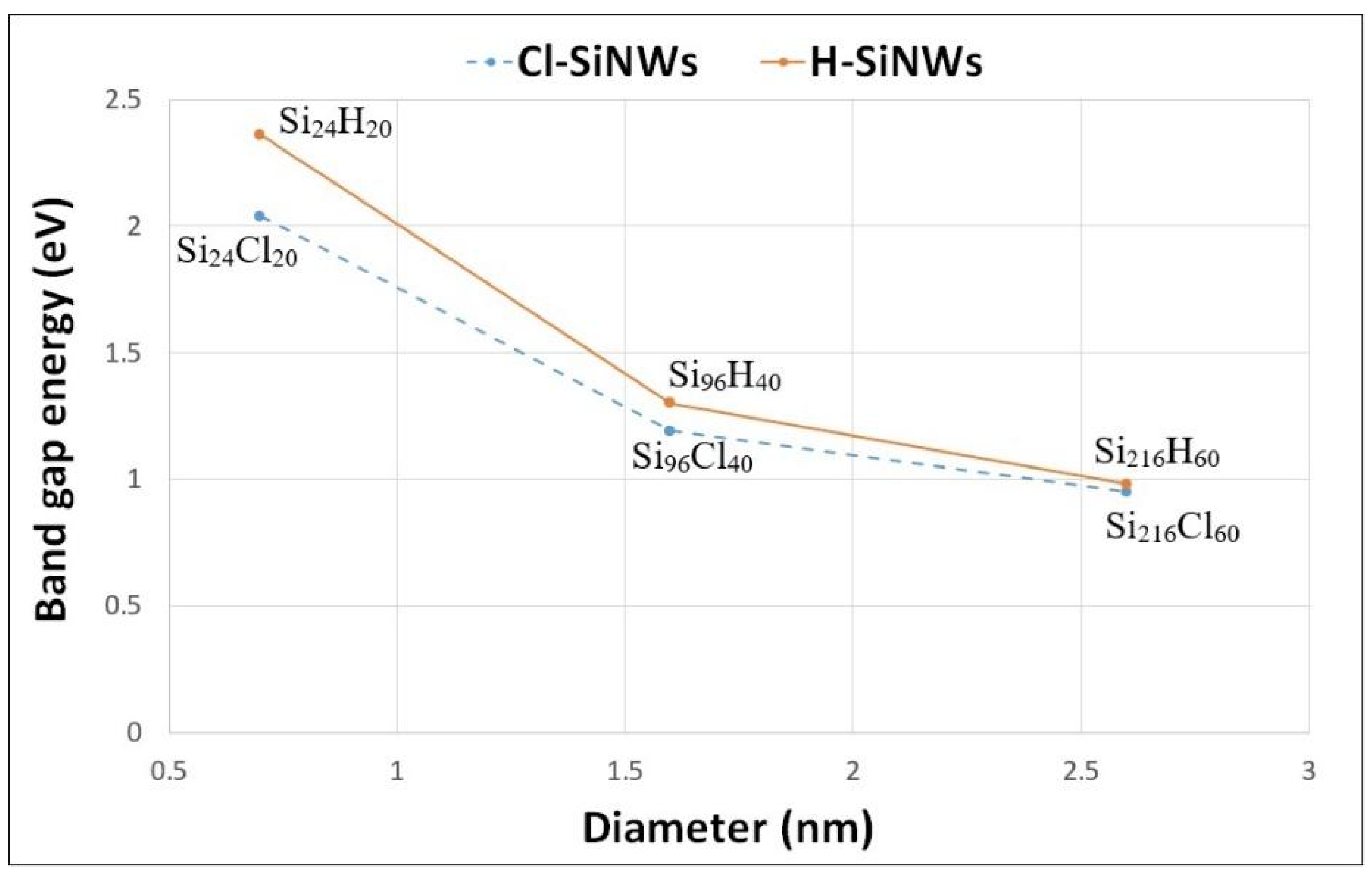
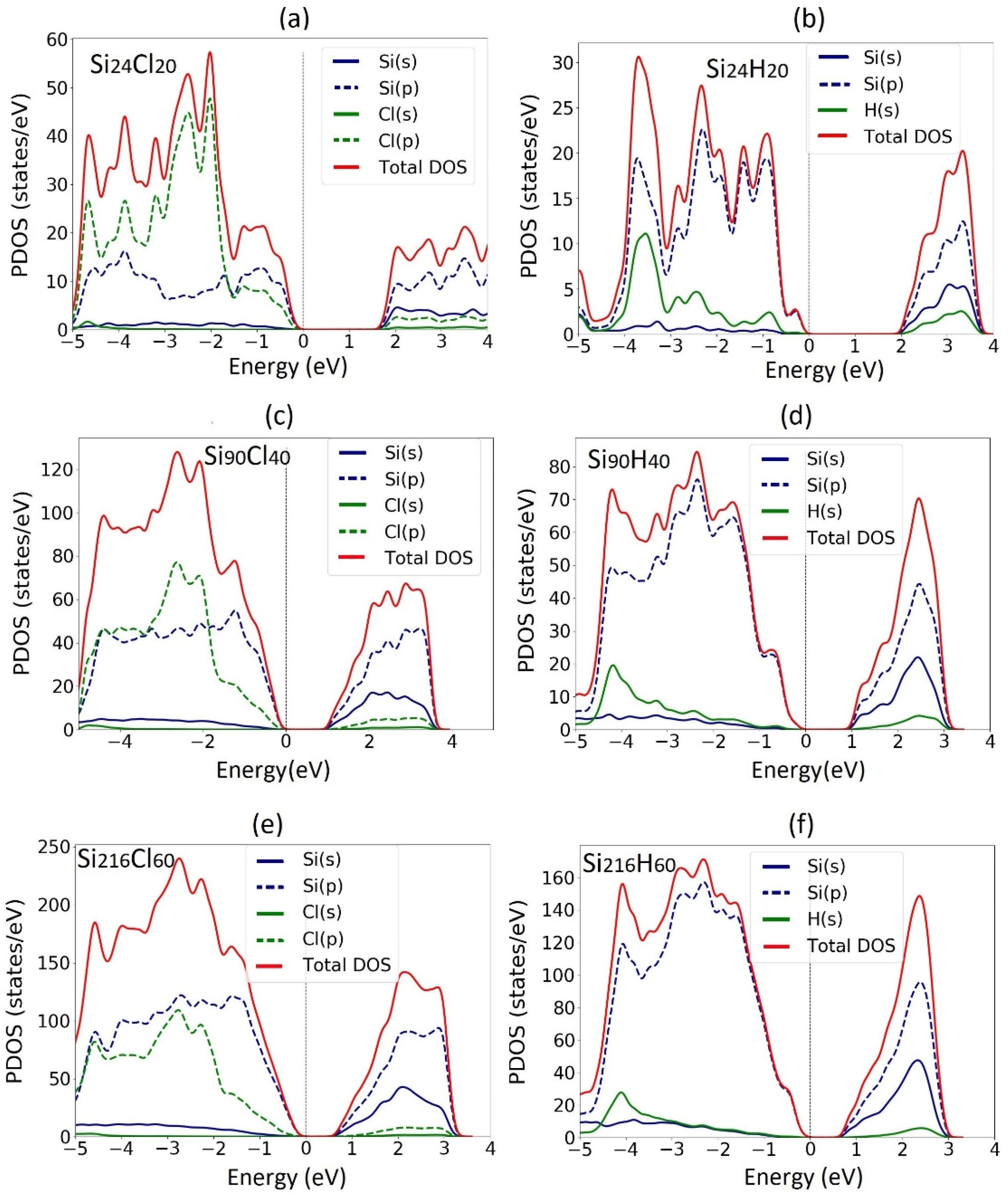
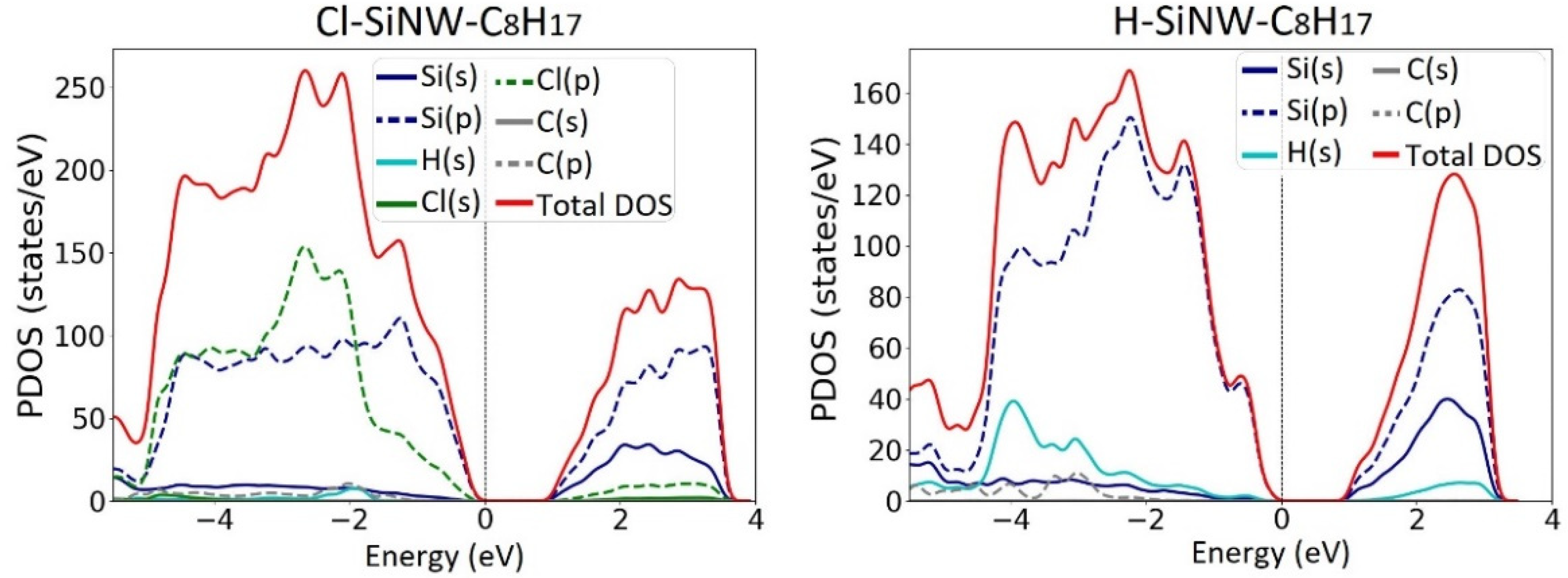
3.1. Electronic Properties of Fully Passivated Cl- and H-SiNWs
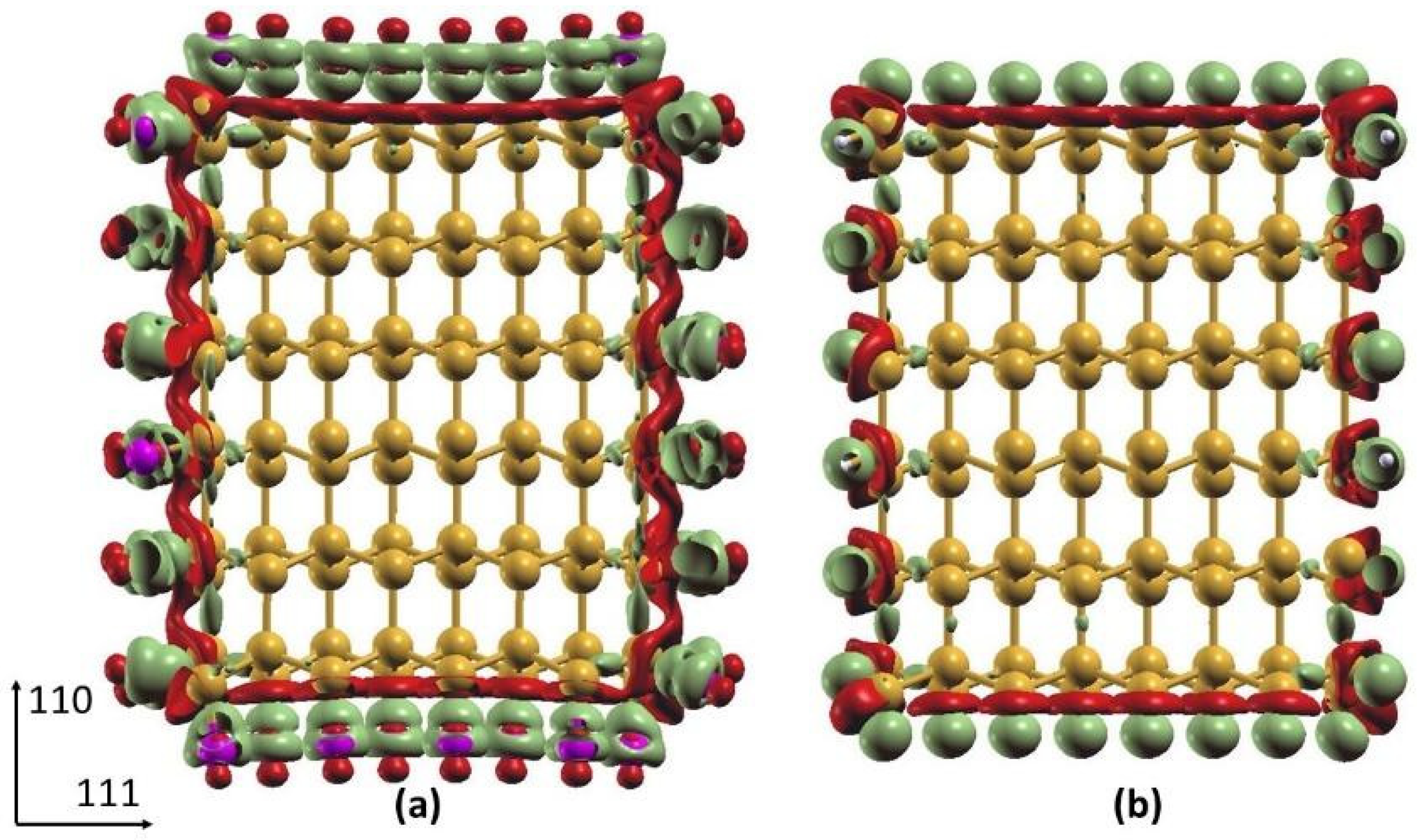
Differential Charge Density of Cl- and H-SiNWs
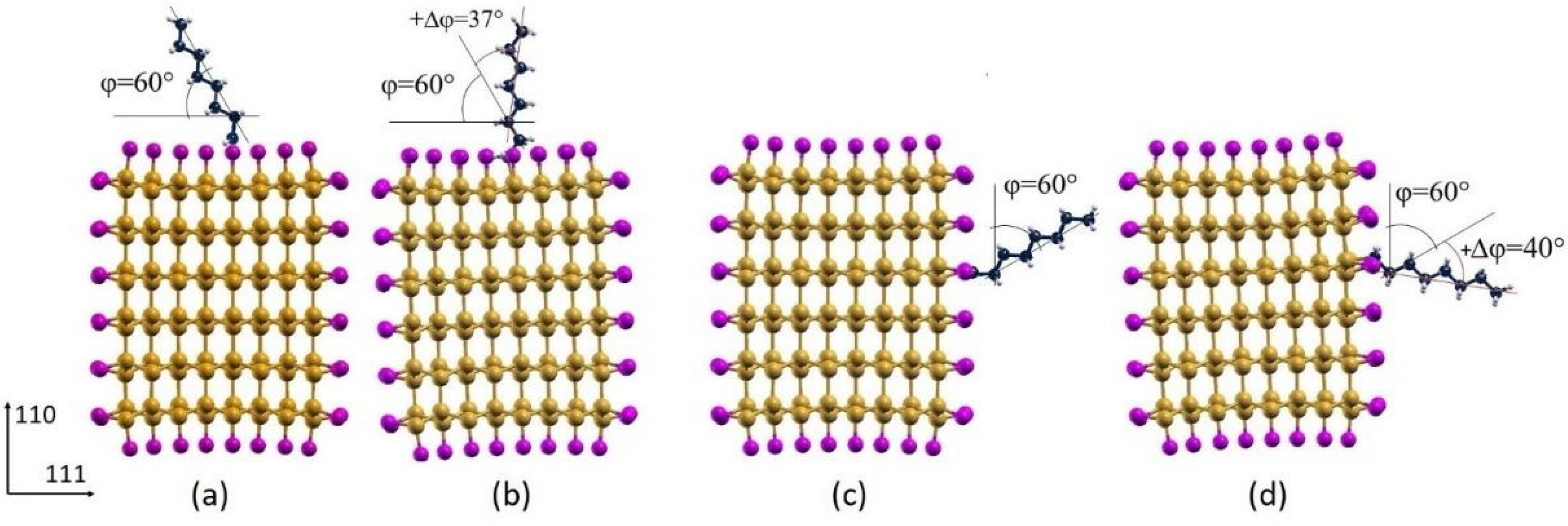
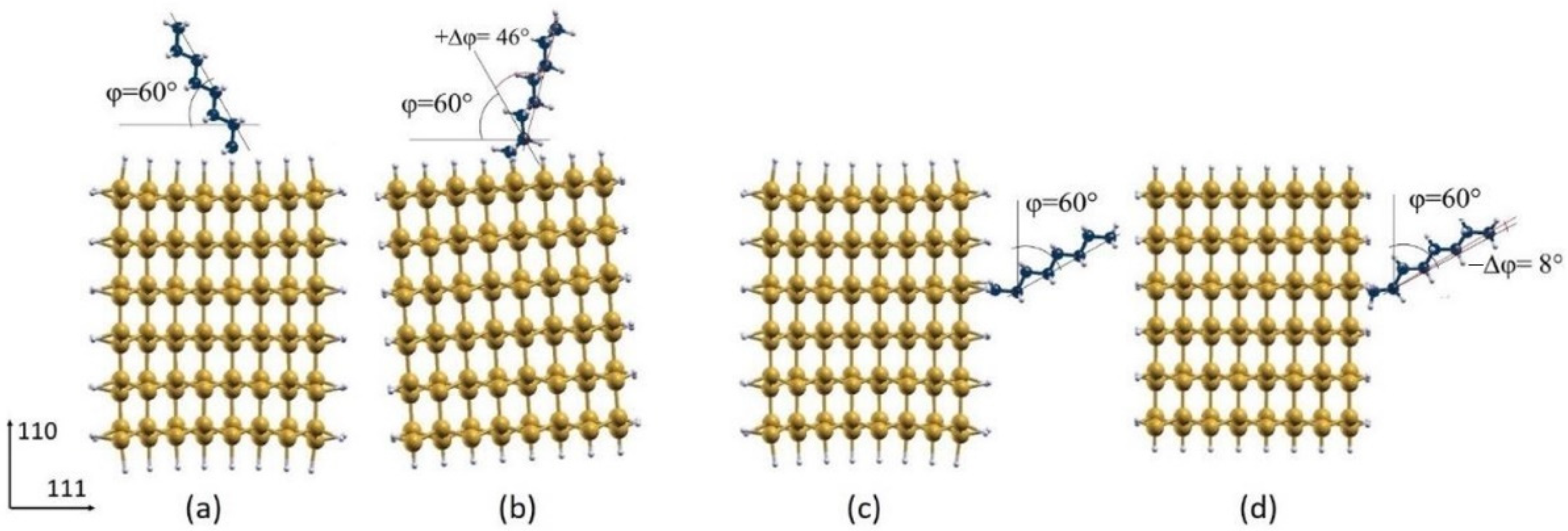
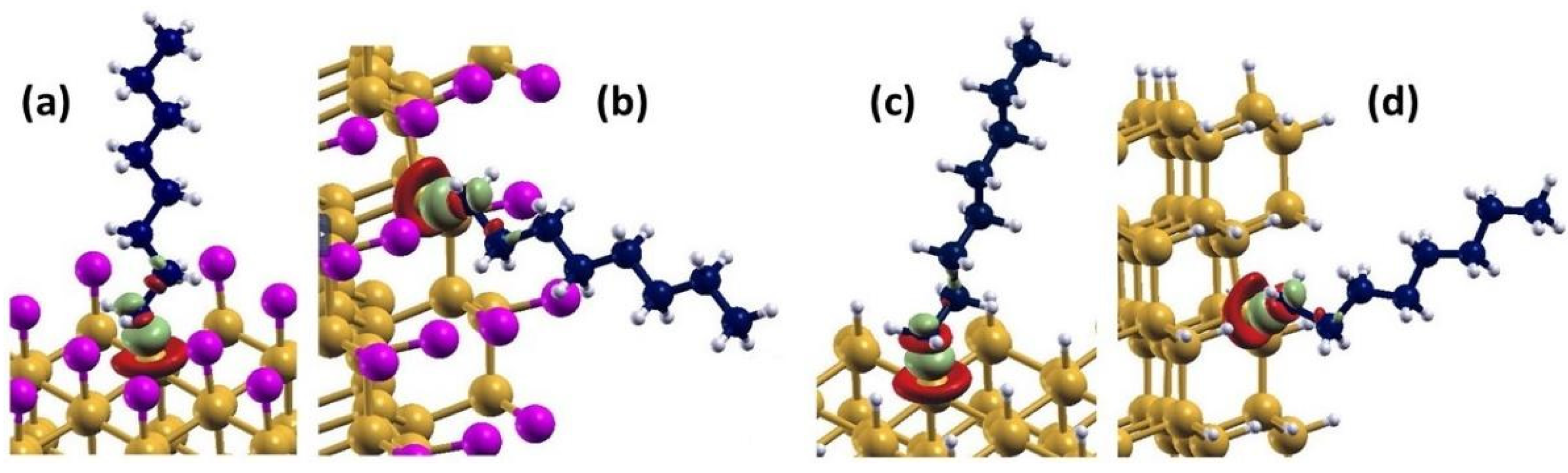
3.2. Octane Moiety Adsorbed on the (111) and (110) Surfaces of H- and Cl- SiNWs
Differential Charge Density of Cl- and H-SiNW-C8H17(111)/(110)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Long, Y.Z.; Yu, M.; Sun, B.; Gu, C.Z.; Fan, Z. Recent advances in large-scale assembly of semiconducting inorganic nanowires and nanofibers for electronics, sensors and photovoltaics. Chem. Soc. Rev. 2012, 41, 4560–4580. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, R.A.; Garozzo, C.; Bongiorno, C.; Di Franco, S.; Italia, M.; Mannino, G.; Scalese, S.; La Magna, A. Molecular doping applied to Si nanowires array based solar cells. Sol. Energy Mater. Sol. Cells 2015, 132, 118–122. [Google Scholar] [CrossRef]
- Sahoo, M.K.; Kale, P. Integration of silocon nanowires in solar cell structure for efficiency enhancement: A review. J. Materiomics. 2019, 5, 34–48. [Google Scholar] [CrossRef]
- Yang, Y.; Yuan, W.; Kang, W.; Ye, Y.; Pan, Q.; Zhang, X.; Ke, Y.; Wang, C.; Qiu, Z.; Tang, Y. A review on silicon nanowire-based anodes for next-generation high-performance lithium-ion batteries from a material-based perspective. Sustain. Energy Fuels 2020, 4, 1577–1594. [Google Scholar] [CrossRef]
- Coffinier, Y. Chapter 3: Rabah Boukherroub. Surface modification of silicon nanowires for biosensing. In Semiconducting Silicon Nanowires for Biomedical Applications, 2nd ed.; Woodhead Publishing: Thorston, UK, 2022; pp. 25–68. [Google Scholar] [CrossRef]
- Izuan, J.; Rashid, A.; Abdullah, J.; Yusof, N.A.; Hajian, R. The Development of Silicon Nanowire as Sensing Material and Its Applications. J. Nanomater. 2013, 2013, 328093. [Google Scholar] [CrossRef] [Green Version]
- Aradi, B.; Ramos, L.E.; Deák, P.; Köhler, T.; Bechstedt, F.; Zhang, R.Q.; Frauenheim, T. Theoretical study of the chemical gap tuning in silicon nanowires. Phys. Rev. B 2006, 76, 035305. [Google Scholar] [CrossRef]
- Schmidt, V.; Wittemann, J.V.; Senz, S.; Gösele, U. Silicon Nanowires: A Review on Aspects of their Growth and their Electrical Properties. Adv. Mater. 2009, 21, 2681–2702. [Google Scholar] [CrossRef]
- de Santiago, F.; Trejo, A.; Miranda, A.; Carvajal, E.; Pérez, L.A.; Cruz-Irisson, M. Band-gap engineering of halogenated silicon nanowires through molecular doping. J. Mol. Model 2017, 23, 314. [Google Scholar] [CrossRef]
- Dass, D. Metallic-semiconducting transition of silicon nanowires by surface passivation. Results Surf. Interfaces 2021, 3, 100009. [Google Scholar] [CrossRef]
- Wu, Y.; Cui, Y.; Huynh, L.; Barrelet, C.J.; Bell, D.C.; Leber, C.M. Controlled Growth and Structures of Molecular-Scale Silicon Nanowires. Nano Lett. 2004, 4, 433–436. [Google Scholar] [CrossRef]
- Bashouti, M.; Stelzner, T.; Christiansen, S.; Haick, H. Covalent Attachment of Alkyl Functionality to 50 nm Silicon Nanowires through a Chlorination/Alkylation Process. J. Phys. Chem. C 2009, 113, 14823–14828. [Google Scholar] [CrossRef] [Green Version]
- Bashouti, M.Y.; Pietsch, M.; Brönstrup, G.; Sivakov, V. Heterojunction based hybrid silicon nanowire solar cell: Surface termination, photoelectron and photoemission spectroscopy study. Prog. Photovolt. 2014, 22, 1050–1061. [Google Scholar] [CrossRef]
- Akbari-Saatlu, M.; Procek, M.; Mattsson, C.; Thungström, G.; Nilsson, H.-E.; Xiong, W.; Xu, B.; Li, Y.; Radamson, H.H. Silicon Nanowires for Gas Sensing: A Review. Nanomaterials 2020, 10, 2215. [Google Scholar] [CrossRef] [PubMed]
- Natan, A.; Kronik, L.; Haick, H.; Tung, R.T. Electrostatic Properties of Ideal and Non-ideal Polar Organic Monolayers: Implications for Electronic Devices. Adv. Mater. 2007, 19, 4103–4117. [Google Scholar] [CrossRef]
- Bashouti, M.Y.; Tung, R.T.; Haick, H. Tuning the Electrical Properties of Si Nanowire Field-Effect Transistors by Molecular Engineering. Small 2009, 5, 2761–2769. [Google Scholar] [CrossRef]
- Yoshitake, M. Work Function and Band Alignment of Electrode Materials: The Art of Interface Potential for Electronic Devices, Solar Cells, and Batteries, 1st ed.; NIMS Monographs, English Edition; Springer: Tokyo, Japan, 2021; pp. 97–112. [Google Scholar]
- Ahoulou, S.; Perret, E.; Nedelec, J.-M. Functionalization and Characterization of Silicon Nanowires for Sensing Applications: A Review. Nanomaterials 2021, 11, 999. [Google Scholar] [CrossRef]
- Bashouti, M.; Stelzner, T.; Berger, A.; Christiansen, S.; Haick, H. Chemical Passivation of Silicon Nanowires with C1–C6 Alkyl Chains through Covalent Si-C Bonds. J. Phys. Chem. 2008, 112, 19168–19172. [Google Scholar] [CrossRef]
- Bashouti, M.; Sardashti, K.; Schmitt, S.; Pietsch, M.; Ristein, J.; Haick, H.; Christiansen, S. Oxide-free hybrid silicon nanowires: From fundamentals to applied nanotechnology. Prog. Surf. Sci. 2013, 88, 39–60. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderbilt, D. Soft Self-Consistent Pseudopotentials in a Generalized Eigenvalue Formalism. Phys. Rev. B Condens. Matter 1990, 41, 7892–7895. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Ciobanu, C.V.; Chan, T.L.; Chuang, F.C.; Wang, C.Z.; Ho, C.M. The structure of ultrathin H-passivated 112. silicon nanowires. J. Phys. Chem. C 2007, 111, 7933–7937. [Google Scholar] [CrossRef]
- Shalabny, A.; Buonocore, F.; Celino, M.; Shalev, G.; Zhang, L.; Wu, W.; Li, P.; Arbiol, J.; Bashouti, M.Y. Semiconductivity Transition in Silicon Nanowires by Hole Transport Layer. Nano Lett. 2020, 20, 8369–8374. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Sim, L.Y.; Da, H.; Jin, H.; Lim, K.; Yang, S.W. Modulation of the work function of silicon nanowire by chemical surface passivation: A DFT study. Theor. Chem. Acc. 2010, 127, 689–695. [Google Scholar] [CrossRef]
- Mohammad, S. Understanding quantum confinement in nanowires: Basics, applications and possible laws. J. Phys. Condens. Matter 2014, 26, 423202. [Google Scholar] [CrossRef] [PubMed]
- Miranda, Á.; Cartoixà, X.; Canadell, E.; Rurali, R. NH3 molecular doping of silicon nanowires grown along the [112], [110], [001], and [111] orientations. Nanoscale Res. Lett. 2012, 7, 308. Available online: http://www.nanoscalereslett.com/content/7/1/308 (accessed on 7 December 2021). [CrossRef] [Green Version]
- Huang, L.; Lu, N.; Yan, J.A.; Chou, M.; Wang, C.; Ho, K. Size-and Strain-Dependent Electronic Structures in H-Passivated Si [112] Nanowires. J. Phys. Chem. C 2008, 112, 15680–15683. [Google Scholar] [CrossRef]
- Haziri, V.; Phal, S.; Boily, J.-F.; Berisha, A.; Tesfalidet, S. Oxygen Interactions with Covalently Grafted 2D Nanometric Carboxyphenyl Thin Films—An Experimental and DFT Study. Coatings 2022, 12, 49. [Google Scholar] [CrossRef]
- Xu, H.; Yang, X.B.; Zhang, C.; Lu, A.J.; Zhang, R.Q. Stabilizing and activating dopants in <112> silicon nanowires by alkene adsorptions: A first-principles study. Appl. Phys. Lett. 2011, 98, 073115. [Google Scholar] [CrossRef]
- Iannone, F.; Ambrosino, F.; Bracco, G.; de Rosa, M.; Funel, A.; Guarnieri, G.; Migliori, S.; Palombi, F.; Ponti, G.; Santoma, G.; et al. CRESCO ENEA HPC clusters: A working example of a multifabric GPFS Spectrum Scale layout. In Proceedings of the 2019 International Conference on High Performance Computing & Simulation (HPCS), Dublin, Ireland, 15–19 July 2019; pp. 1051–1052. [Google Scholar] [CrossRef]
- Mariano, A.; D’Amato, D.; Ambrosino, F.; Aprea, G.; Buonocore, F.; Celino, M.; Colavincenzo, A.; Fina, M.; Funel, A.; Giusepponi, S.; et al. Fast Access to Remote Objects 2.0. A renewed gateway to ENEAGRID distributed computing resources. Future Gener. Comput. Syst. 2019, 94, 920–928. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H-SiNWs | (eV/atom) | Cl-SiNWs | (eV/atom) |
|---|---|---|---|
| Si24H20 | −0.0025 | Si24Cl20 | −0.73 |
| Si96H40 | −0.035 | Si96Cl40 | −0.51 |
| Si216H60 | −0.068 | Si216Cl60 | −0.41 |
| H-SiNWs | Eg (eV) | Cl-SiNWs | Eg (eV) | d (nm) |
|---|---|---|---|---|
| Si24H20 | 2.36 | Si24Cl20 | 2.04 | 0.7 |
| Si96H40 | 1.30 | Si96Cl40 | 1.19 | 1.7 |
| Si216H60 | 0.98 | Si216Cl60 | 0.95 | 2.6 |
| NW Structure | BDE (kJ/Mol) |
|---|---|
| Cl-Si-C8H17 (111) | 733.85 |
| Cl-Si-C8H17 (110) | 716.47 |
| H-Si-C8H17 (111) | 637.30 |
| H-Si-C8H17 (110) | 632.67 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrucci, B.; Buonocore, F.; Giusepponi, S.; Shalabny, A.; Bashouti, M.Y.; Celino, M. Ab Initio Study of Octane Moiety Adsorption on H- and Cl-Functionalized Silicon Nanowires. Nanomaterials 2022, 12, 1590. https://doi.org/10.3390/nano12091590
Ferrucci B, Buonocore F, Giusepponi S, Shalabny A, Bashouti MY, Celino M. Ab Initio Study of Octane Moiety Adsorption on H- and Cl-Functionalized Silicon Nanowires. Nanomaterials. 2022; 12(9):1590. https://doi.org/10.3390/nano12091590
Chicago/Turabian StyleFerrucci, Barbara, Francesco Buonocore, Simone Giusepponi, Awad Shalabny, Muhammad Y. Bashouti, and Massimo Celino. 2022. "Ab Initio Study of Octane Moiety Adsorption on H- and Cl-Functionalized Silicon Nanowires" Nanomaterials 12, no. 9: 1590. https://doi.org/10.3390/nano12091590