
Influence of Group-IVA Doping on Electronic and Optical Properties of ZnS Monolayer: A First-Principles Study

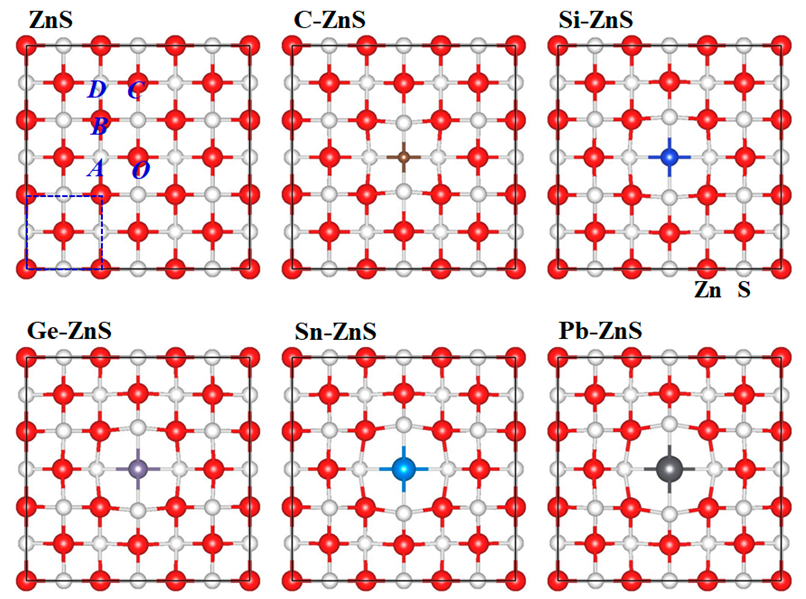{kind=link}
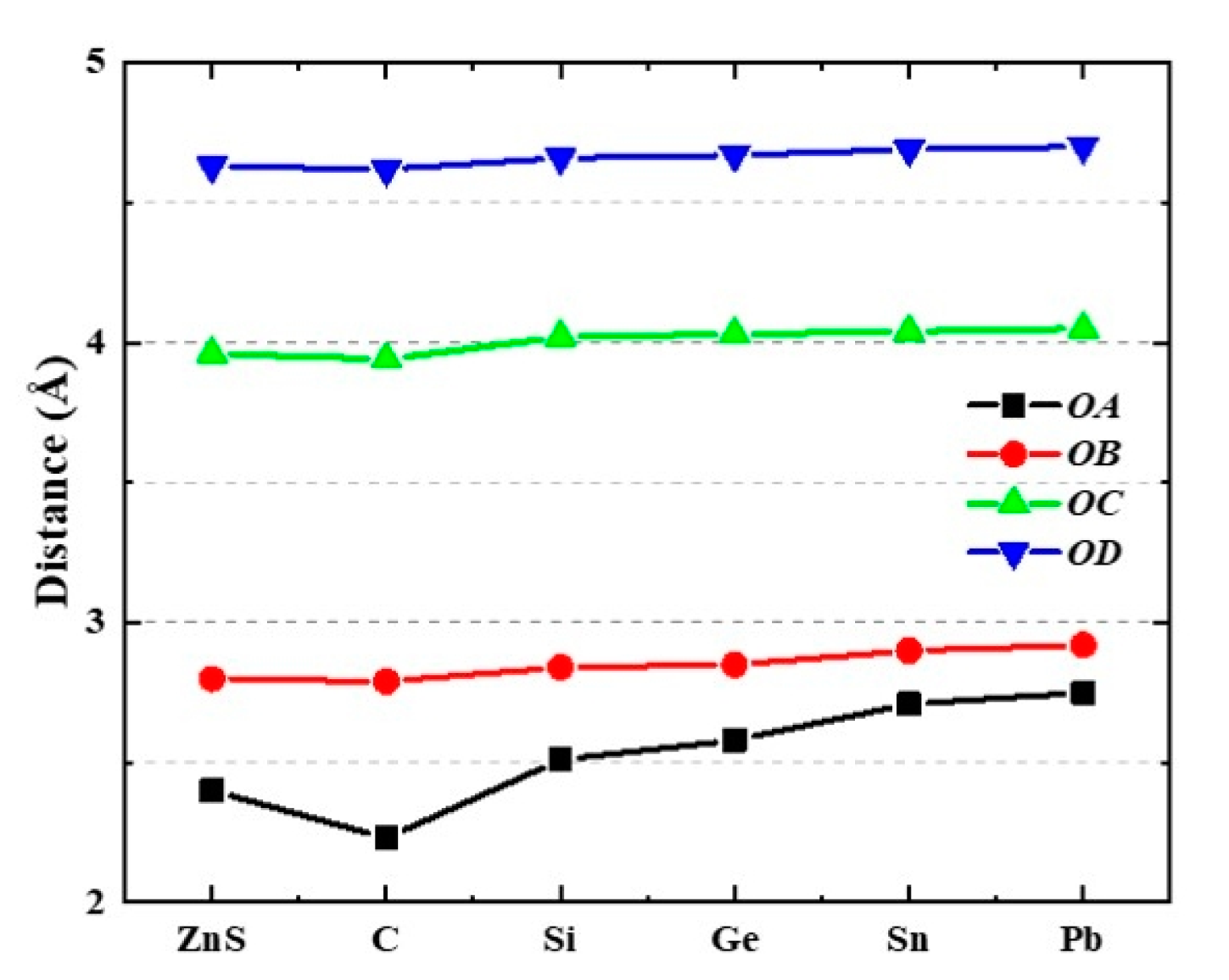{kind=link}
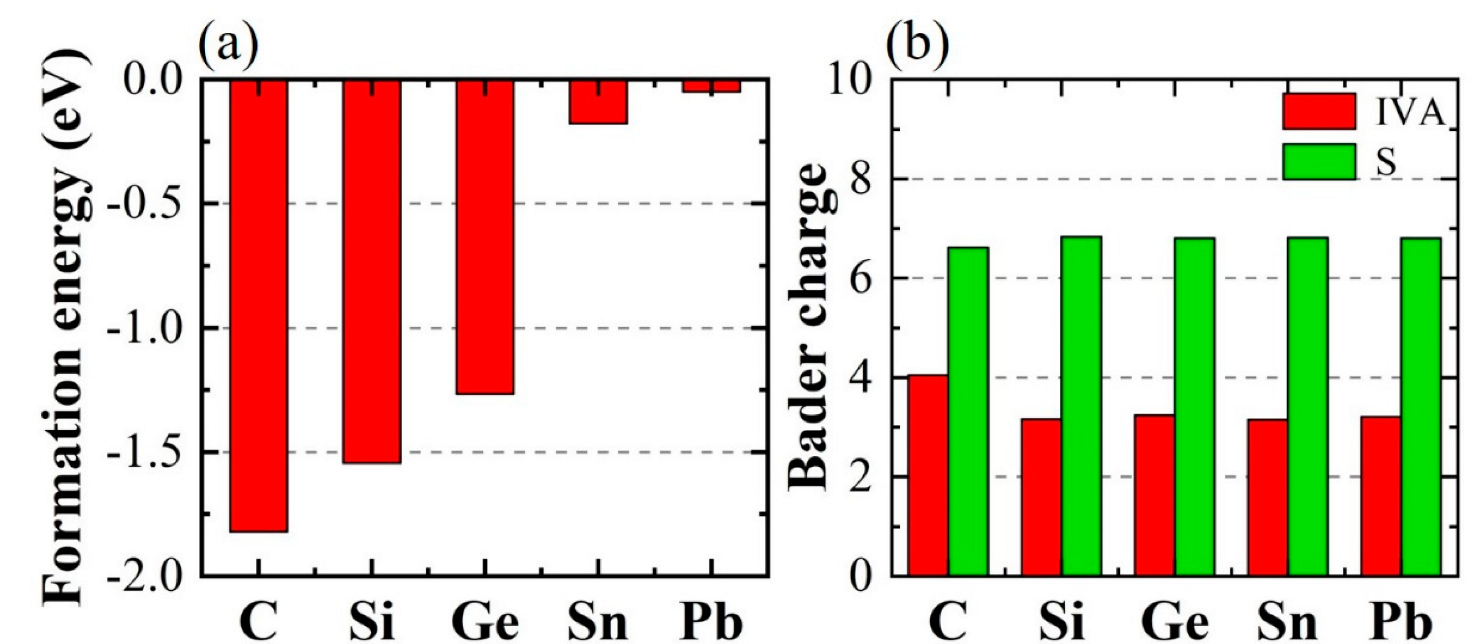{kind=link}
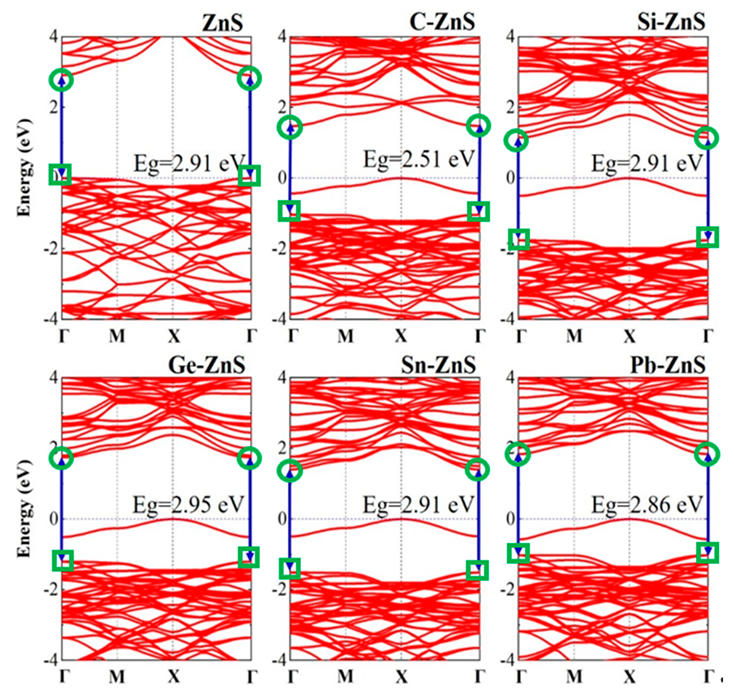{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Details
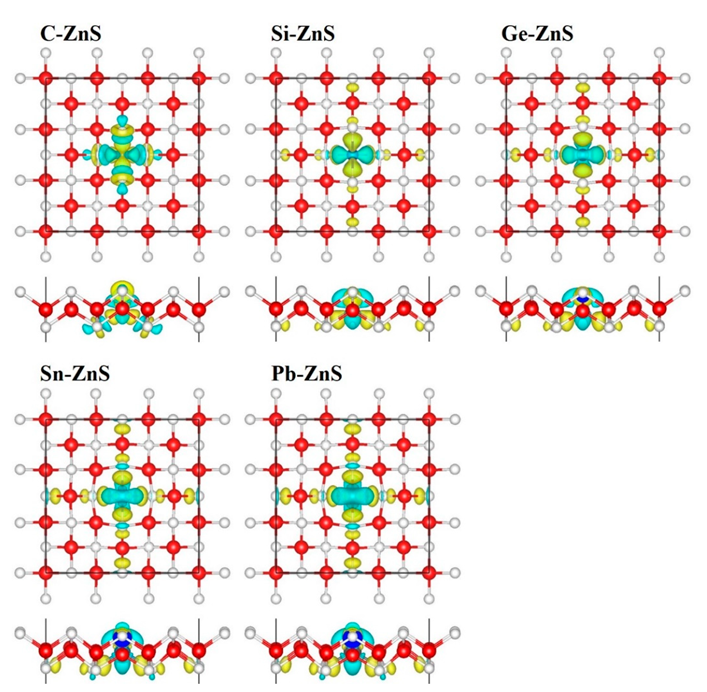
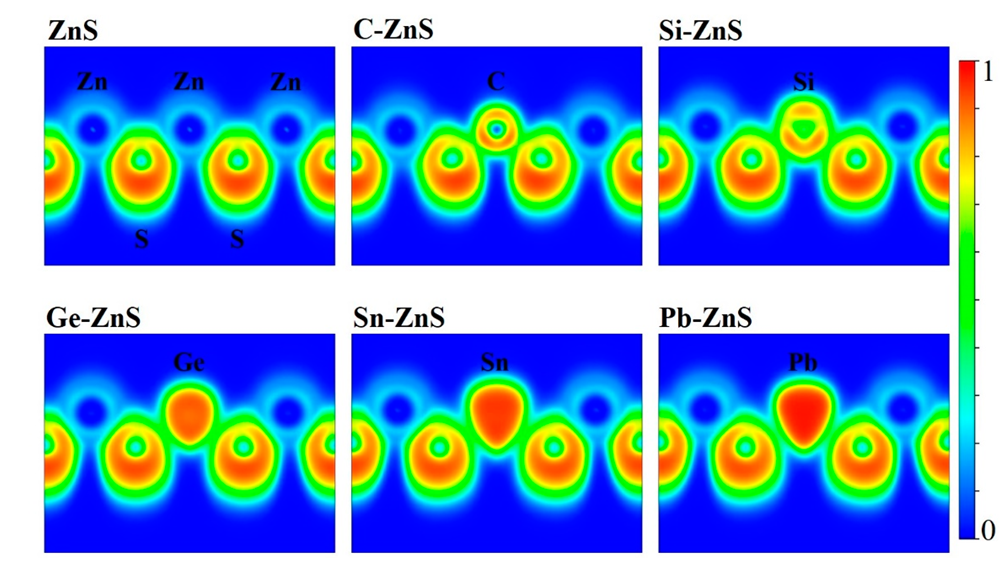
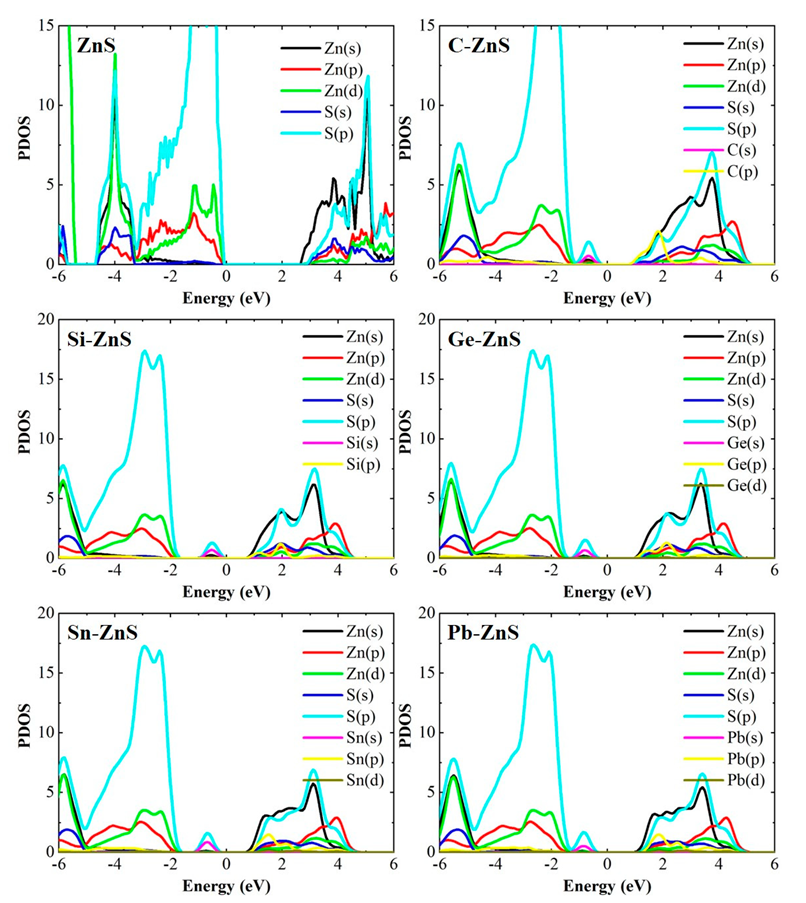
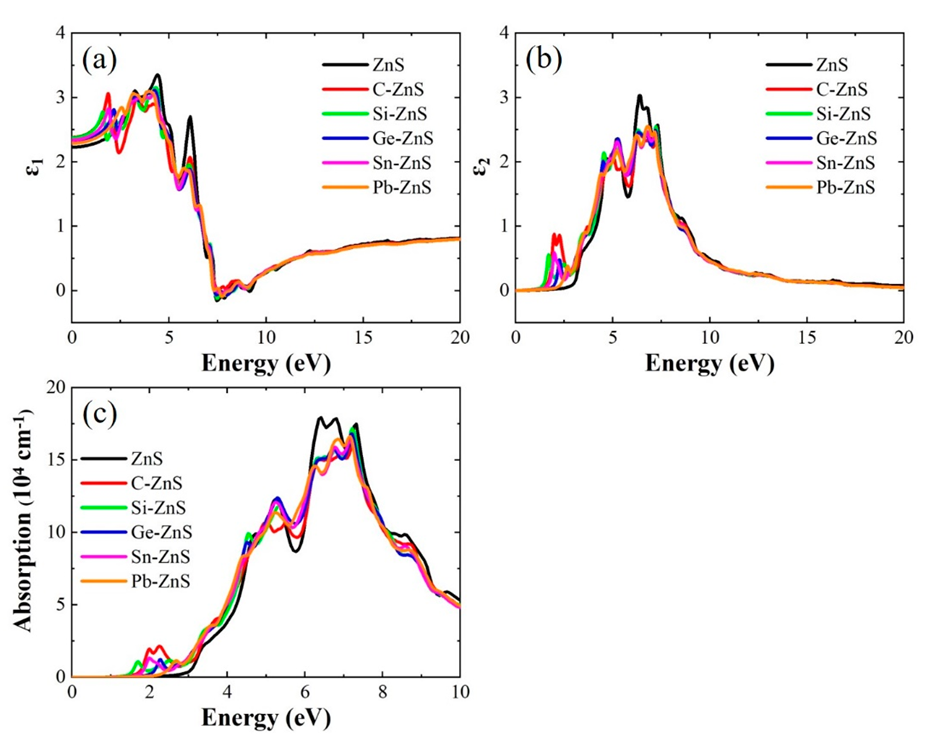
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.H.; Kalantar-Zadeh, K.; Kis, A.; Coleman, J.N.; Strano, M.S. Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nat. Nanotechnol. 2012, 7, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Kong, X.; Hu, Z.-X.; Yang, F.; Ji, W. High-mobility transport anisotropy and linear dichroism in few-layer black phosphorus. Nat. Commun. 2014, 5, 4475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Cai, X.; Yi, S.; Chen, J.; Dai, Y.; Niu, C.; Guo, Z.; Xie, M.; Liu, F.; Cho, J.-H.; et al. Multivalency-driven formation of Te-based monolayer materials: A combined first-principles and experimental study. Phys. Rev. Lett. 2017, 119, 106101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.K.; Hennig, R.G. Computational prediction of two-dimensional group-IV mono-chalcogenides. Appl. Phys. Lett. 2014, 105, 042103. [Google Scholar] [CrossRef]
- Kamal, C.; Chakrabarti, A.; Ezawa, M. Direct band gaps in group IV-VI monolayer materials: Binary counterparts of phosphorene. Phys. Rev. B 2016, 93, 125428. [Google Scholar] [CrossRef] [Green Version]
- Choi, W.; Choudhary, N.; Han, G.H.; Park, J.; Akinwande, D.; Lee, Y.H. Recent development of two-dimensional transition metal dichalcogenides and their applications. Mater. Today 2017, 20, 116–130. [Google Scholar] [CrossRef]
- Li, Q.; Meng, J.; Li, Z. Recent progress on Schottky sensors based on two-dimensional transition metal dichalcogenides. J. Mater. Chem. A 2022, 10, 8107–8128. [Google Scholar] [CrossRef]
- Yu, S.; Tang, J.; Wang, Y.; Xu, F.; Li, X.; Wang, X. Recent advances in two-dimensional ferromagnetism: Strain-, doping-, structural- and electric field-engineering toward spintronic applications. Sci. Technol. Adv. Mater. 2022, 23, 140–160. [Google Scholar] [CrossRef]
- Sun, Y.; Sun, Z.; Gao, S.; Cheng, H.; Liu, Q.; Piao, J.; Yao, T.; Wu, C.; Hu, S.; Wei, S.; et al. Fabrication of flexible and freestanding zinc chalcogenide single layers. Nat. Commun. 2012, 3, 1057. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, N.; Tang, Z.-R.; Xu, Y.-J. Graphene transforms wide band gap ZnS to a visible light photocatalyst. The new role of graphene as a macromolecular photosensitizer. ACS Nano 2012, 6, 9777–9789. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Zhai, T.; Gautam, U.K.; Li, L.; Wu, L.; Bando, Y.; Golberg, D. ZnS nanostructures: From synthesis to applications. Prog. Mater. Sci. 2011, 56, 175–287. [Google Scholar] [CrossRef]
- Hinuma, Y.; Grüneis, A.; Kresse, G.; Oba, F. Band alignment of semiconductors from density-functional theory and many-body perturbation theory. Phys. Rev. B 2014, 90, 155405. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.X.; Zapien, J.A.; Chen, W.W.; Lifshitz, Y.; Lee, S.T.; Meng, X.M. Lasing in ZnS nanowires grown on anodic aluminum oxide templates. Appl. Phys. Lett. 2004, 85, 2361–2363. [Google Scholar] [CrossRef]
- Huang, X.; Li, J.; Fu, H. The first covalent organic-inorganic networks of hybrid chalcogenides: Structures that may lead to a new type of quantum wells. J. Am. Chem. Soc. 2000, 122, 8789–8790. [Google Scholar] [CrossRef]
- Huang, X.; Li, J.; Zhang, Y.; Mascarenhas, A. From 1D chain to 3D network: Tuning hybrid II-VI nanostructures and their optical properties. J. Am. Chem. Soc. 2003, 125, 7049–7055. [Google Scholar] [CrossRef]
- Huang, X.; Li, J. From single to multiple atomic layers: A unique approach to the systematic tuning of structures and properties of inorganic-organic hybrid nanostructured semiconductors. J. Am. Chem. Soc. 2007, 129, 3157–3162. [Google Scholar] [CrossRef]
- Es-Smairi, A.; Fazouan, N.; Atmani, E.H.; Khuili, M.; Maskar, E. Al doping effect on optoelectronic and thermoelectric properties of h-ZnS monolayer: A DFT approach. Appl. Phys. A 2021, 127, 698. [Google Scholar] [CrossRef]
- Lashgari, H.; Boochani, A.; Shekaari, A.; Solaymani, S.; Sartipi, E.; Mendi, R.T. Electronic and optical properties of 2D graphene-like ZnS: DFT calculations. Appl. Surf. Sci. 2016, 369, 76–81. [Google Scholar] [CrossRef]
- Krainara, N.; Limtrakul, J.; Illas, F.; Bromley, S.T. Structural and electronic bistability in ZnS single sheets and single-walled nanotubes. Phys. Rev. B 2011, 83, 233305. [Google Scholar] [CrossRef]
- Chaurasiya, R.; Dixit, A.; Pandey, R. Strain-driven thermodynamic stability and electronic transitions in ZnX (X = O, S, Se, and Te) monolayers. J. Appl. Phys. 2019, 125, 082540. [Google Scholar] [CrossRef]
- Jafari, M.; Alvani, K. Effect of doping chromium on electronic and magnetic properties of ZnS monolayer: A DFT study. Mater. Res. Express 2019, 6, 0850b5. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, L.; Xiao, B.; Cheng, Y.; Zhang, J. The structure and electronic properties of crimson phosphorus. Appl. Phys. Lett. 2019, 115, 163101. [Google Scholar] [CrossRef]
- Hogan, C.; Lechifflart, P.; Brozzesi, S.; Voronovich-Solonevich, S.; Melnikov, A.; Flammini, R.; Sanna, S.; Holtgrewe, K. Theoretical study of stability, epitaxial formation, and phase transformations of two-dimensional pnictogen allotropes. Phys. Rev. B 2021, 104, 245421. [Google Scholar] [CrossRef]
- Tong, C.-J.; Zhang, H.; Zhang, Y.-N.; Liu, H.; Liu, L.-M. New manifold two-dimensional single-layer structures of zinc-blende compounds. J. Mater. Chem. A 2014, 2, 17971–17978. [Google Scholar] [CrossRef]
- Unsal, E.; Senger, R.T.; Sahin, H. Stable monolayer α-phase of CdTe: Strain-dependent properties. J. Mater. Chem. C 2017, 5, 12249–12255. [Google Scholar] [CrossRef] [Green Version]
- Naseri, M.; Bafekry, A.; Faraji, M.; Hoat, D.M.; Fadlallah, M.M.; Ghergherehchi, M.; Sabbaghi, N.; Gogova, D. Two-dimensional buckled tetragonal cadmium chalcogenides including CdS, CdSe, and CdTe monolayers as photo-catalysts for water splitting. Phys. Chem. Chem. Phys. 2021, 23, 12226–12232. [Google Scholar] [CrossRef]
- Li, L.; Li, P.; Lu, N.; Dai, J.; Zeng, X.C. Simulation evidence of hexagonal-to-tetragonal ZnSe structure transition: A monolayer material with a wide-range tunable direct bandgap. Adv. Sci. 2015, 2, 1500290. [Google Scholar] [CrossRef]
- Liu, B.; Su, W.-S.; Wu, B.-R. A first-principles study of electronic and optical properties of the tetragonal phase of monolayer ZnS modulated by biaxial strain. RSC Adv. 2022, 12, 6166–6173. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, X. Alloy engineering of electronic and optical properties of tetragonal monolayer zinc chalcogenides. J. Alloys Compd. 2017, 695, 1392–1396. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple (vol 77, pg 3865, 1996). Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188. [Google Scholar] [CrossRef]
- Patel, M.J.; Raval, D.; Gupta, S.K.; Gajjar, P.N. First-principles study of Mn-doped and Nb-doped CsPbCl3 monolayers as an absorber layer in solar cells. J. Phys. Chem. Lett. 2021, 12, 7319–7327. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, B.; Su, W.-S.; Wu, B.-R. Influence of Group-IVA Doping on Electronic and Optical Properties of ZnS Monolayer: A First-Principles Study. Nanomaterials 2022, 12, 3898. https://doi.org/10.3390/nano12213898
Liu B, Su W-S, Wu B-R. Influence of Group-IVA Doping on Electronic and Optical Properties of ZnS Monolayer: A First-Principles Study. Nanomaterials. 2022; 12(21):3898. https://doi.org/10.3390/nano12213898
Chicago/Turabian StyleLiu, Bin, Wan-Sheng Su, and Bi-Ru Wu. 2022. "Influence of Group-IVA Doping on Electronic and Optical Properties of ZnS Monolayer: A First-Principles Study" Nanomaterials 12, no. 21: 3898. https://doi.org/10.3390/nano12213898