
Effects of Process and Formulation Parameters on Submicron Polymeric Particles Produced by a Rapid Emulsion-Diffusion Method

 ,
,
Abstract
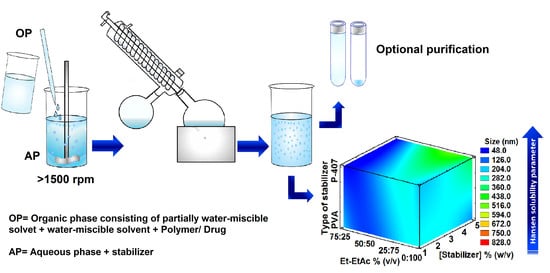
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Experimental Methods
2.2.1. Pre-Optimization of the Method to Produce Nanoparticles
2.2.2. Nanoparticle Preparation and Experimental Design
2.2.3. Particle Size, Charge and pH Determination
2.2.4. Determination of Solubility Parameters of the Nanoparticles Components
2.2.5. Statistical Analysis
3. Results and Discussion
3.1. Pre-Optimization of the Rapid Emulsion-Diffusion Method
3.2. Influence of the Process and Formulation Parameters
3.2.1. Effect of the Solvent Blend (OP) Composition/Ratio on Particle Size and PdI
3.2.2. Effect of the Composition of Solvent Blend and Its OP Ratio on Zeta Potential and pH
3.2.3. Effect of the Stirring Rate on Particle Size and PdI
3.2.4. Effect of the Polymer Concentration on Particle Size, PdI and Zeta Potential
3.2.5. Effect of the Stabilizers and Their Concentration on Particle Size and PdI
3.2.6. Effect of the Stabilizers and Their Concentration on Zeta Potential and pH
3.2.7. Effect of the Addition Order of the OP and AP in the Emulsification Step on the Size and Zeta Potential
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Leroux, J.C.; Allemann, E.; Doelker, E.; Gurny, R. New approach for the preparation of nanoparticles by an emulsification-diffusion method. Eur. J. Pharm. Biopharm. 1995, 41, 14–18. [Google Scholar]
- Mora-Huertas, C.E.; Fessi, H.; Elaissari, A. Influence of process and formulation parameters on the formation of submicron particles by solvent displacement and emulsification-diffusion methods critical comparison. Adv. Colloid Interface Sci. 2011, 163, 90–122. [Google Scholar] [CrossRef] [PubMed]
- Crucho, C.I.C.; Barros, M.T. Polymeric nanoparticles: A study on the preparation variables and characterization methods. Mater. Sci. Eng. C 2017, 80, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Almoustafa, H.A.; Alshawsh, M.A.; Chik, Z. Technical aspects of preparing PEG-PLGA nanoparticles as carrier for chemotherapeutic agents by nanoprecipitation method. Int. J. Pharm. 2017, 533, 275–284. [Google Scholar] [CrossRef]
- Krishnaswamy, K.; Orsat, V. Sustainable Delivery Systems through Green Nanotechnology. In Nano- and Microscale Drug Delivery Systems; Grumezescu, A.M., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 17–32. [Google Scholar]
- Allouche, J. Synthesis of organic and bioorganic nanoparticles: An overview of the preparation methods. In Nanomaterials: A Danger or a Promise? A Chemical and Biological Perspective; Springer: London, UK, 2013; pp. 27–74. [Google Scholar]
- Vanderhoff, J.W.; El-Aasser, M.S.; Ugelstad, J. Polymer Emulsification Process. U.S. Patent 4,177,177, 4 December 1979. [Google Scholar]
- Quintanar-Guerrero, D.; Allémann, E.; Fessi, H.; Doelker, E. Pseudolatex preparation using a novel emulsion-diffusion process involving direct displacement of partially water-miscible solvents by distillation. Int. J. Pharm. 1999, 188, 155–164. [Google Scholar] [CrossRef]
- Trotta, M.; Debernardi, F.; Caputo, O. Preparation of solid lipid nanoparticles by a solvent emulsification–diffusion technique. Int. J. Pharm. 2003, 257, 153–160. [Google Scholar] [CrossRef]
- Hariharan, S.; Bhardwaj, V.; Bala, I.; Sitterberg, J.; Bakowsky, U.; Ravi Kumar, M.N. Design of estradiol loaded PLGA nanoparticulate formulations: A potential oral delivery system for hormone therapy. Pharm. Res. 2006, 23, 184–195. [Google Scholar] [CrossRef]
- Sahana, D.K.; Mittal, G.; Bhardwaj, V.; Kumar, M.N.V.R. PLGA nanoparticles for oral delivery of hydrophobic drugs: Influence of organic solvent on nanoparticle formation and release behavior in vitro and in vivo using estradiol as a model drug. J. Pharm. Sci. 2008, 97, 1530–1542. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.K.; Swarnakar, N.K.; Godugu, C.; Singh, R.P.; Jain, S. The effect of the oral administration of polymeric nanoparticles on the efficacy and toxicity of tamoxifen. Biomaterials 2011, 32, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Galindo-Pérez, M.J.; Quintanar-Guerrero, D.; Cornejo-Villegas, M.d.l.Á.; Zambrano-Zaragoza, M.d.l.L. Optimization of the emulsification-diffusion method using ultrasound to prepare nanocapsules of different food-core oils. LWT 2018, 87, 333–341. [Google Scholar] [CrossRef]
- Chen, C.; Yang, W.; Wang, D.T.; Chen, C.L.; Zhuang, Q.Y.; Kong, X.D. A modified spontaneous emulsification solvent diffusion method for the preparation of curcumin-loaded PLGA nanoparticles with enhanced in vitro anti-tumor activity. Front. Mater. Sci. 2014, 8, 332–342. [Google Scholar] [CrossRef]
- Cheerarot, O.; Baimark, Y. Biodegradable silk fibroin/chitosan blend microparticles prepared by emulsification-diffusion method. e-Polymers 2015, 15, 67. [Google Scholar] [CrossRef]
- Mainardes, R.M.; Evangelista, R.C. Praziquantel-loaded PLGA nanoparticles: Preparation and characterization. J. Microencapsul. 2005, 22, 13–24. [Google Scholar] [CrossRef]
- Manchanda, R.; Fernandez-Fernandez, A.; Nagesetti, A.; McGoron, A.J. Preparation and characterization of a polymeric (PLGA) nanoparticulate drug delivery system with simultaneous incorporation of chemotherapeutic and thermo-optical agents. Colloids Surf. B Biointerfaces 2010, 75, 260–267. [Google Scholar] [CrossRef]
- Poletto, F.S.; Fiel, L.A.; Donida, B.; Ré, M.I.; Guterres, S.S.; Pohlmann, A.R. Controlling the size of poly (hydroxybutyrate-co-hydroxyvalerate) nanoparticles prepared by emulsification–diffusion technique using ethanol as surface agent. Colloids Surf. A Physicochem. Eng. Asp. 2008, 324, 105–112. [Google Scholar] [CrossRef]
- USPC. United States Pharmacopeia and National Formulary (USP43-NF38); United States Pharmacopeial Convention: Rockville, MD, USA, 2021; p. 6712. [Google Scholar]
- Singh-Joy, S.D.; McLain, V.C. Safety assessment of poloxamers 101, 105, 108, 122, 123, 124, 181, 182, 183, 184, 185, 188, 212, 215, 217, 231, 234, 235, 237, 238, 282, 284, 288, 331, 333, 334, 335, 338, 401, 402, 403, and 407, poloxamer 105 benzoate, and poloxamer 182 dibenzoate as used in cosmetics. Int. J. Toxicol. 2008, 27, 93–128. [Google Scholar]
- FDA. Inactive Ingredient Database, 2021st ed.; FDA/Center for Drug Evaluation and Research: Silver Spring, MD, USA, 2021.
- Süß, S.; Sobisch, T.; Peukert, W.; Lerche, D.; Segets, D. Determination of Hansen parameters for particles: A standardized routine based on analytical centrifugation. Adv. Powder Technol. 2018, 29, 1550–1561. [Google Scholar] [CrossRef]
- Gårdebjer, S.; Andersson, M.; Engström, J.; Restorp, P.; Persson, M.; Larsson, A. Using Hansen solubility parameters to predict the dispersion of nanoparticles in polymeric films. Polym. Chem. 2016, 7, 1756–1764. [Google Scholar] [CrossRef]
- Fujiwara, N.; Nishida, T.; Yamamoto, H. Adaptation of Hansen solubility parameter in evaluating transparency of composite materials. Heliyon 2019, 5, e02833. [Google Scholar] [CrossRef] [Green Version]
- Shah, D.O. Surfactant Science Series. In Fine Particles: Synthesis, Characterization, and Mechanisms of Growth; Sugimoto, T., Ed.; Springer Nature: Cham, Switzerland, 2002; Volume 92, p. 179. [Google Scholar]
- Li, Z.; Tao, W.; Zhang, D.; Wu, C.; Song, B.; Wang, S.; Wang, T.; Hu, M.; Liu, X.; Wang, Y.; et al. The studies of PLGA nanoparticles loading atorvastatin calcium for oral administration in vitro and in vivo. Asian J. Pharm. Sci. 2017, 12, 285–291. [Google Scholar] [CrossRef]
- Birnbaum, D.T.; Kosmala, J.D.; Henthorn, D.B.; Brannon-Peppas, L. Controlled release of beta-estradiol from PLAGA microparticles: The effect of organic phase solvent on encapsulation and release. J. Control Release 2000, 65, 375–387. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Huggins, M.L. The Solubility of Nonelectrolytes. By Joel H. Hildebrand and Robert S. Scott. J. Phys. Chem. 1951, 55, 619–620. [Google Scholar] [CrossRef]
- Hansen, C.M. Hansen Solubility Parameters: A User’s Handbook, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2007. [Google Scholar]
- Van Krevelen, D.W.; Te Nijenhuis, K. (Eds.) Comprehensive Tables. In Properties of Polymers, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 889–953. [Google Scholar]
- Madsen, C.G.; Skov, A.; Baldursdottir, S.; Rades, T.; Jorgensen, L.; Medlicott, N.J. Simple measurements for prediction of drug release from polymer matrices-Solubility parameters and intrinsic viscosity. Eur. J. Pharm. BioPharm. 2015, 92, 1–7. [Google Scholar] [CrossRef]
- Reichardt, C.; Welton, T. (Eds.) Appendix A. Properties, Purification, and Use of Organic Solvents. In Solvents and Solvent Effects in Organic Chemistry, 4th ed.; Wiley-VCH: Weinheim, Germany, 2011; pp. 549–586. [Google Scholar]
- Reichardt, C.; Welton, T. (Eds.) Empirical Parameters of Solvent Polarity. In Solvents and Solvent Effects in Organic Chemistry; Wiley-VCH: Weinheim, Germany, 2010; pp. 425–508. [Google Scholar]
- Wongsawa, T.; Koonsang, T.; Kunthakudee, N.; Prapasawat, T.; Maneeintr, K.; Pancharoen, U. The experimental investigations on viscosity, surface tension, interfacial tension and solubility of the binary and ternary systems for tributyl phosphate (TBP) extractant in various organic solvents with water: Thermodynamic NRTL model and molecular interaction approach. J. Mol. Liq. 2018, 251, 229–237. [Google Scholar]
- Yaws, C.L.; Richmond, P.C. Chapter 21-Surface tension—Organic compounds. In Thermophysical Properties of Chemicals and Hydrocarbons; Yaws, C.L., Ed.; William Andrew Publishing: Norwich, NY, USA, 2009; pp. 686–781. [Google Scholar]
- Kitak, T.; Dumičić, A.; Planinšek, O.; Šibanc, R.; Srčič, S. Determination of Solubility Parameters of Ibuprofen and Ibuprofen Lysinate. Molecules 2015, 20, 21549–21568. [Google Scholar] [CrossRef]
- Smallwood, I.M. (Ed.) Front Matter. In Handbook of Organic Solvent Properties; Butterworth-Heinemann: Oxford, UK, 1996; pp. 1–326. [Google Scholar]
- Rossini, E.; Bochevarov, A.D.; Knapp, E.W. Empirical Conversion of pK (a) Values between Different Solvents and Interpretation of the Parameters: Application to Water, Acetonitrile, Dimethyl Sulfoxide, and Methanol. ACS Omega 2018, 3, 1653–1662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manallack, D.T.; Prankerd, R.J.; Yuriev, E.; Oprea, T.I.; Chalmers, D.K. The significance of acid/base properties in drug discovery. Chem. Soc. Rev. 2013, 42, 485–496. [Google Scholar] [CrossRef] [Green Version]
- Radtke, V.; Stoica, D.; Leito, I.; Camões, F.; Krossing, I.; Anes, B.; Roziková, M.; Deleebeeck, L.; Veltzé, S.; Näykki, T.; et al. A unified pH scale for all solvents: Part I-intention and reasoning (IUPAC Technical Report). Pure Appl. Chem. 2021, 93, 1049–1060. [Google Scholar] [CrossRef]
- Yoo, J.W.; Mitragotri, S. Polymer particles that switch shape in response to a stimulus. Proc. Natl. Acad. Sci. USA 2010, 107, 11205–11210. [Google Scholar] [CrossRef] [Green Version]
- Moustafine, R.I.; Salachova, A.R.; Frolova, E.S.; Kemenova, V.A.; Van den Mooter, G. Interpolyelectrolyte complexes of Eudragit E PO with sodium alginate as potential carriers for colonic drug delivery: Monitoring of structural transformation and composition changes during swellability and release evaluating. Drug. Dev. Ind. Pharm. 2009, 35, 1439–1451. [Google Scholar] [CrossRef]
- Patra, C.N.; Priya, R.; Swain, S.; Kumar Jena, G.; Panigrahi, K.C.; Ghose, D. Pharmaceutical significance of Eudragit: A review. Future J. Pharm. Sci. 2017, 3, 33–45. [Google Scholar] [CrossRef]
- Serjeant, E.P.; Dempsey, B. Ionization Constants of Organic Acids in Aqueous Solution; Pergamon Press: Oxford, UK, 1979. [Google Scholar]
- Kim, H.; Babu, C.R.; Burgess, D.J. Quantification of protonation in organic solvents using solution NMR spectroscopy: Implication in salt formation. Int. J. Pharm. 2013, 448, 123–131. [Google Scholar] [CrossRef]
- Wypych, G.A. (Ed.) 3.7-Generic Solvents. In Databook of Green Solvents; ChemTech Publishing: Toronto, ON, Canada, 2019; pp. 237–402. [Google Scholar]
- Rao, V.M.; Engh, K.; Qiu, Y. Design of pH-independent controlled release matrix tablets for acidic drugs. Int. J. Pharm. 2003, 252, 81–86. [Google Scholar] [CrossRef]
- Tang, Z.G.; Black, R.A.; Curran, J.M.; Hunt, J.A.; Rhodes, N.P.; Williams, D.F. Surface properties and biocompatibility of solvent-cast poly[-caprolactone] films. Biomaterials 2004, 25, 4741–4748. [Google Scholar] [CrossRef] [PubMed]
- Van Krevelen, D.W.; Te Nijenhuis, K. (Eds.) Chapter 11-Electrical Properties. In Properties of Polymers, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 319–354. [Google Scholar]
- Kwon, H.Y.; Lee, J.Y.; Choi, S.W.; Jang, Y.; Kim, J.H. Preparation of PLGA nanoparticles containing estrogen by emulsification–diffusion method. Colloids Surf. A Physicochem. Eng. Asp. 2001, 182, 123–130. [Google Scholar] [CrossRef]
- Galindo-Rodríguez, S.A.; Puel, F.; Briançon, S.; Allémann, E.; Doelker, E.; Fessi, H. Comparative scale-up of three methods for producing ibuprofen-loaded nanoparticles. Eur. J. Pharm. Sci. 2005, 25, 357–367. [Google Scholar] [CrossRef]
- Song, K.C.; Lee, H.S.; Choung, I.Y.; Cho, K.I.; Ahn, Y.; Choi, E.J.J.C.; Physicochemical, S.A.; Aspects, E. The effect of type of organic phase solvents on the particle size of poly (d, l-lactide-co-glycolide) nanoparticles. Colloids Surf. A Physicochem. Eng. Asp. 2006, 276, 162–167. [Google Scholar] [CrossRef]
- Suksiriworapong, J.; Rungvimolsin, T.; Agomol, A.; Junyaprasert, V.B.; Chantasart, D. Development and characterization of lyophilized diazepam-loaded polymeric micelles. AAPS PharmSciTech 2014, 15, 52–64. [Google Scholar] [CrossRef] [Green Version]
- Domínguez-Delgado, C.; Fuentes-Prado, E.; Escobar-Chávez, J.; Vidal-Romero, G.; Rodríguez Cruz, I.; Díaz-Torres, R. Chitosan and Pluronic® F-127: Pharmaceutical Applications. In Encyclopedia of Biomedical Polymers and Polymeric Biomaterials; Mishra, M., Ed.; Taylor and Francis Group LLC: Boca Raton, FL, USA; London, UK; New York, NY, USA, 2016; pp. 1513–1535. [Google Scholar]
- Zhang, R.; Somasundaran, P. Advances in adsorption of surfactants and their mixtures at solid/solution interfaces. Adv. Colloid Interface Sci. 2006, 123, 213–229. [Google Scholar] [CrossRef]
- Hirsjärvi, S.; Peltonen, L.; Hirvonen, J. Surface pressure measurements in particle interaction and stability studies of poly (lactic acid) nanoparticles. Int. J. Pharm. 2008, 348, 153–160. [Google Scholar] [CrossRef]
- Fink, J.K. (Ed.) Chapter 2: Poly (vinyl alcohol). In Handbook of Engineering and Specialty Thermoplastics, Water Soluble Polymers; Scrivener Publishing LLC: Boston, MA, USA, 2011; Volume 2, pp. 39–68. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent Type of OP | Saturation | Emulsification | Dilution (Water) | EVP | NP Type/Ref. | ||
|---|---|---|---|---|---|---|---|
| Phases Addition | Mixing | S | |||||
| Standard emulsification-diffusion method | |||||||
| Partially water-miscible solvents | + | OP → AP | High-shear stirring | + | + | +/F | NSP [1] |
| Modifications of the emulsification-diffusion method | |||||||
| Partially water-miscible solvents | + | OP → AP | Low/high shear stirring | + | − | + | NSP [8] |
| Partially water-miscible solvents | + (47 °C) | OP → AP | High-shear stirring (47 ± 2 °C) | + | + | + | SLN [9] |
| Miscible, non-miscible and partially miscible solvents in water (individual or combined) | − | OP → AP (AP: glucose–ACE) | Pre-stirring (1000 rpm/3 h) High-shear stirring (15,000 rpm/5 min) | + | + | + | NSP [11] |
| Partially water-miscible solvent | − | OP → AP | Pre-stirring (3 h) High-shear stirring (15,000 rpm/5 min) | + | + (water bath 40 °C) | + | [10] NSP |
| Partially water-miscible solvent | − | OP → AP | Pre-stirring (1000 rpm/20 min) → High-shear stirring (10,000–15,000 rpm) or Sonication (60 % amplitude/1 min) | + | + | + | NSP [12] |
| Partially water-miscible solvent | + | OP → AP | Ultrasound (2 min), ice bath | + | + | + (40 °C) | NC [13] |
| Two organic phases: 1. Partially water-miscible gas + drug/ 2. Water-miscible solvent + polymer | − | Two steps: 1 = OP1 → OP2 2 = OP2 → AP 2 mL/min | Magnetic stirring | + | + | + (3 h) | NC [14] |
| Partially water-miscible solvent | − | AP → OP | Magnetic stirring | ˗ | − | C | MCP [15] |
| Modifications of the emulsification-solvent evaporation method | |||||||
| Partially miscible or non-miscible solvents in water | − | OP → AP | Ultrasound (55 W/1 min) | + | − | + | NSP [16] |
| Blends of solvents miscible and non-miscible in water | − | OP → AP | Ultrasound (50 W/30 s, in an ice bath) | + | − | + | NSP [17] |
| Blends of solvents miscible and non-miscible in water | − | Two steps: 1: OP → AP1 2: AP1 → AP2 | Step 1: High stirring (17,500 rpm/5 min), Step 2: Magnetic stirring (40 °C/40 min). | + | − | + (40 °C) | NSP, NC and NE [18] |
| Modification of the emulsification-diffusion method proposed in this study | |||||||
| Blends of solvents miscible and partially miscible in water | − | OP → AP or AP → OP (2 mL/min) | Low/medium shear stirring (>1500–8000 rpm) | + | − | + (30 °C) | NSP |
| Batch | Stabilizer % (w/v) | PLGA Type, % (w/v) | AP (mL) | OP % (v/v) mL | Stirring | Ratio OP:AP |
|---|---|---|---|---|---|---|
| a | PVA (2.0) | (85:15), 1 | 0 | Et-PEG (90:10) 20 | * | (1:0) |
| 0 | Et-PEG (80:20) 20 | * | ||||
| (85:15), 1.4 | 0 | Et-PEG-THF (57.14:14.29:28.57) 28 | * | (1.4:0) | ||
| (85:15), 1.8 | 0 | Et-PEG-THF (44.44:11.11:44.44) 36 | * | (1.8:0) | ||
| (85:15), 1.4 | 40 | Et-PEG-THF (36.36:9.509:54.54) 44 | Magnetic | (1.05:0.95) | ||
| b | PVA (2.0) | (85:15), 2.2 | 40 | Et-MC (50:50) 20 | Magnetic | (1:2) |
| c | PVA (0.4545) | (50:50), 1 | 44 | EtAc-Et (71.43:28.57) 20 | Magnetic | (0.77:1.22) |
| d | PVA (2.0) | (50:50), 1 | 40 | THF-PEG (80:20) 20 | Magnetic | (1:2) |
| e | PVA (2.0) | (50:50), 1 | 40 | THF (100) 20 | Magnetic | (1:2) |
| f | PVA (2.0) | (50:50), 1 | 40 | THF-EtAc-PEG (70:25:5) 20 | Magnetic | (1:2) |
| g | PVA (2.0) | (50:50), 1 | 40 | THF-EtAc (75:25) 20 | Magnetic 10 min + UT 8000 rpm, 10 min | (1:2) |
| h | PVA (2.0) | (50:50), 0.5 | 20 | THF (100) 20 | Magnetic | (1:1) |
| i | PVA (2.0) | (50:50), 0.5 | 40 | THF (100) 20 | Magnetic | (1:2) |
| j | PVA (2.0) | (50:50), 1 | 40 | THF-EtAc (50:50) 20 | UT, 8000 rpm * | (1:2) |
| Design | Polymer (mg) | OP Ratio (v/v) | Stabilizer (% w/v) | Stirring Rate (rpm) | Factors | Levels | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | PLGA (50:50) (200 mg) | - | PVA (2%) | 8000 a | OP ratio | 75:25 | 50:50 | 25:75 | 0:100 | ||
| Type of OP | THF-EtAc | ACE-MEK | |||||||||
| 2 | Eudragit® E 100 (−-) | ACE-EtAc | PVA (2%) | 2000 b | [Polymer] | 200 mg | 400 mg | 800 mg | |||
| OP ratio | 75:25 | 50:50 | 25:75 | 0:100 | |||||||
| 3 | Eudragit® E 100 (200 mg) | - | PVA (2%) | 2000 b | water miscible solvent in the OP | ACE-EtAc | Et-EtAC | ||||
| OP ratio | 75:25 | 50:50 | 25:75 | 0:100 | |||||||
| 4 | Eudragit® E 100 (200 mg) | Et-EtAc | - | 2000 b | [Stabilizer] | 1% (w/v) | 2% (w/v) | 5% (w/v) | |||
| Type of stabilizer | PVA | P-407 | |||||||||
| OP ratio | 75:25 | 50:50 | 25:75 | 0:100 | |||||||
| 5 | Eudragit® E 100 (200 mg) | MEK (100) | PVA (2%) | - | Stirring rate | 500 | 1500 | 2000 | 3000 | ||
| (rpm) | |||||||||||
| 6 | Eudragit® E 100 (400 mg) | ACE-MEK | PVA (2%) | 1500 b | OP ratio | 75:25 | 50:50 | 25:75 | 0:100 | ||
| 7 | PLGA (50:50) (200 mg) | ACE-MEK (50:50) | PVA (2%) | 8000 a | AP:OP order of addition | OP added to the AP AP added to the OP | |||||
| 8 | Eudragit® E 100 (400 mg) | MEK (100) | - | 1500 b | [PVA] | 1% (w/v) | 2% (w/v) | ||||
| 9 | PLGA (50:50) (200 mg) | ACE-MEK (75:25) | - | 2000 | [P-407] | 0.25% (w/v) | 0.5% (w/v) | 1% (w/v) | |||
| 10 | PLGA (50:50) (200 mg) | ACE-EtAc | P-407 (5%) | 2000 | OP ratio | 25:75 | 50:50 | ||||
| 11 | PLGA (50:50) (200 mg) | - | P-407 (1%) | 2000 | OP ratio | 25:75 | 75:25 | ||||
| Type of OP | ACE-EtAc | ACE-MEK | |||||||||
| 12 | PLGA (50:50) (200 mg) | ACE-MEK (75:25) | (1%) | 2000 | Type of stabilizer | PVA | P-407 | ||||
| Pure Solvents and Solvent Blends | Hansen Solubility Parameter (MPa1/2) | |||
|---|---|---|---|---|
| δD | δP | δH | δHaSP | |
| ACE 100% | 15.5 | 10.4 | 7 | 19.9 |
| EtAc 100% | 15.2 | 5.3 | 9.2 | 18.5 |
| MEK 100% | 15.9 | 9 | 5.1 | 19.0 |
| THF 100% | 16.8 | 5.7 | 8 | 19.5 |
| Et 100% | 15.8 | 8.8 | 19.5 | 26.6 |
| ACE-MEK (0:100) | 15.9 | 9.0 | 5.1 | 19.0 |
| ACE-MEK (25:75) | 15.8 | 9.4 | 5.6 | 19.2 |
| ACE-MEK (50:50) | 15.7 | 9.7 | 6.1 | 19.4 |
| ACE-MEK (75:25) | 15.6 | 10.1 | 6.5 | 19.7 |
| THF-EtAc (0:100) | 15.2 | 5.3 | 9.2 | 18.6 |
| THF-EtAc (25:75) | 15.6 | 5.4 | 8.9 | 18.8 |
| THF-EtAc (50:50) | 16.0 | 5.5 | 8.6 | 19.0 |
| THF-EtAc (75:25) | 16.4 | 5.6 | 8.3 | 19.2 |
| ACE-EtAc (0:100) | 15.2 | 5.3 | 9.2 | 18.6 |
| ACE-EtAc (25:75) | 15.3 | 6.6 | 8.7 | 18.7 |
| ACE-EtAc (50:50) | 15.4 | 7.9 | 8.1 | 19.0 |
| ACE-EtAc (75:25) | 15.4 | 9.1 | 7.6 | 19.4 |
| Et-EtAc (0:100) | 15.2 | 5.3 | 9.2 | 18.6 |
| Et-EtAc (25:75) | 15.4 | 6.2 | 11.8 | 20.3 |
| Et-EtAc (50:50) | 15.5 | 7.1 | 14.4 | 22.3 |
| Et-EtAc (75:25) | 15.7 | 7.9 | 16.9 | 24.4 |
| Solvent Blend or OP | [Polymer] | [Stabilizer] % (w/v) | Correlation Equation for Particle Size a | r2 |
|---|---|---|---|---|
| ACE-MEK (0:100 to 75:25) | PLGA (50:50), 200 mg | PVA 2% | y = −427.93x + 8556.4 | 0.8432 |
| THF-EtAc (0:100 to 75:25) | y = −157.57x + 3201.8 | 0.7699 | ||
| ACE-EtAc (0:100 to 75:25) | Eudragit® E100, 200 mg | y = −124.29x + 2590.2 | 0.8197 | |
| Eudragit® E100, 400 mg | y = −130.23x + 2723.1 | 0.2000 | ||
| Eudragit® E100, 800 mg | y = 8135.6x − 144,556 | 0.0227 | ||
| ACE-MEK (0:100 to 75:25) | Eudragit® E100, 400 mg | y = −739.64x + 14,788 | 0.1503 | |
| Et-EtAc (0:100 to 75:25) | Eudragit® E100, 200 mg | PVA 1% | y = −2.7458x + 238.35 | 0.0281 |
| PVA 2% | y = −20.285x + 642.2 | 0.9037 | ||
| PVA 5% | y = −7.8519x + 338.56 | 0.1376 | ||
| P-407 1% | y = −35.846x + 917.08 | 0.9016 | ||
| P-407 2% | y = −26.586x + 726.96 | 0.3481 | ||
| P-407 5% | y = −64.851x + 1686.3 | 0.2465 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domínguez-Delgado, C.L.; Akhtar, Z.; Awuah-Mensah, G.; Wu, B.; Smyth, H.D.C. Effects of Process and Formulation Parameters on Submicron Polymeric Particles Produced by a Rapid Emulsion-Diffusion Method. Nanomaterials 2022, 12, 229. https://doi.org/10.3390/nano12020229
Domínguez-Delgado CL, Akhtar Z, Awuah-Mensah G, Wu B, Smyth HDC. Effects of Process and Formulation Parameters on Submicron Polymeric Particles Produced by a Rapid Emulsion-Diffusion Method. Nanomaterials. 2022; 12(2):229. https://doi.org/10.3390/nano12020229
Chicago/Turabian StyleDomínguez-Delgado, Clara Luisa, Zubia Akhtar, Godfrey Awuah-Mensah, Braden Wu, and Hugh David Charles Smyth. 2022. "Effects of Process and Formulation Parameters on Submicron Polymeric Particles Produced by a Rapid Emulsion-Diffusion Method" Nanomaterials 12, no. 2: 229. https://doi.org/10.3390/nano12020229