
First-Principles Molecular Dynamics Simulations on Water–Solid Interface Behavior of H2O-Based Atomic Layer Deposition of Zirconium Dioxide

Abstract
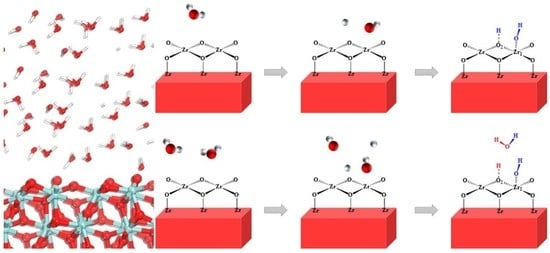
:
1. Introduction
2. Computational Details
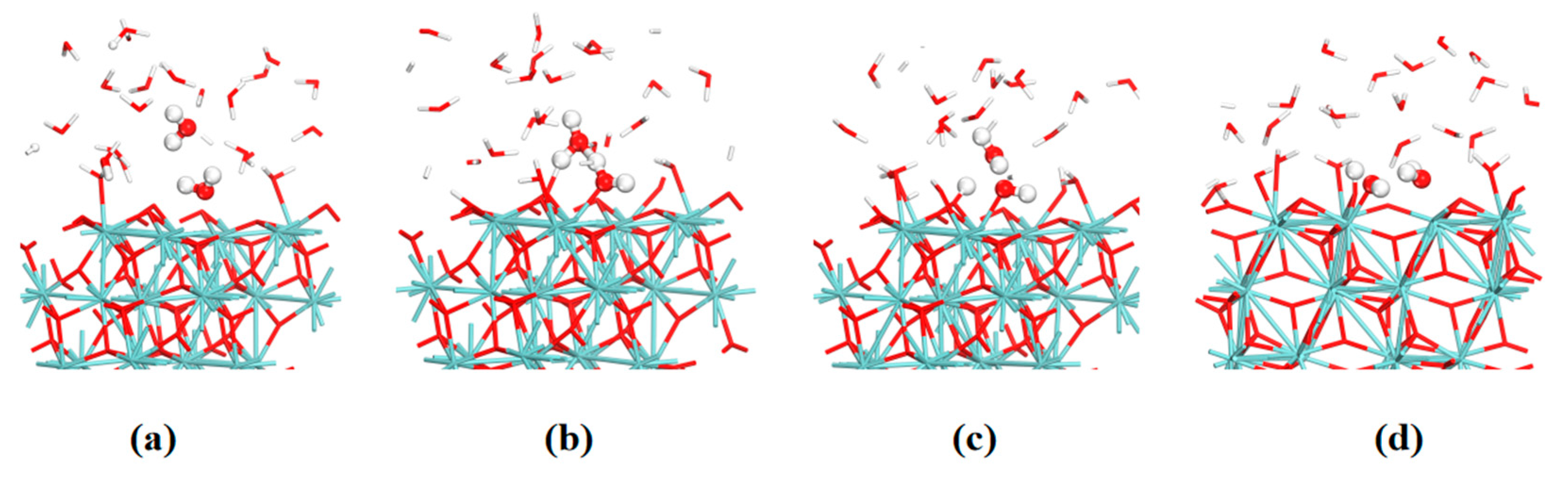
3. Results and Discussion
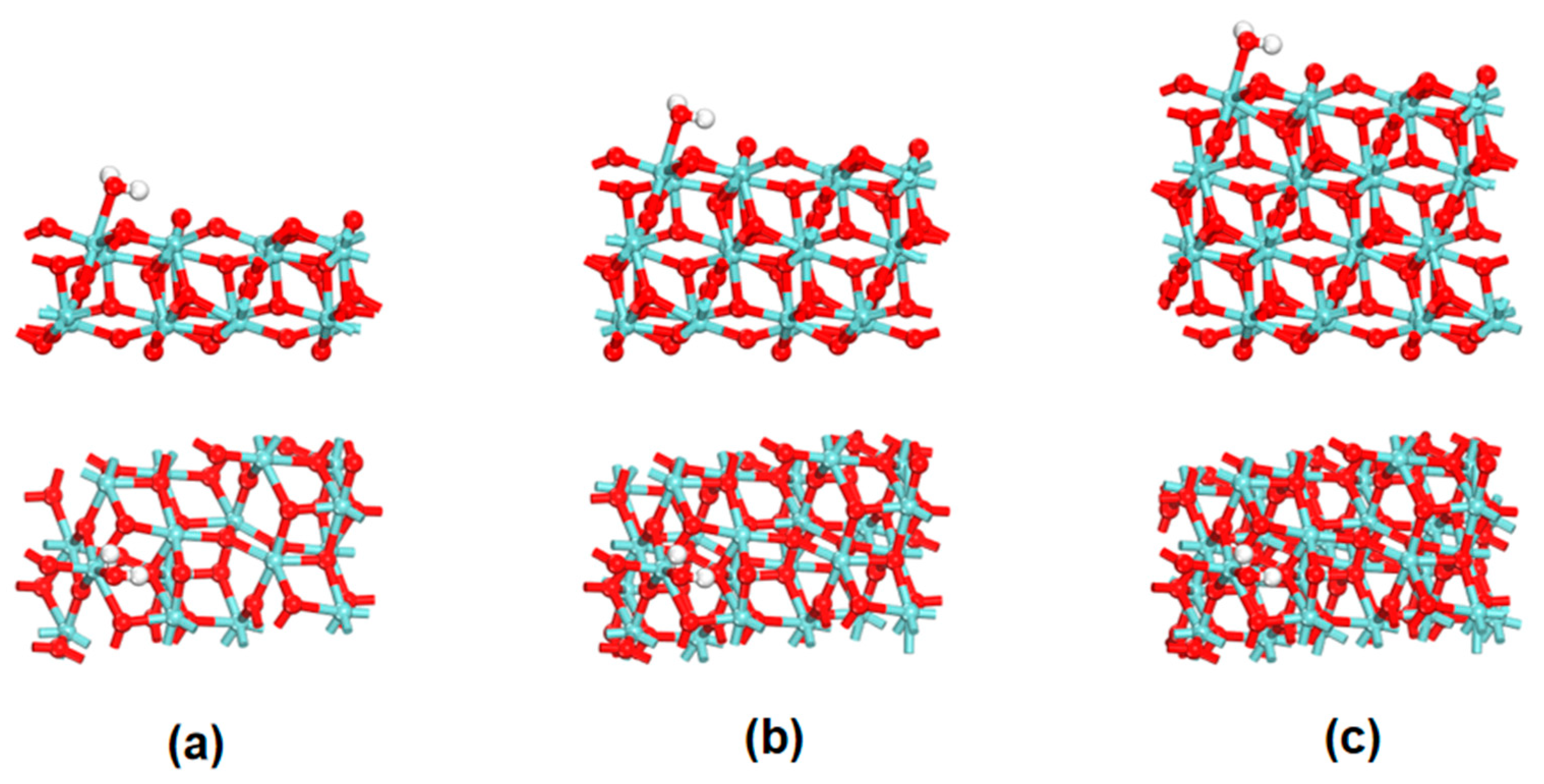
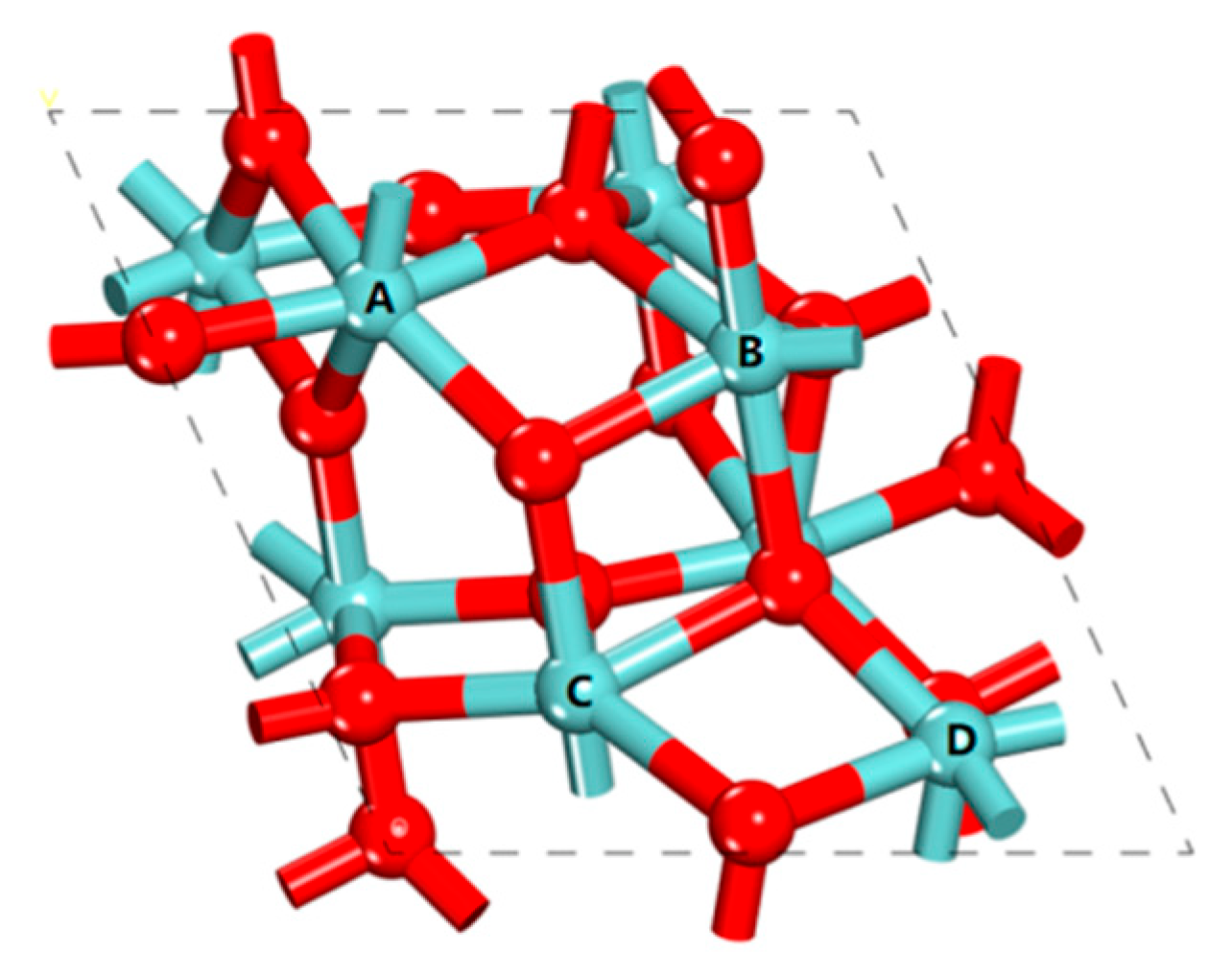
3.1. Surface Model
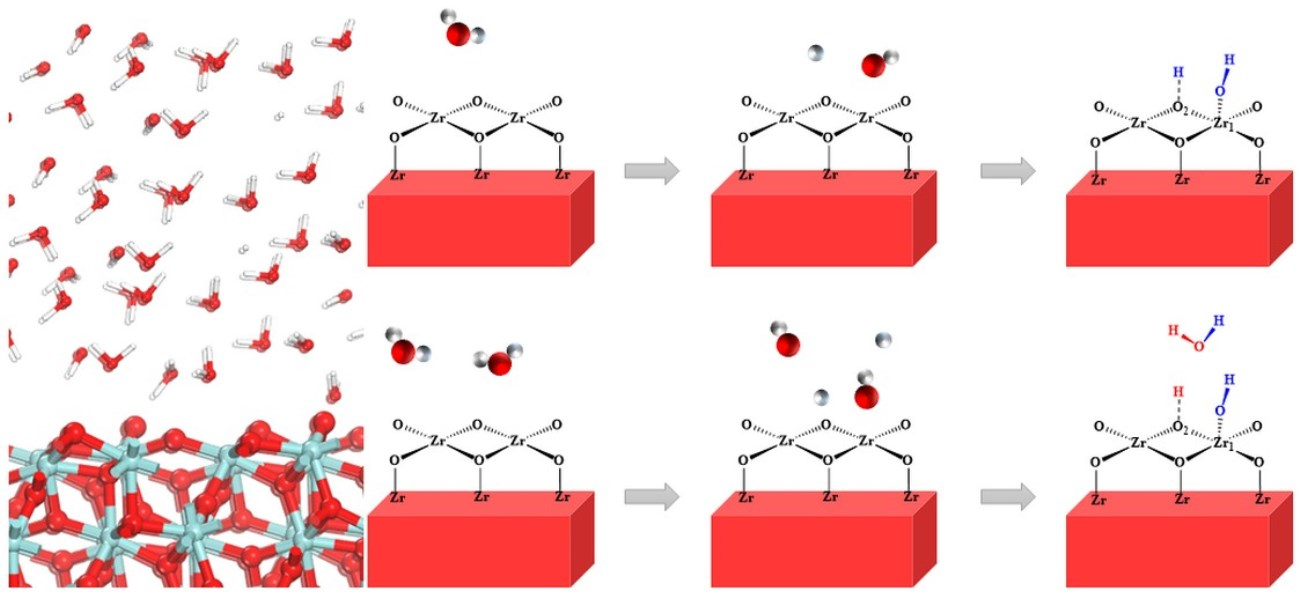
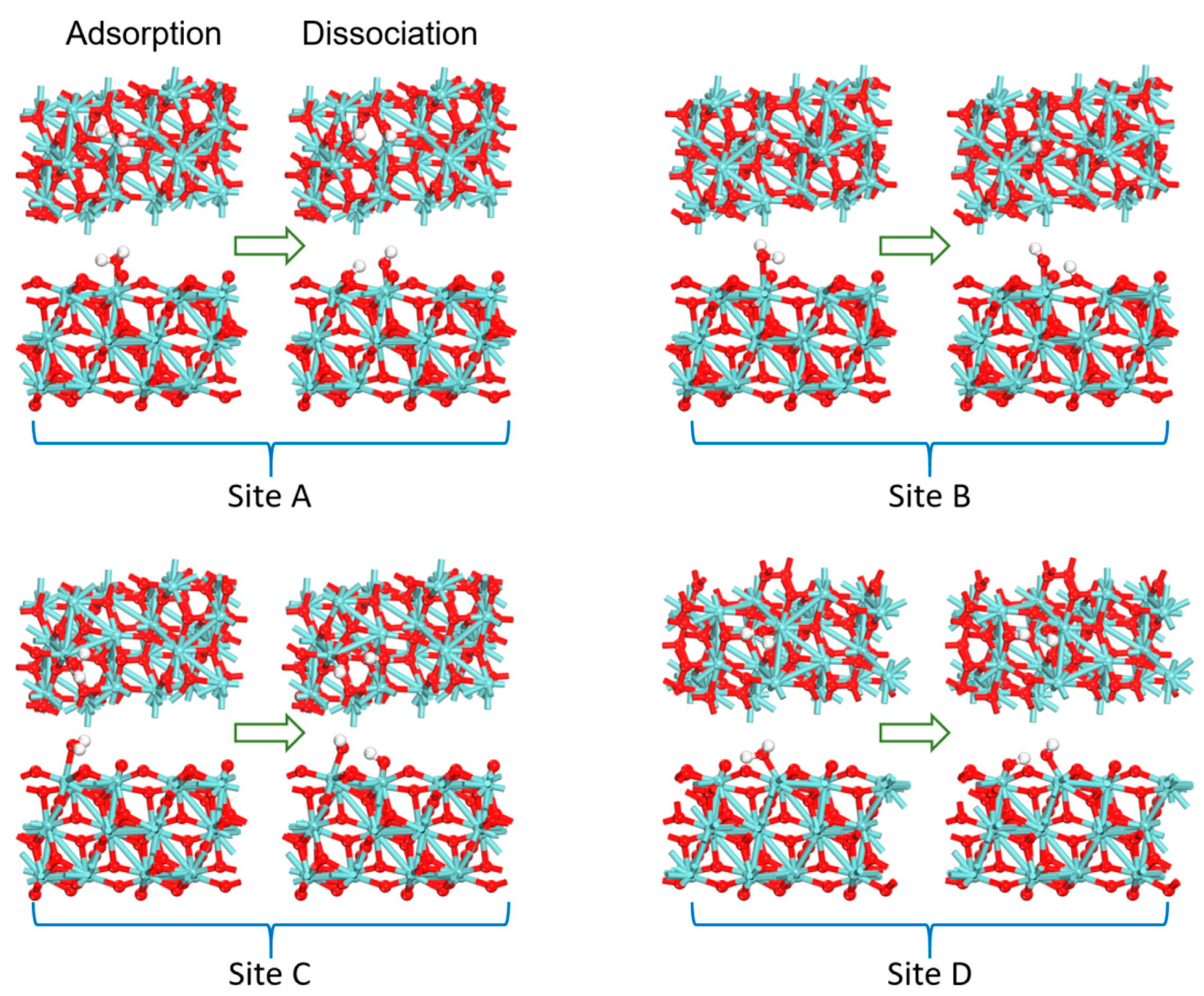
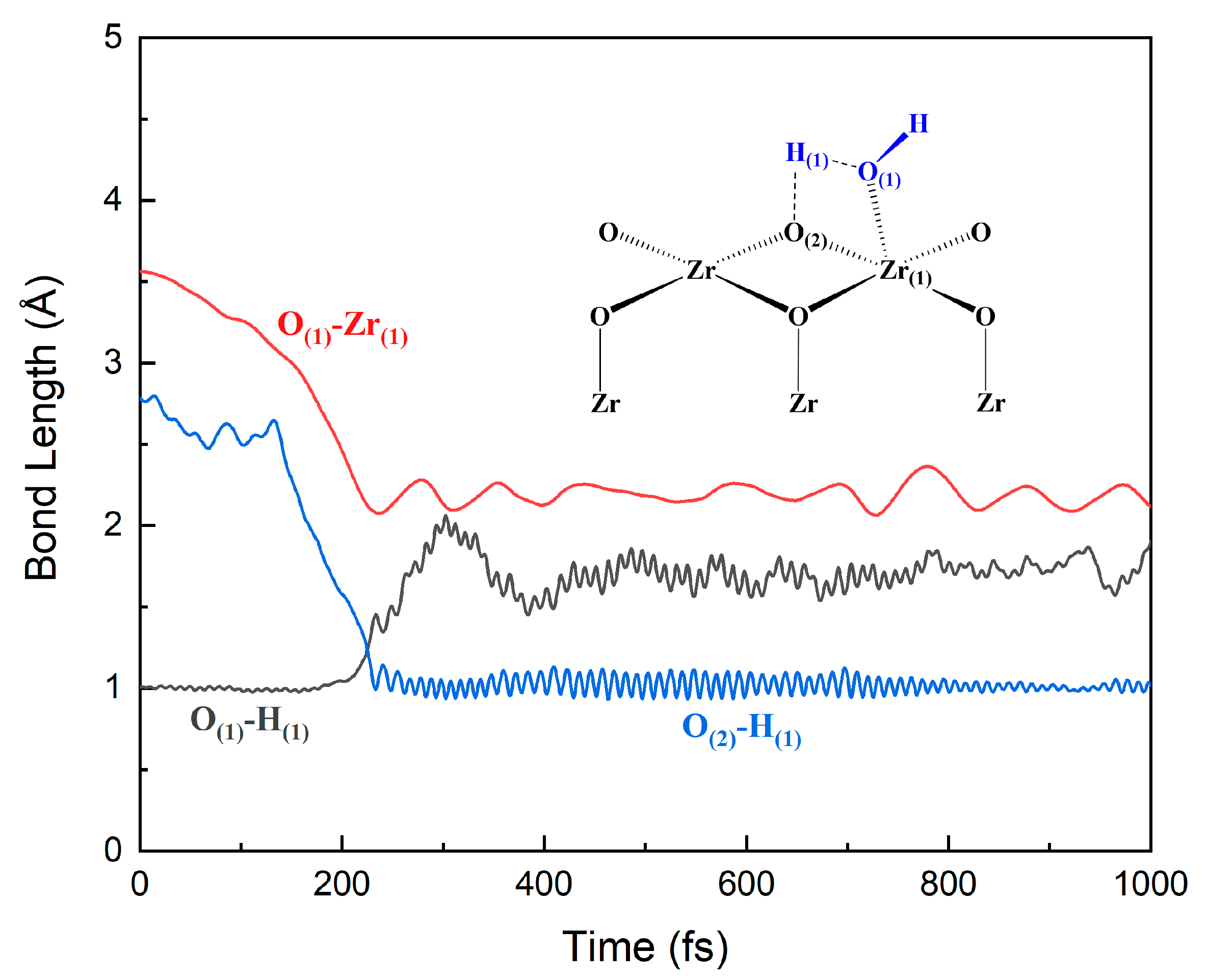
3.2. Adsorption and Dissociation of a Water Molecule on the ZrO2 (111) Surface
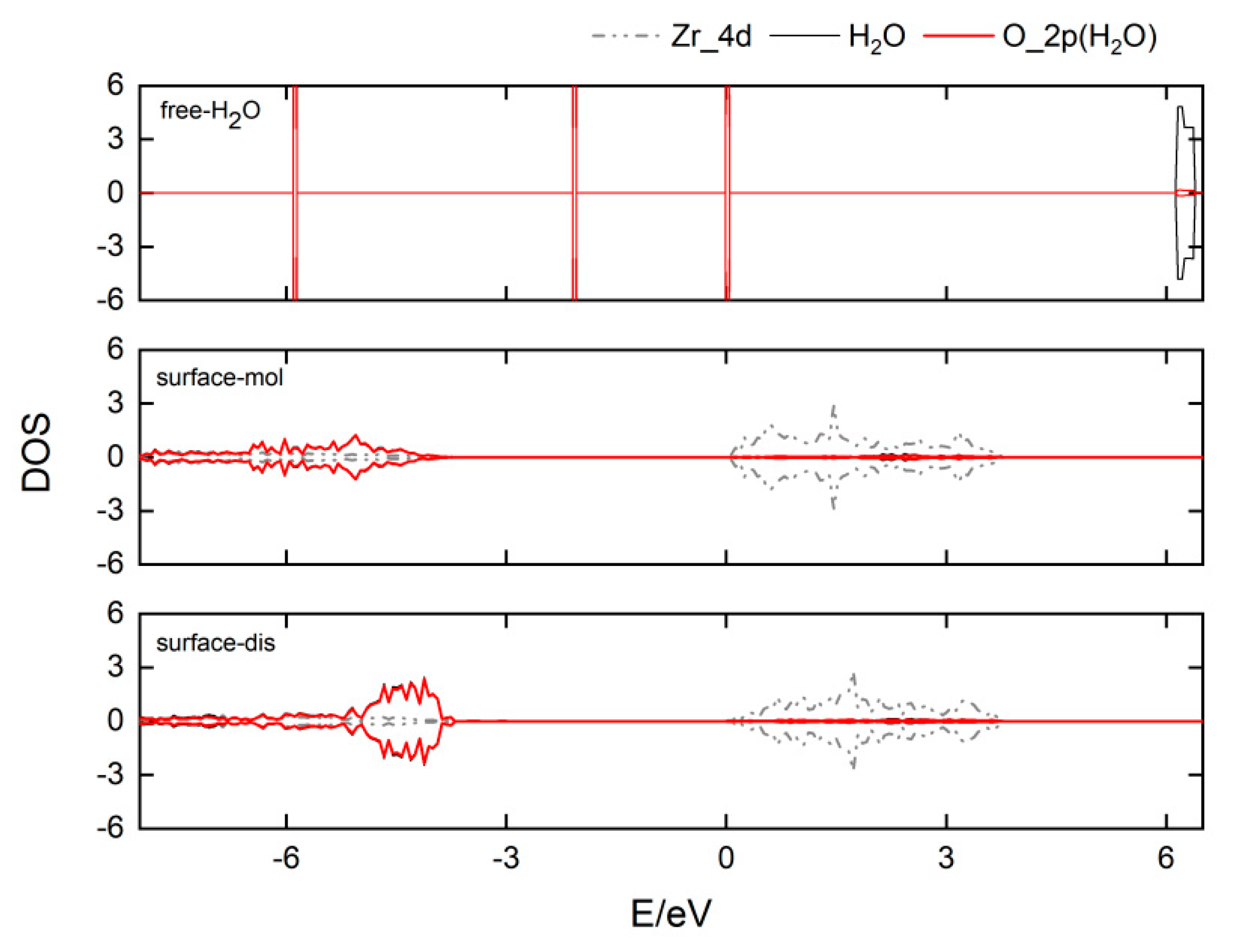
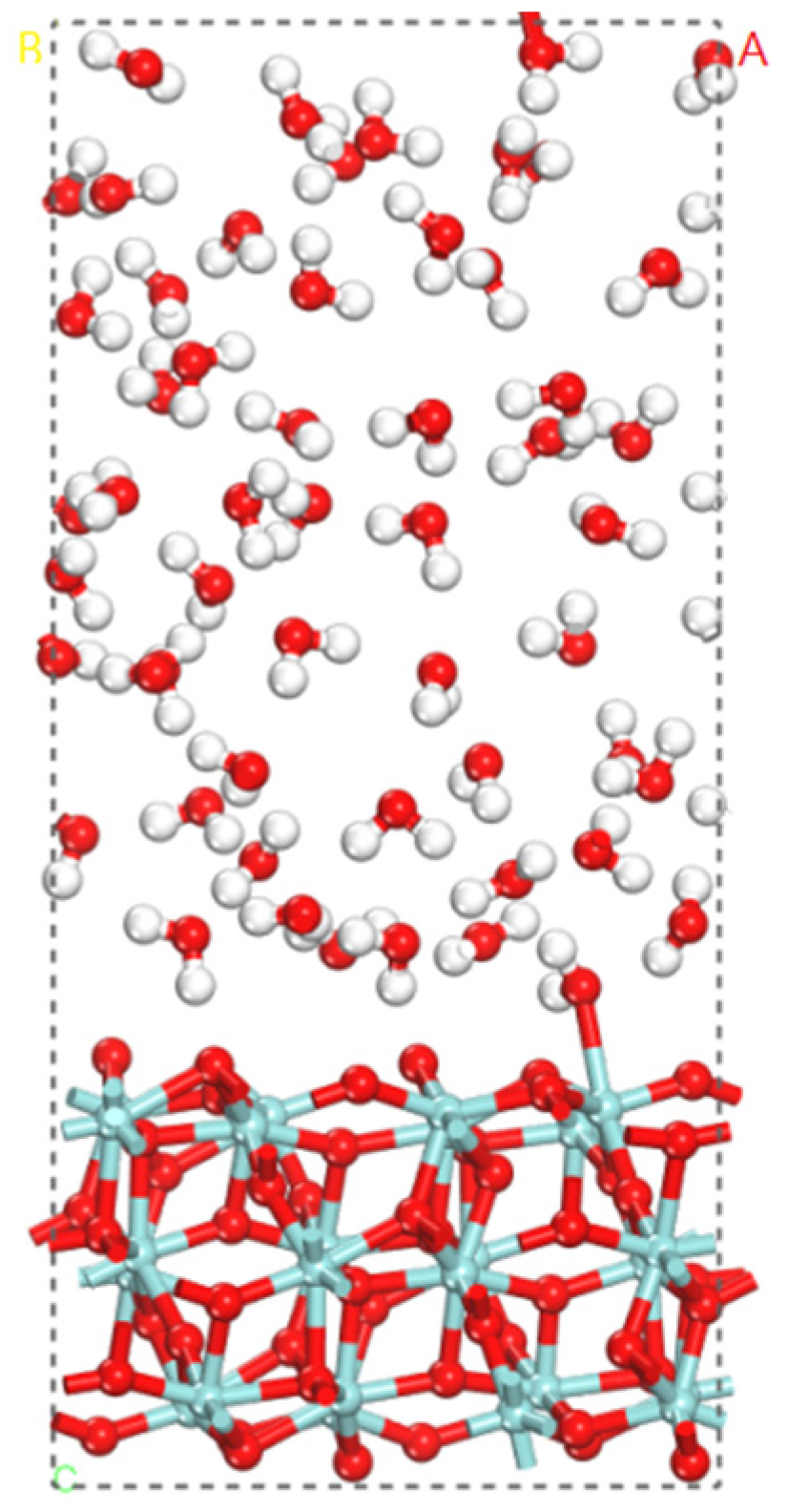
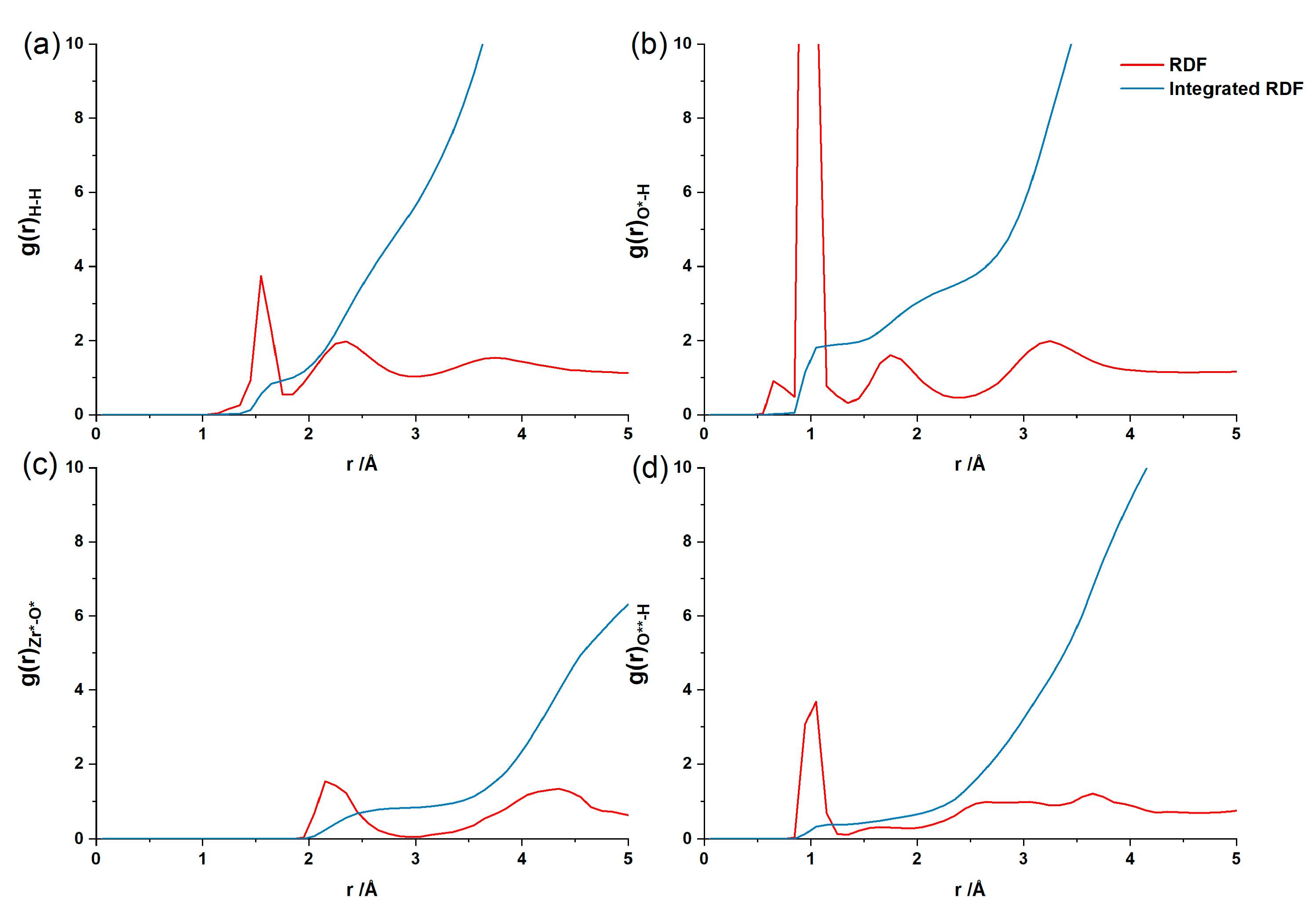
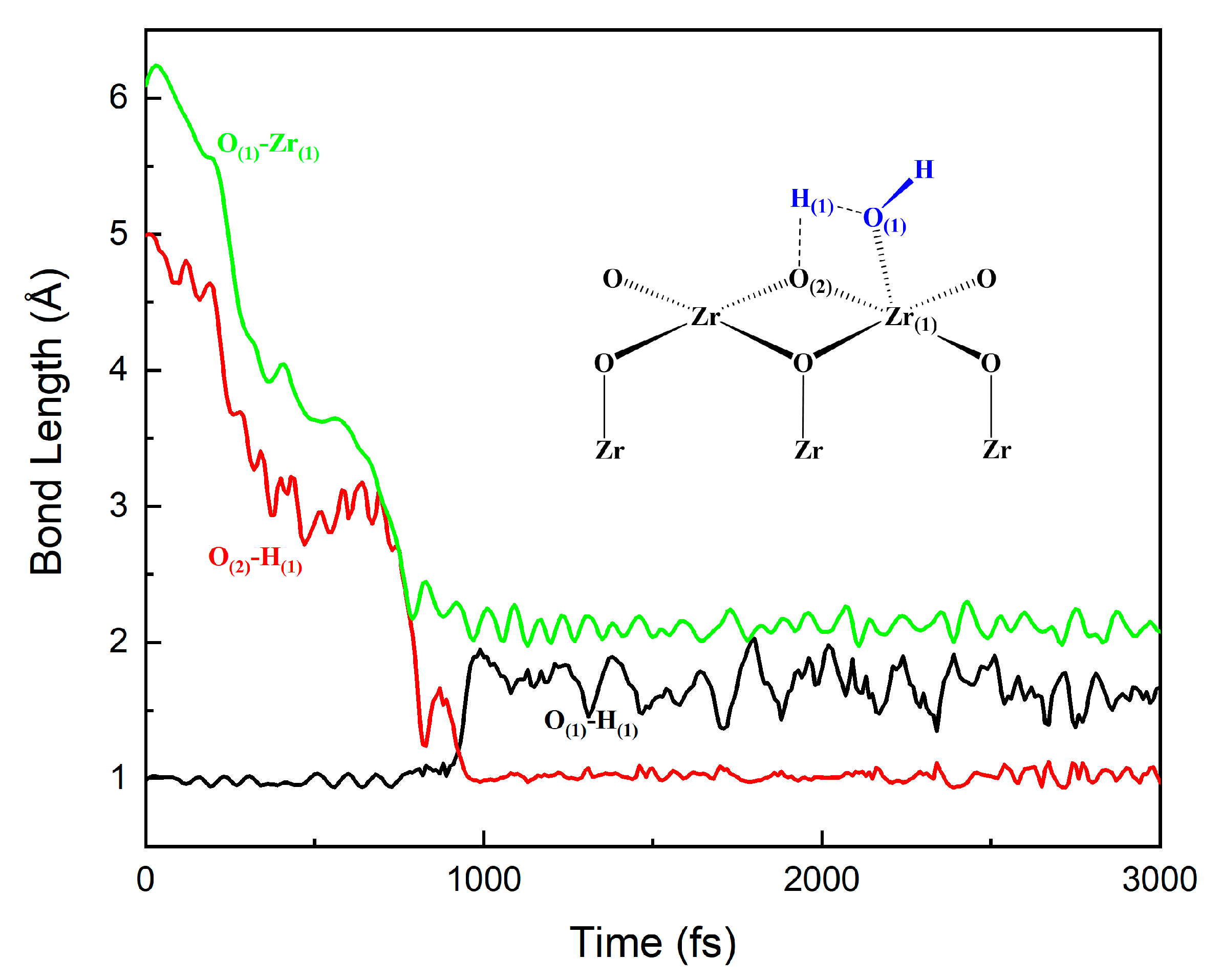
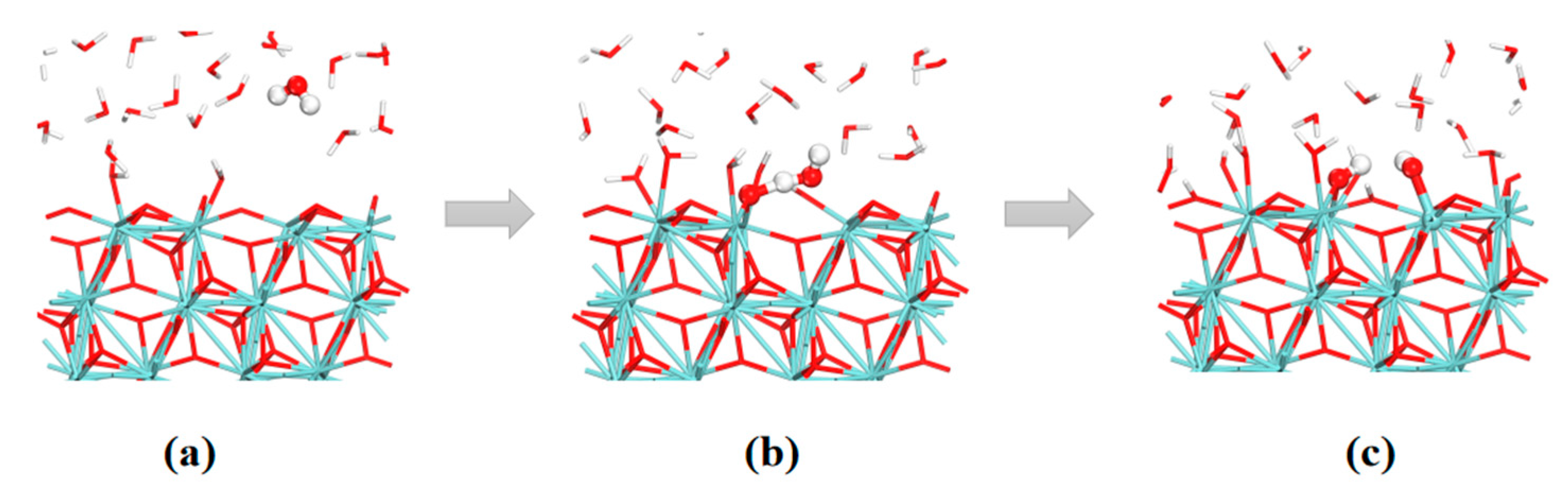
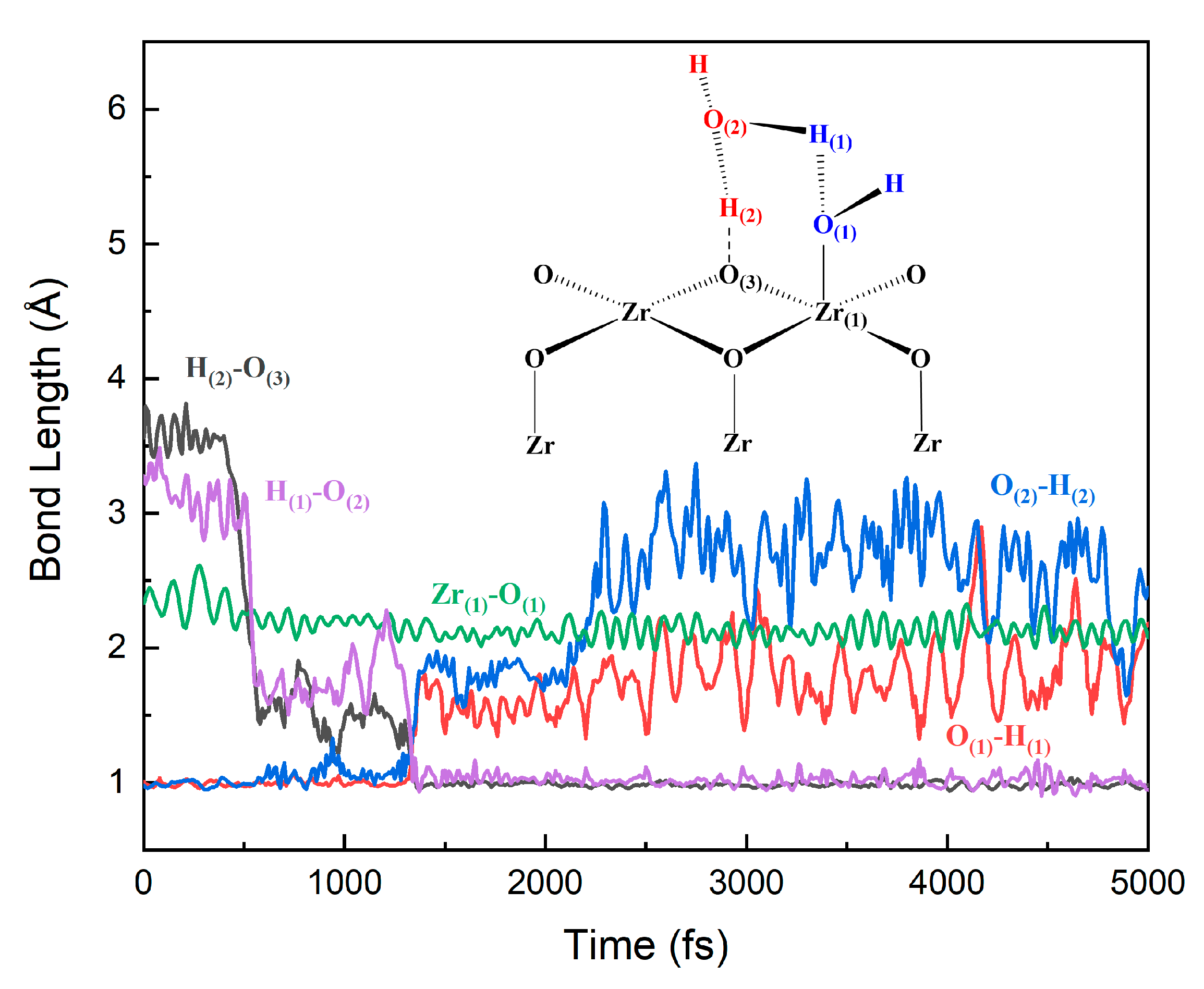
3.3. The Water–Solid Interface Reaction of ZrO2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Zaera, F. The surface chemistry of thin film atomic layer deposition (ALD) processes for electronic device manufacturing. J. Mater. Chem. 2008, 18, 3521–3526. [Google Scholar] [CrossRef]
- Rolison, D.R.; Long, J.W.; Lytle, J.C.; Fischer, A.E.; Rhodes, C.P.; McEvoy, T.M.; Bourg, M.E.; Lubers, A.M. Multifunctional 3D nanoarchitectures for energy storage and conversion. Chem. Soc. Rev. 2008, 38, 226–252. [Google Scholar] [CrossRef]
- Marichy, C.; Bechelany, M.; Pinna, N. Atomic Layer Deposition of Nanostructured Materials for Energy and Environmental Applications. Adv. Mater. 2012, 24, 1017–1032. [Google Scholar] [CrossRef]
- O’Neill, B.J.; Jackson, D.H.K.; Lee, J.; Canlas, C.; Stair, P.C.; Marshall, C.L.; Elam, J.W.; Kuech, T.F.; Dumesic, J.A.; Huber, G.W. Catalyst Design with Atomic Layer Deposition. ACS Catal. 2015, 5, 1804–1825. [Google Scholar] [CrossRef] [Green Version]
- Palmstrom, A.F.; Santra, P.K.; Bent, S.F. Atomic layer deposition in nanostructured photovoltaics: Tuning optical, electronic and surface properties. Nanoscale 2015, 7, 12266–12283. [Google Scholar] [CrossRef]
- Asundi, A.S.; Raiford, J.A.; Bent, S.F. Opportunities for Atomic Layer Deposition in Emerging Energy Technologies. ACS Energy Lett. 2019, 4, 908–925. [Google Scholar] [CrossRef]
- Gaskell, J.M.; Jones, A.C.; Chalker, P.R.; Werner, M.; Aspinall, H.C.; Taylor, S.; Taechakumput, P.; Heys, P.N. Deposition of lanthanum zirconium dioxide high-κ films by liquid injection ALD and MOCVD. Chem. Vap. Depos. 2007, 13, 684–690. [Google Scholar] [CrossRef]
- Dezelah, C.L., IV; Niinistö, J.; Kukli, K.; Munnik, F.; Lu, J.; Ritala, M.; Leskelä, M.; Niinistö, L. The atomic layer deposition of HfO2 and ZrO2 using advanced metallocene precursors and H2O as the oxygen source. Chem. Vap. Depos. 2008, 14, 358–365. [Google Scholar] [CrossRef]
- Kaipio, M.; Blanquart, T.; Banerjee, M.; Xu, K.; Niinistö, J.; Longo, V.; Mizohata, K.; Devi, A.; Ritala, M.; Leskelä, M. Atomic layer deposition of TiO2 and ZrO2 thin films using heteroleptic guanidinate precursors. Chem. Vap. Depos. 2014, 20, 209–216. [Google Scholar] [CrossRef]
- Jung, J.-S.; Lee, S.-K.; Hong, C.-S.; Shin, J.-H.; Kim, J.-M.; Kang, J.-G. Atomic layer deposition of ZrO2 thin film on Si(100) using {η5:η1-Cp(CH2)3NMe}Zr(NMe2)2/O3 as precursors. Thin Solid Film. 2015, 589, 831–837. [Google Scholar] [CrossRef]
- Kanomata, K.; Tokoro, K.; Imai, T.; Pansila, P.; Miura, M.; Ahmmad, B.; Kubota, S.; Hirahara, K.; Hirose, F. Room-temperature atomic layer deposition of ZrO2 using tetrakis(ethylmethylamino)zirconium and plasma-excited humidified argon. Appl. Surf. Sci. 2016, 387, 497–502. [Google Scholar] [CrossRef] [Green Version]
- Mahuli, N.; Cavanagh, A.S.; George, S.M. Atomic layer deposition of hafnium and zirconium oxyfluoride thin films. J. Vac. Sci. Technol. A Vac. Surf. Film. 2021, 39, 022403. [Google Scholar] [CrossRef]
- Xu, W.; Lemaire, P.C.; Sharma, K.; Gasvoda, R.J.; Hausmann, D.M.; Agarwal, S. Mechanism for growth initiation on aminosilane-functionalized SiO2 during area-selective atomic layer deposition of ZrO2. J. Vac. Sci. Technol. A Vac. Surf. Film. 2021, 39, 032402. [Google Scholar] [CrossRef]
- Walter, E.J.; Lewis, S.P.; Rappe, A.M. First principles study of carbon monoxide adsorption on zirconia-supported copper. Surf. Sci. 2001, 495, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Grau-Crespo, R.; Hernández, N.C.; Sanz, J.F.; de Leeuw, N.H. Theoretical Investigation of the Deposition of Cu, Ag, and Au Atoms on the ZrO2(111) Surface. J. Phys. Chem. C 2007, 111, 10448–10454. [Google Scholar] [CrossRef]
- Gennard, S.; Corà, F.; Catlow, C.R.A. Comparison of the Bulk and Surface Properties of Ceria and Zirconia by ab Initio Investigations. J. Phys. Chem. B 1999, 103, 10158–10170. [Google Scholar] [CrossRef]
- Jung, C.; Ishimoto, R.; Tsuboi, H.; Koyama, M.; Endou, A.; Kubo, M.; Del Carpio, C.A.; Miyamoto, A. Interfacial properties of ZrO2 supported precious metal catalysts: A density functional study. Appl. Catal. A Gen. 2006, 305, 102–109. [Google Scholar] [CrossRef]
- Jung, K.T.; Bell, A.T. The effects of synthesis and pretreatment conditions on the bulk structure and surface properties of zirconia. J. Mol. Catal. A Chem. 2000, 163, 27–42. [Google Scholar] [CrossRef]
- Camellone, M.F.; Ribeiro, F.N.; Szabová, L.; Tateyama, Y.; Fabris, S. Catalytic Proton Dynamics at the Water/Solid Interface of Ceria-Supported Pt Clusters. J. Am. Chem. Soc. 2016, 138, 11560–11567. [Google Scholar] [CrossRef]
- Bu, Y.; Cui, T.; Zhao, M.; Zheng, W.; Gao, W.; Jiang, Q. Evolution of Water Structures on Stepped Platinum Surfaces. J. Phys. Chem. C 2018, 122, 604–611. [Google Scholar] [CrossRef]
- Zhang, X.; Hu, X.; Lei, H.; Hu, J.; Zhang, Y. Mechanical force-induced polymerization and depolymerization of F-actin at water/solid interfaces. Nanoscale 2016, 8, 6008–6013. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Cheng, F.; Binks, B.P.; Yang, H. pH-responsive gas–water–solid interface for multiphase catalysis. J. Am. Chem. Soc. 2015, 137, 15015–15025. [Google Scholar] [CrossRef] [PubMed]
- Shaat, M. Viscosity of Water Interfaces with Hydrophobic Nanopores: Application to Water Flow in Carbon Nanotubes. Langmuir 2017, 33, 12814–12819. [Google Scholar] [CrossRef] [PubMed]
- Yali, Y.; Chunhai, L.; Juan, H.; Yi, L.; Wenkai, C. Density functional theory study of H2O adsorption and decomposition. Chin. J. Catal. 2009, 30, 328. [Google Scholar]
- Iskandarova, I.; Knizhnik, A.; Rykova, E.; Bagatur’Yants, A.; Potapkin, B.; Korkin, A. First-principle investigation of the hydroxylation of zirconia and hafnia surfaces. Microelectron. Eng. 2003, 69, 587–593. [Google Scholar] [CrossRef]
- Korhonen, S.T.; Calatayud, M.; Krause, A.O.I. Stability of Hydroxylated (1‾11) and (1‾01) Surfaces of Monoclinic Zirconia: A Combined Study by DFT and Infrared Spectroscopy. J. Phys. Chem. C 2008, 112, 6469–6476. [Google Scholar] [CrossRef]
- Korhonen, S.T.; Calatayud, M.; Krause, A.O.I. Structure and Stability of Formates and Carbonates on Monoclinic Zirconia: A Combined Study by Density Functional Theory and Infrared Spectroscopy. J. Phys. Chem. C 2008, 112, 16096–16102. [Google Scholar] [CrossRef]
- Piskorz, W.; Gryboś, J.; Zasada, F.; Cristol, S.; Paul, J.-F.; Adamski, A.; Sojka, Z. Periodic DFT and Atomistic Thermodynamic Modeling of the Surface Hydration Equilibria and Morphology of Monoclinic ZrO2 Nanocrystals. J. Phys. Chem. C 2011, 115, 24274–24286. [Google Scholar] [CrossRef]
- Xia, G.-J.; Wang, Y.-G. Dynamic Simulation on Surface Hydration and Dehydration of Monoclinic Zirconia. Chin. J. Chem. Phys. 2022, 35, 629–638. [Google Scholar] [CrossRef]
- Cerrato, G.; Bordiga, S.; Barbera, S.; Morterra, C. Surface characterization of monoclinic ZrO2: I. Morphol-ogy, FTIR spectral features, and computer modelling. Appl. Surf. Sci. 1997, 115, 53–65. [Google Scholar] [CrossRef]
- Whittle, K.; Lumpkin, G.; Ashbrook, S. Neutron diffraction and MAS NMR of Cesium Tungstate defect pyrochlores. J. Solid State Chem. 2006, 179, 512–521. [Google Scholar] [CrossRef]
- Vienna Ab initio Simulation Package (VASP) 6.2. Available online: https://www.vasp.at/ (accessed on 12 October 2021).
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Li, L.; Li, Y.; Guo, X.; Zhang, Y.-F.; Chen, W.-K. Adsorption and dissociation of water on HfO2 (111) and (110) surfaces. Acta Phys.-Chim. Sin. 2013, 29, 937–945. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Layer | dO–Zr/(Å) | dO–H/(Å) | ∠HOH/(°) | Eads/(kcal/mol) |
|---|---|---|---|---|
| 2 | 2.418 | 0.976/0.994 | 103.6 | −18.3 |
| 3 | 2.404 | 0.976/0.995 | 103.6 | −18.5 |
| 4 | 2.402 | 0.977/0.993 | 103.8 | −18.7 |
| Site | dO–Zr/(Å) | dO–H/(Å) | ∠HOH/(°) | Eads/(kcal/mol) |
|---|---|---|---|---|
| A | 2.364 | 0.979/1.004 | 104.4 | −20.8 |
| B | 2.394 | 0.974/0.998 | 107.8 | −18.6 |
| C | 2.354 | 0.977/1.000 | 106.4 | −19.5 |
| D | 2.234 | 0.974/1.067 | 107.9 | −34.0 |
| Site | dO–Zr/(Å) | dO–H/(Å) | ∠HOH/(°) | Eads/(kcal/mol) |
|---|---|---|---|---|
| A | 2.080 | 0.972/1.642 | 109.9 | −19.3 |
| B | 2.085 | 0.969/1.691 | 136.5 | −16.5 |
| C | 2.073 | 0.973/1.774 | 106.7 | −12.3 |
| D | 2.091 | 0.972/1.465 | 110.8 | −33.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, R.; Zhou, Z.; Wang, Y.; Xiao, H.; Xu, L.; Ding, Y.; Li, X.; Li, A.; Fang, G. First-Principles Molecular Dynamics Simulations on Water–Solid Interface Behavior of H2O-Based Atomic Layer Deposition of Zirconium Dioxide. Nanomaterials 2022, 12, 4362. https://doi.org/10.3390/nano12244362
Xu R, Zhou Z, Wang Y, Xiao H, Xu L, Ding Y, Li X, Li A, Fang G. First-Principles Molecular Dynamics Simulations on Water–Solid Interface Behavior of H2O-Based Atomic Layer Deposition of Zirconium Dioxide. Nanomaterials. 2022; 12(24):4362. https://doi.org/10.3390/nano12244362
Chicago/Turabian StyleXu, Rui, Zhongchao Zhou, Yingying Wang, Hongping Xiao, Lina Xu, Yihong Ding, Xinhua Li, Aidong Li, and Guoyong Fang. 2022. "First-Principles Molecular Dynamics Simulations on Water–Solid Interface Behavior of H2O-Based Atomic Layer Deposition of Zirconium Dioxide" Nanomaterials 12, no. 24: 4362. https://doi.org/10.3390/nano12244362