
Binding Capabilities of Different Genetically Engineered pVIII Proteins of the Filamentous M13/Fd Virus and Single-Walled Carbon Nanotubes


Abstract
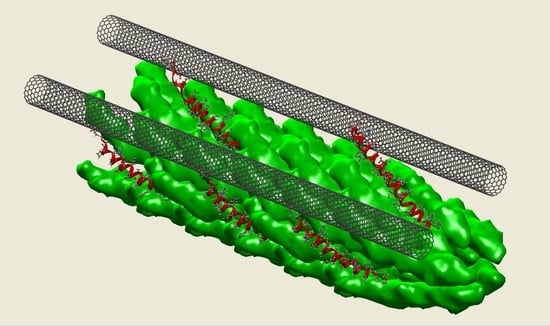
:
1. Introduction
2. Results and Discussion
2.1. Selecting SWNT-Binding Candidates

2.2. Engineered Virus Quantification
2.3. SWNT-Binding Peptides Display Verification
2.4. Virus Thermal and Sonication Stability
2.5. SWNT–Virus Binding Characterization
3. Conclusions and Summary
4. Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cao, Y.; Cong, S.; Cao, X.; Wu, F.; Liu, Q.; Amer, M.R.; Zhou, C. Review of Electronics Based on Single-Walled Carbon Nanotubes. Top. Curr. Chem. 2017, 375, 75. [Google Scholar] [CrossRef] [PubMed]
- De Volder, M.F.; Tawfick, S.H.; Baughman, R.H.; Hart, A.J. Carbon nanotubes: Present and future commercial applications. Science 2013, 339, 535–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, T.; Nag, A.; Mukhopadhyay, S.C.; Xu, Y. Carbon nanotubes and its gas-sensing applications: A review. Sens. Actuators A-Phys. 2019, 291, 107–143. [Google Scholar] [CrossRef]
- Koo, J.H.; Song, J.K.; Kim, D.H. Solution-processed thin films of semiconducting carbon nanotubes and their application to soft electronics. Nanotechnology 2019, 30, 132001. [Google Scholar] [CrossRef]
- Liu, L.; Ma, W.; Zhang, Z. Macroscopic carbon nanotube assemblies: Preparation, properties, and potential applications. Small 2011, 7, 1504–1520. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.; Flahaut, E.; Golzio, M. Overview of Carbon Nanotubes for Biomedical Applications. Materials 2019, 12, 624. [Google Scholar] [CrossRef] [Green Version]
- Sireesha, M.; Babu, V.J.; Ramakrishna, S. Functionalized carbon nanotubes in bio-world: Applications, limitations and future directions. Mater. Sci. Eng. B-Adv. Funct. Solid-State Mater. 2017, 223, 43–63. [Google Scholar] [CrossRef]
- Wang, L.; Liu, H.; Konik, R.M.; Misewich, J.A.; Wong, S.S. Carbon nanotube-based heterostructures for solar energy applications. Chem. Soc. Rev. 2013, 42, 8134–8156. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, J.-Q.; Qian, W.-Z.; Zhang, Y.; Wei, F. The road for nanomaterials industry: A review of carbon nanotube production, post-treatment, and bulk applications for composites and energy storage. Small 2013, 9, 1237–1265. [Google Scholar] [CrossRef]
- Rao, R.; Pint, C.L.; Islam, A.E.; Weatherup, R.S.; Hofmann, S.; Meshot, E.R.; Wu, F.; Zhou, C.; Dee, N.; Amama, P.B.; et al. Carbon Nanotubes and Related Nanomaterials: Critical Advances and Challenges for Synthesis toward Mainstream Commercial Applications. ACS Nano 2018, 12, 11756–11784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Gong, C.; Yuan, X.; Wei, G. Controlling the Self-Assembly of Biomolecules into Functional Nanomaterials through Internal Interactions and External Stimulations: A Review. Nanomaterials 2019, 9, 285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuai, C.J.; Xu, Y.; Feng, P.; Wang, G.; Xiong, S.; Peng, S. Antibacterial polymer scaffold based on mesoporous bioactive glass loaded with in situ grown silver. Chem. Eng. J. 2019, 374, 304–315. [Google Scholar] [CrossRef]
- Shuai, C.J.; Liu, G.F.; Yang, Y.W.; Qi, F.W.; Peng, S.P.; Yang, W.J.; He, C.X.; Wang, G.Y.; Qian, G.W. A strawberry-like Ag-decorated barium titanate enhances piezoelectric and antibacterial activities of polymer scaffold. Nano Energy 2020, 74, 104825. [Google Scholar] [CrossRef]
- Mallakpour, S.; Soltanian, S. Surface functionalization of carbon nanotubes: Fabrication and applications. RSC Adv. 2016, 6, 109916–109935. [Google Scholar] [CrossRef]
- Yang, Y.; Zheng, M.; Jagota, A. Learning to predict single-wall carbon nanotube-recognition DNA sequences. NPJ Comput. Mater. 2019, 5, 3. [Google Scholar] [CrossRef]
- Kuang, Z.; Kim, S.N.; Crookes-Goodson, W.J.; Farmer, B.L.; Naik, R.R. Biomimetic chemosensor: Designing peptide recognition elements for surface functionalization of carbon nanotube field effect transistors. ACS Nano 2010, 4, 452–458. [Google Scholar] [CrossRef]
- Tsyboulski, D.A.; Bakota, E.L.; Witus, L.S.; Rocha, J.D.; Hartgerink, J.D.; Weisman, R.B. Self-assembling peptide coatings designed for highly luminescent suspension of single-walled carbon nanotubes. J. Am. Chem. Soc. 2008, 130, 17134–17140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witus, L.S.; Rocha, J.D.R.; Yuwono, V.M.; Paramonov, S.E.; Weisman, R.B.; Hartgerink, J.D. Peptides that non-covalently functionalize single-walled carbon nanotubes to give controlled solubility characteristics. J. Mater. Chem. 2007, 17, 1909–1915. [Google Scholar] [CrossRef]
- Panhuis, M.; Gowrisanker, S.; Vanesko, D.J.; Mire, C.A.; Jia, H.; Xie, H.; Baughman, R.H.; Musselman, I.H.; Gnade, B.E.; Dieckmann, G.R.; et al. Nanotube network transistors from peptide-wrapped single-walled carbon nanotubes. Small 2005, 1, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Andarias, J.; Mejias, S.H.; Sakurai, T.; Matsuda, W.; Seki, S.; Feixas, F.; Osuna, S.; Atienza, C.; Martin, N.; Cortajarena, A.L. Toward Bioelectronic Nanomaterials: Photoconductivity in Protein-Porphyrin Hybrids Wrapped around SWCNT. Adv. Funct. Mater. 2018, 28, 1704031. [Google Scholar] [CrossRef]
- Kase, D.; Kulp, J.L., 3rd; Yudasaka, M.; Evans, J.S.; Iijima, S.; Shiba, K. Affinity selection of peptide phage libraries against single-wall carbon nanohorns identifies a peptide aptamer with conformational variability. Langmuir 2004, 20, 8939–8941. [Google Scholar] [CrossRef]
- Su, Z.; Leung, T.; Honek, J.F. Conformational selectivity of peptides for single-walled carbon nanotubes. J. Phys. Chem. B 2006, 110, 23623–23627. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.F.; Jain, D.; Burke, P. Nanotube-Peptide Interactions on a Silicon Chip. J. Phys. Chem. C 2009, 113, 3978–3985. [Google Scholar] [CrossRef]
- Pender, M.J.; Sowards, L.A.; Hartgerink, J.D.; Stone, M.O.; Naik, R.R. Peptide-mediated formation of single-wall carbon nanotube composites. Nano Lett. 2006, 6, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Humphreys, E.S.; Chung, S.Y.; Delduco, D.F.; Lustig, S.R.; Wang, H.; Parker, K.N.; Rizzo, N.W.; Subramoney, S.; Chiang, Y.M.; et al. Peptides with selective affinity for carbon nanotubes. Nat. Mater. 2003, 2, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Kulp, J.L., 3rd; Shiba, K.; Evans, J.S. Probing the conformational features of a phage display polypeptide sequence directed against single-walled carbon nanohorn surfaces. Langmuir 2005, 21, 11907–11914. [Google Scholar] [CrossRef] [PubMed]
- Kriplani, U.; Kay, B.K. Selecting peptides for use in nanoscale materials using phage-displayed combinatorial peptide libraries. Curr. Opin. Biotechnol. 2005, 16, 470–475. [Google Scholar] [CrossRef]
- Baneyx, F.; Schwartz, D.T. Selection and analysis of solid-binding peptides. Curr. Opin. Biotechnol. 2007, 18, 312–317. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Mui, K.; Daub, E.; Leung, T.; Honek, J. Single-walled carbon nanotube binding peptides: Probing tryptophan’s importance by unnatural amino acid substitution. J. Phys. Chem. B 2007, 111, 14411–14417. [Google Scholar] [CrossRef]
- Moon, J.S.; Kim, W.G.; Kim, C.; Park, G.T.; Heo, J.; Yoo, S.Y.; Oh, J.W. M13 Bacteriophage-Based Self-Assembly Structures and Their Functional Capabilities. Mini Rev. Org. Chem. 2015, 12, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kehoe, J.W.; Kay, B.K. Filamentous phage display in the new millennium. Chem. Rev. 2005, 105, 4056–4072. [Google Scholar] [CrossRef] [PubMed]
- Petrenko, V.A. Landscape Phage: Evolution from Phage Display to Nanobiotechnology. Viruses 2018, 10, 311. [Google Scholar] [CrossRef] [Green Version]
- Moradi, M.; Li, Z.; Qi, J.; Xing, W.; Xiang, K.; Chiang, Y.M.; Belcher, A.M. Improving the capacity of sodium ion battery using a virus-templated nanostructured composite cathode. Nano Lett. 2015, 15, 2917–2921. [Google Scholar] [CrossRef] [PubMed]
- Dang, X.; Yi, H.; Ham, M.H.; Qi, J.; Yun, D.S.; Ladewski, R.; Strano, M.S.; Hammond, P.T.; Belcher, A.M. Virus-templated self-assembled single-walled carbon nanotubes for highly efficient electron collection in photovoltaic devices. Nat. Nanotechnol. 2011, 6, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Ghosh, D.; Ham, M.H.; Qi, J.; Barone, P.W.; Strano, M.S.; Belcher, A.M. M13 phage-functionalized single-walled carbon nanotubes as nanoprobes for second near-infrared window fluorescence imaging of targeted tumors. Nano Lett. 2012, 12, 1176–1183. [Google Scholar] [CrossRef] [Green Version]
- Bardhan, N.M.; Ghosh, D.; Belcher, A.M. Carbon nanotubes as in vivo bacterial probes. Nat. Commun. 2014, 5, 4918. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, D.; Bagley, A.F.; Na, Y.J.; Birrer, M.J.; Bhatia, S.N.; Belcher, A.M. Deep, noninvasive imaging and surgical guidance of submillimeter tumors using targeted M13-stabilized single-walled carbon nanotubes. Proc. Natl. Acad. Sci. USA 2014, 111, 13948–13953. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.Y.; Byeon, H.H.; Jang, C.; Choi, J.H.; Choi, I.S.; Jung, Y.; Kim, W.; Chang, J.; Yi, H. Hydrodynamic assembly of conductive nanomesh of single-walled carbon nanotubes using biological glue. Adv. Mater. 2015, 27, 922–928. [Google Scholar] [CrossRef]
- Byeon, H.H.; Lee, S.W.; Lee, E.H.; Kim, W.; Yi, H. Biologically templated assembly of hybrid semiconducting nanomesh for high performance field effect transistors and sensors. Sci. Rep. 2016, 6, 35591. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.W.; Lee, K.Y.; Song, Y.W.; Choi, W.K.; Chang, J.; Yi, H. Direct Electron Transfer of Enzymes in a Biologically Assembled Conductive Nanomesh Enzyme Platform. Adv. Mater. 2016, 28, 1577–1584. [Google Scholar] [CrossRef]
- Lee, S.W.; Kang, T.H.; Lee, S.K.; Lee, K.Y.; Yi, H. Hydrodynamic Layer-by-Layer Assembly of Transferable Enzymatic Conductive Nanonetworks for Enzyme-Sticker-Based Contact Printing of Electrochemical Biosensors. ACS Appl. Mater. Interfaces 2018, 10, 36267–36274. [Google Scholar] [CrossRef] [PubMed]
- Byeon, H.H.; Lee, W.C.; Kim, W.; Kim, S.K.; Kim, W.; Yi, H. Bio-fabrication of nanomesh channels of single-walled carbon nanotubes for locally gated field-effect transistors. Nanotechnology 2017, 28, 025304. [Google Scholar] [CrossRef] [PubMed]
- Merryman, A.E.; Sabaraya, I.V.; Rowles, L.S., 3rd; Toteja, A.; Carrillo, S.I.; Sabo-Attwood, T.; Saleh, N.B. Interaction between functionalized multiwalled carbon nanotubes and MS2 bacteriophages in water. Sci. Total Environ. 2019, 670, 1140–1145. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.X.; Kong, B.S.; Yang, S.B.; Youn, S.C.; Park, S.; Joo, T.; Jung, H.T. Effect of SWNT Defects on the Electron Transfer Properties in P3HT/SWNT Hybrid Materials. Adv. Funct. Mater. 2008, 18, 2659–2665. [Google Scholar] [CrossRef]
- Zeinabad, H.A.; Zarrabian, A.; Saboury, A.A.; Alizadeh, A.M.; Falahati, M. Interaction of single and multi wall carbon nanotubes with the biological systems: Tau protein and PC12 cells as targets. Sci. Rep. 2016, 6, 26508. [Google Scholar] [CrossRef]
- Lee, Y.J.; Yi, H.; Kim, W.J.; Kang, K.; Yun, D.S.; Strano, M.S.; Ceder, G.; Belcher, A.M. Fabricating genetically engineered high-power lithium-ion batteries using multiple virus genes. Science 2009, 324, 1051–1055. [Google Scholar] [CrossRef] [Green Version]
- Nam, K.T.; Peelle, B.R.; Lee, S.W.; Belcher, A.M. Genetically driven assembly of nanorings based on the M13 virus. Nano Lett. 2004, 4, 23–27. [Google Scholar] [CrossRef]
- Lee, S.W.; Mao, C.B.; Flynn, C.E.; Belcher, A.M. Ordering of quantum dots using genetically engineered viruses. Science 2002, 296, 892–895. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Chiang, C.Y.; Lee, S.K.; Gao, Y.; Hu, E.L.; De Yoreo, J.; Belcher, A.M. Programmable assembly of nanoarchitectures using genetically engineered viruses. Nano Lett. 2005, 5, 1429–1434. [Google Scholar] [CrossRef]
- Hemminga, M.A.; Vos, W.L.; Nazarov, P.V.; Koehorst, R.B.M.; Wolfs, C.; Spruijt, R.B.; Stopar, D. Viruses: Incredible nanomachines. New advances with filamentous phages. Eur. Biophys. J. Biophys. Lett. 2010, 39, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Iannolo, G.; Minenkova, O.; Petruzzelli, R.; Cesareni, G. Modifying Filamentous Phage Capsid: Limits in the Size of the Major Capsid Protein. J. Mol. Biol. 1995, 248, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Loset, G.A.; Roos, N.; Bogen, B.; Sandlie, I. Expanding the versatility of phage display II: Improved affinity selection of folded domains on protein VII and IX of the filamentous phage. PLoS ONE 2011, 6, e17433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeri, A.C.; Mesleh, M.F.; Nevzorov, A.A.; Opella, S.J. Structure of the coat protein in fd filamentous bacteriophage particles determined by solid-state NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2003, 100, 6458–6463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, G.; Gershoni, J.M. A general insert label for peptide display on chimeric filamentous bacteriophages. Anal. Biochem. 2012, 420, 68–72. [Google Scholar] [CrossRef]
- Schier, R.; Bye, J.; Apell, G.; McCall, A.; Adams, G.P.; Malmqvist, M.; Weiner, L.M.; Marks, J.D. Isolation of high-affinity monomeric human Anti-c-erbB-2 single chain Fv using affinity-driven selection. J. Mol. Biol. 1996, 255, 28–43. [Google Scholar] [CrossRef]
- Thevenet, P.; Shen, Y.; Maupetit, J.; Guyon, F.; Derreumaux, P.; Tuffery, P. PEP-FOLD: An updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012, 40, W288–W293. [Google Scholar] [CrossRef] [Green Version]
- Enshell-Seijffers, D.; Smelyanski, L.; Gershoni, J.M. The rational design of a ‘type 88’ genetically stable peptide display vector in the filamentous bacteriophage fd. Nucleic Acids Res. 2001, 29, E50. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Pruitt, K.D.; Schoch, C.L.; Sherry, S.T.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2021, 49, D92–D96. [Google Scholar] [CrossRef]
- Freund, N.T.; Enshell-Seijffers, D.; Gershoni, J.M. Phage display selection, analysis, and prediction of B cell epitopes. Curr. Protoc. Immunol. 2009, 86, 9.8.1–9.8.30. [Google Scholar] [CrossRef]
- Kanaya, S.; Kinouchi, M.; Abe, T.; Kudo, Y.; Yamada, Y.; Nishi, T.; Mori, H.; Ikemura, T. Analysis of codon usage diversity of bacterial genes with a self-organizing map (SOM): Characterization of horizontally transferred genes with emphasis on the E. coli O157 genome. Gene 2001, 276, 89–99. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Clone | V4 | V19 | V23 | V28 | Native |
|---|---|---|---|---|---|
| Titer (viruses/mL) | 6 × 1010 | 1.2 × 1012 | 1.5 × 1012 | 5 × 1011 | 1.26 × 1012 |
| Titer standard error | 0.47 | 1.16 | 0.72 | 1.74 | 0.52 |
| Relative titer | 1 | 20 | 25 | 8.3 | 21 |
| Relative protein concentration | 1 | 2.3 | 1.1 | 1.2 | 1.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sweedan, A.; Cohen, Y.; Yaron, S.; Bashouti, M.Y. Binding Capabilities of Different Genetically Engineered pVIII Proteins of the Filamentous M13/Fd Virus and Single-Walled Carbon Nanotubes. Nanomaterials 2022, 12, 398. https://doi.org/10.3390/nano12030398
Sweedan A, Cohen Y, Yaron S, Bashouti MY. Binding Capabilities of Different Genetically Engineered pVIII Proteins of the Filamentous M13/Fd Virus and Single-Walled Carbon Nanotubes. Nanomaterials. 2022; 12(3):398. https://doi.org/10.3390/nano12030398
Chicago/Turabian StyleSweedan, Amro, Yachin Cohen, Sima Yaron, and Muhammad Y. Bashouti. 2022. "Binding Capabilities of Different Genetically Engineered pVIII Proteins of the Filamentous M13/Fd Virus and Single-Walled Carbon Nanotubes" Nanomaterials 12, no. 3: 398. https://doi.org/10.3390/nano12030398