
Steam Activation of Acid-Chars for Enhanced Textural Properties and Pharmaceuticals Removal

Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of Acid-Chars
2.2. Activation of Acid-Char Samples
2.3. Characterization of the Materials
2.4. Batch Adsorption Experiments
3. Results and Discussion
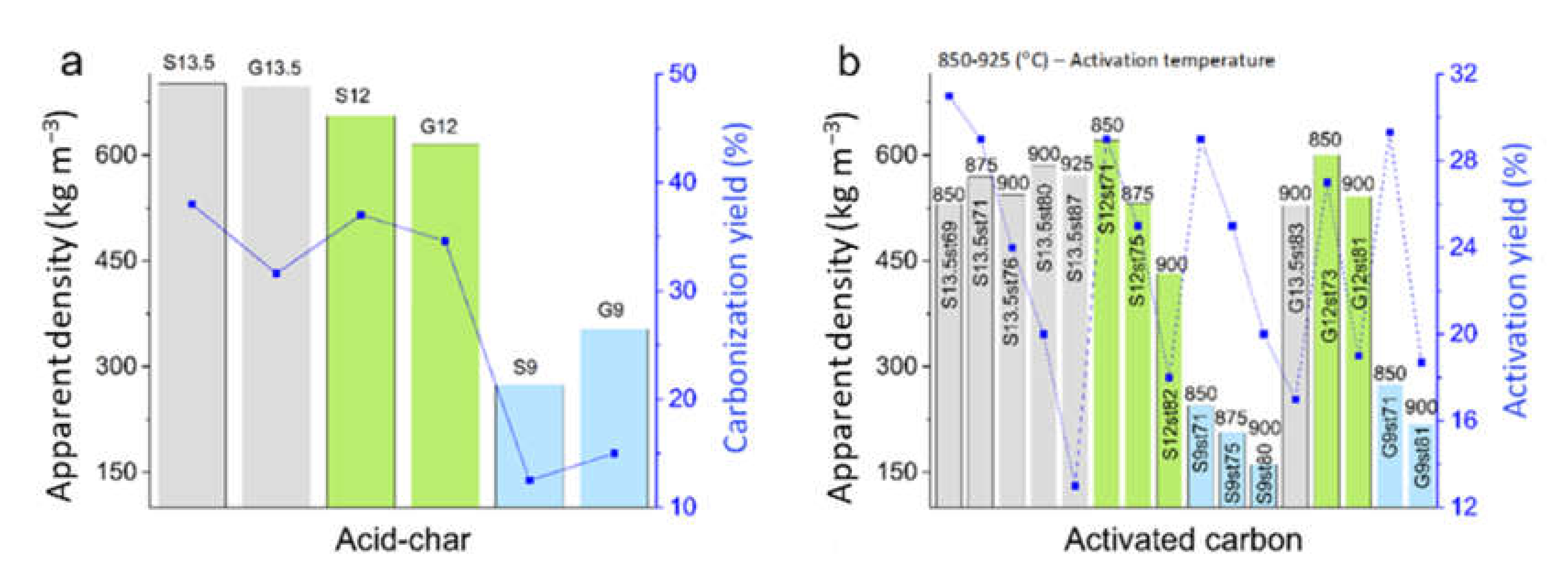
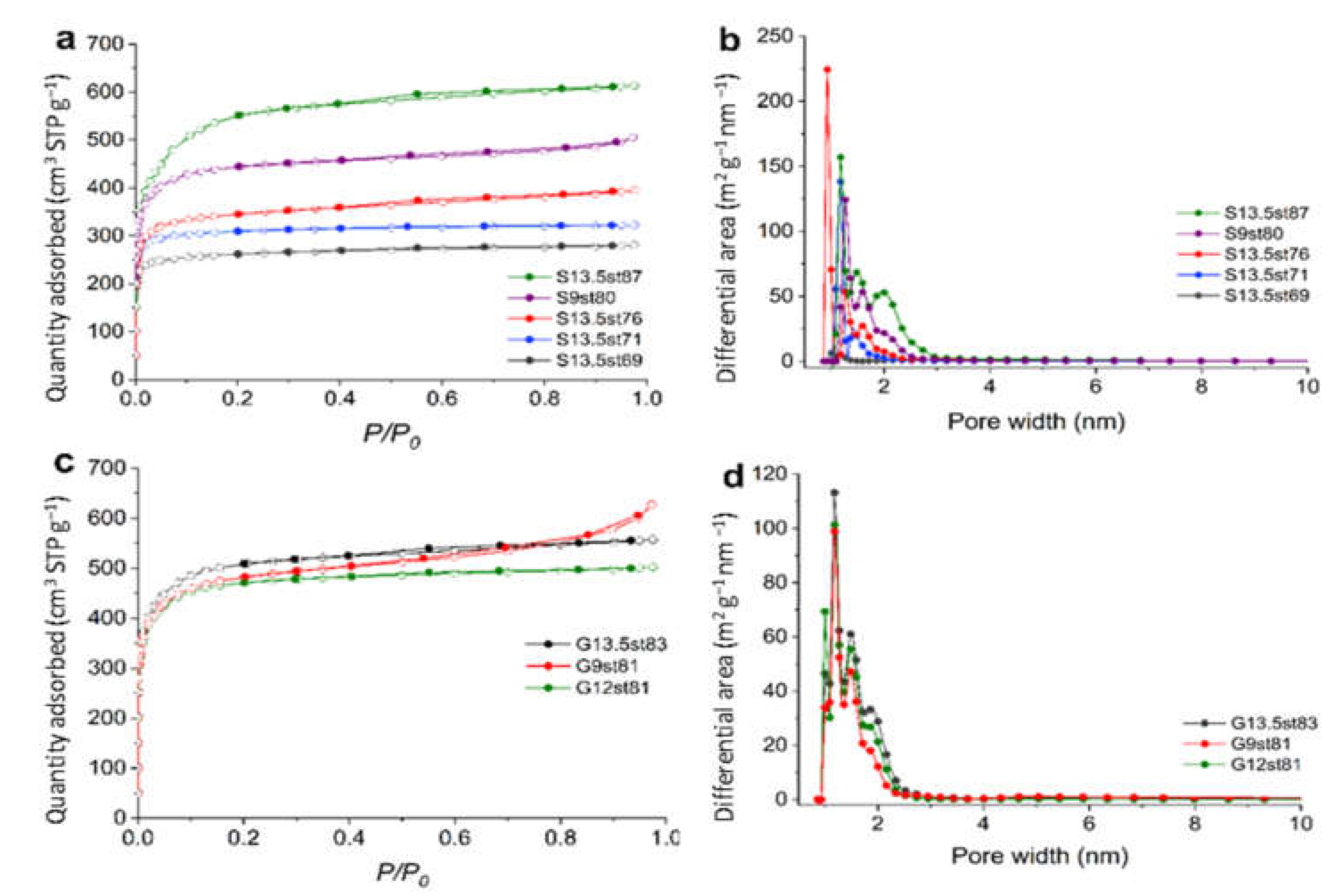
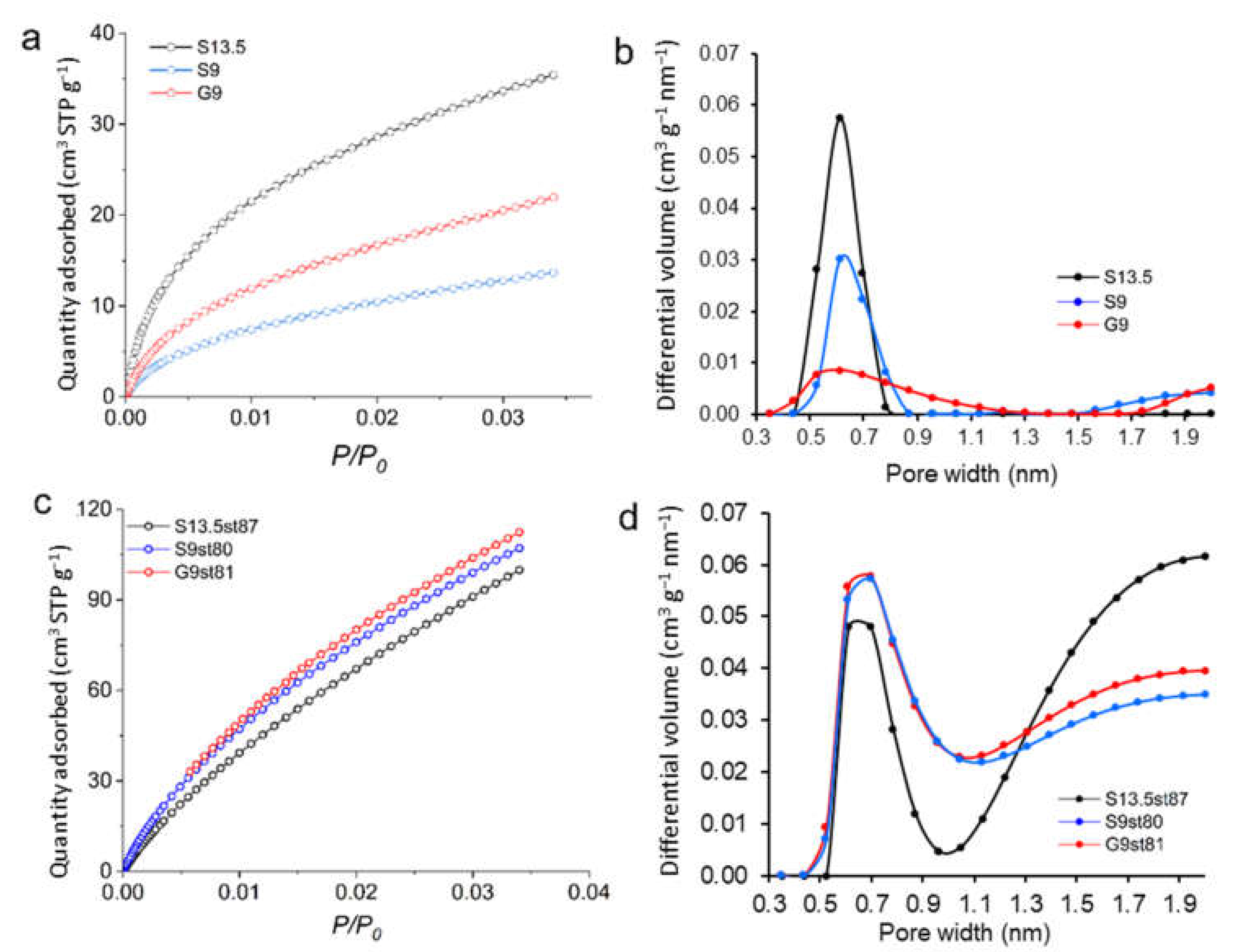
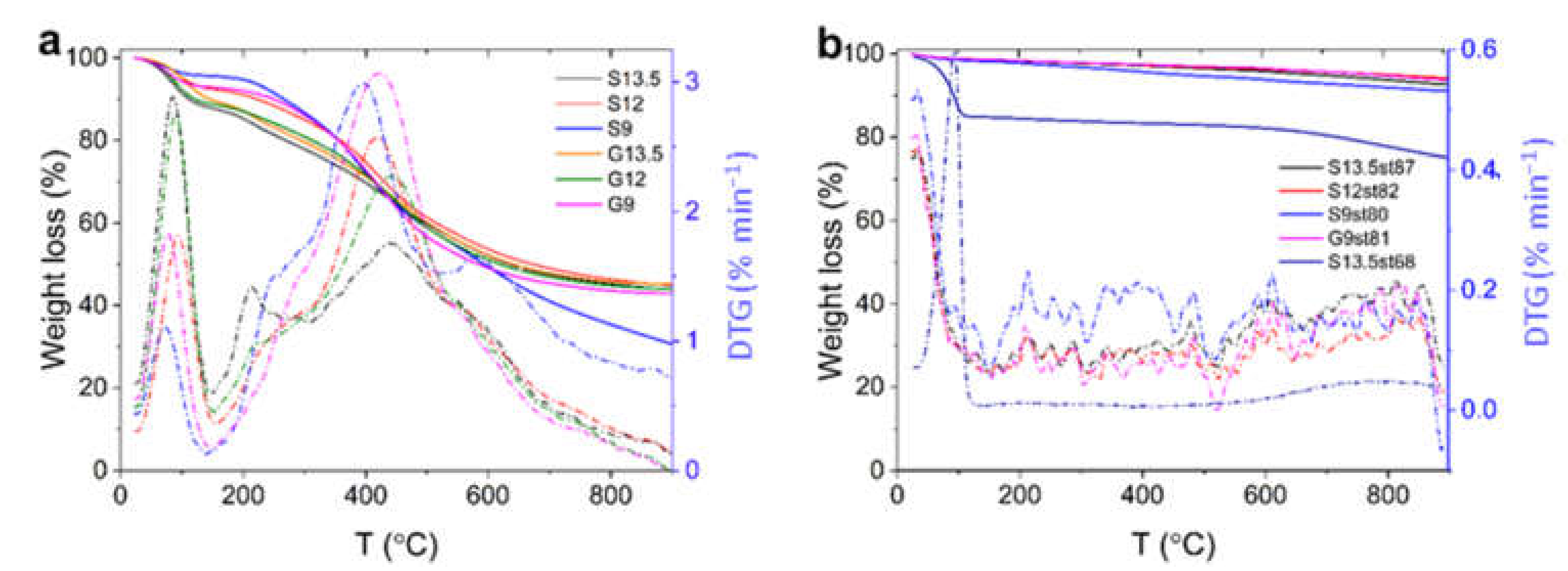
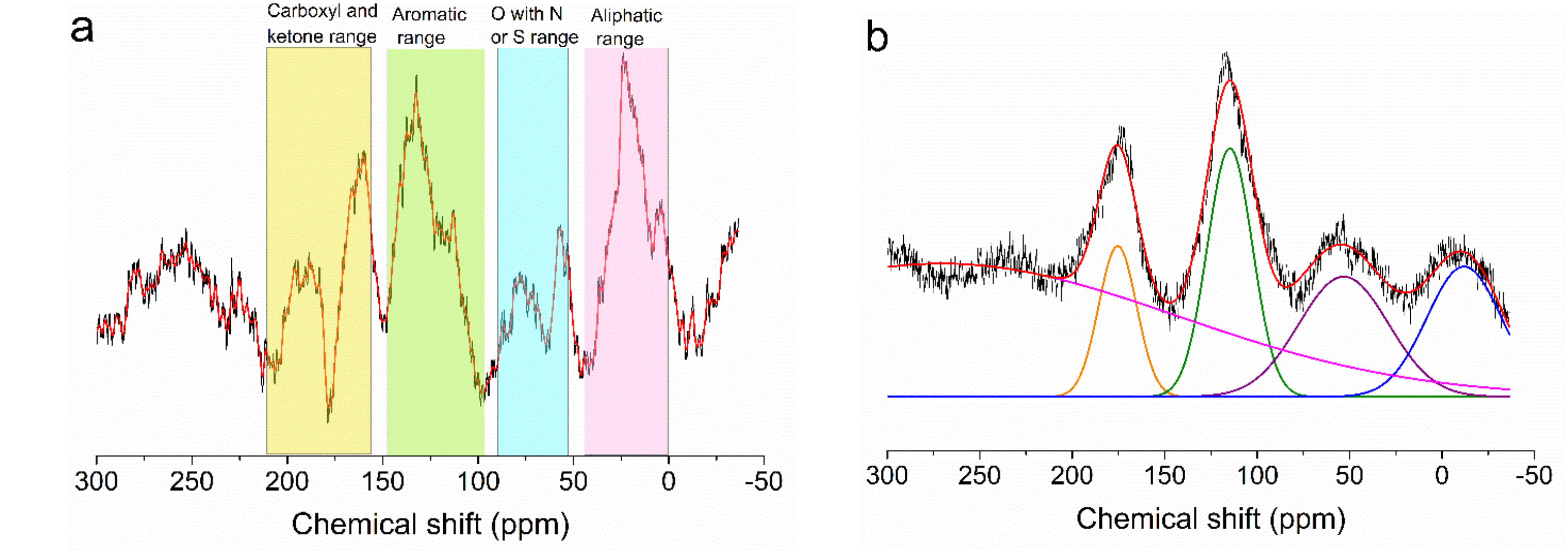
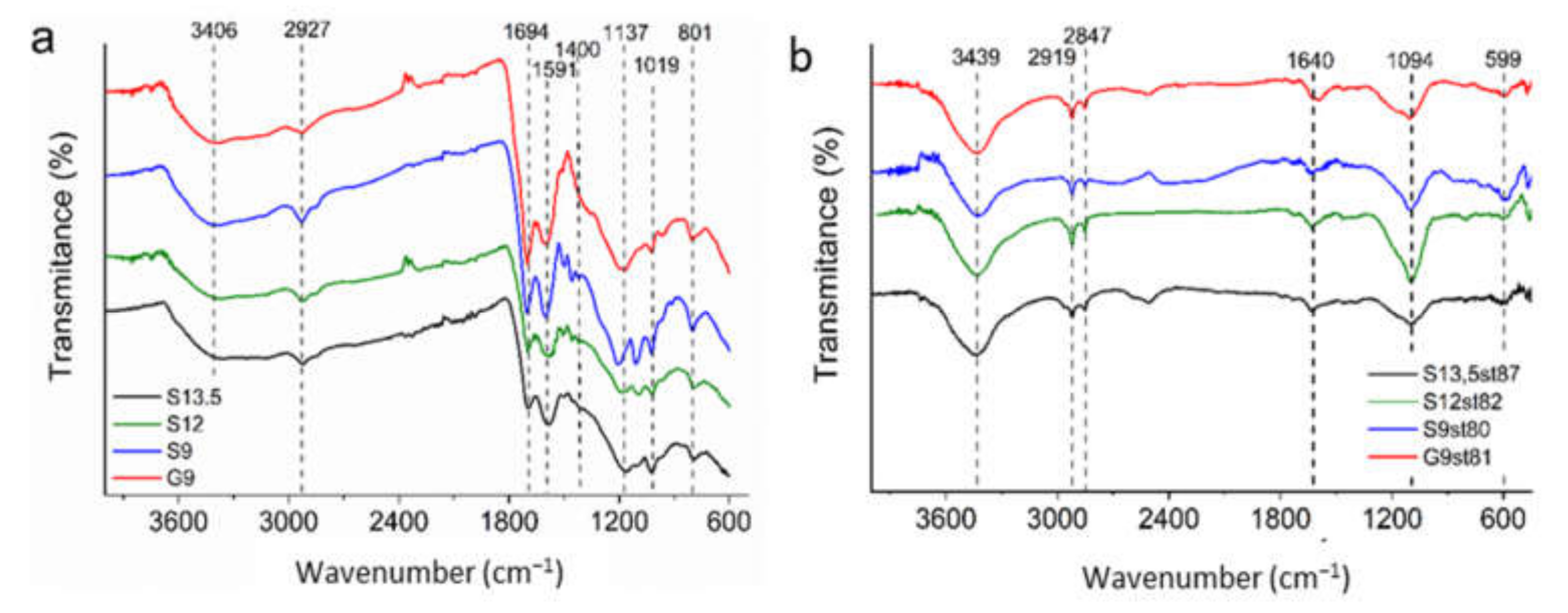
3.1. Characterization of Acid-Char and Activated Carbon Samples
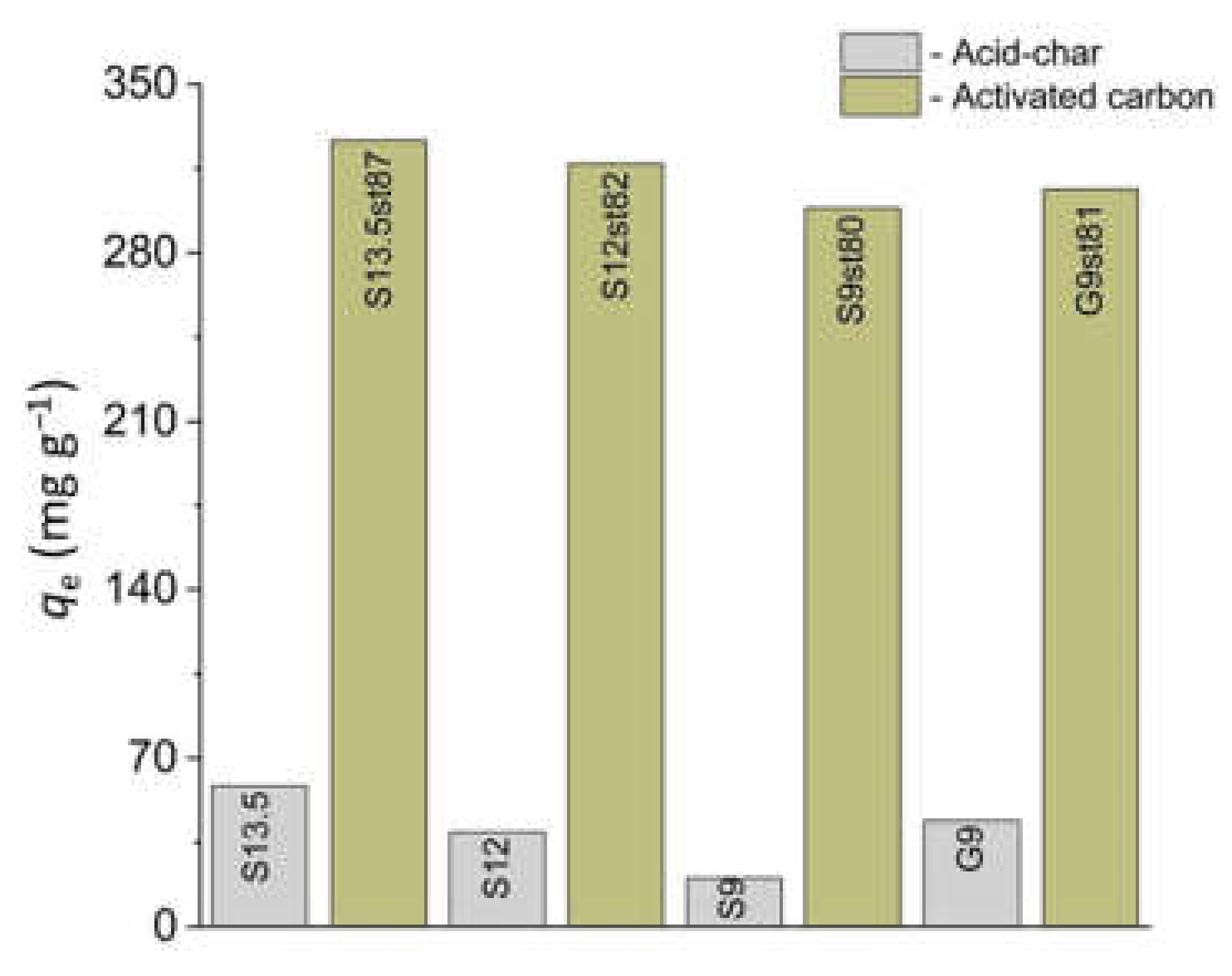
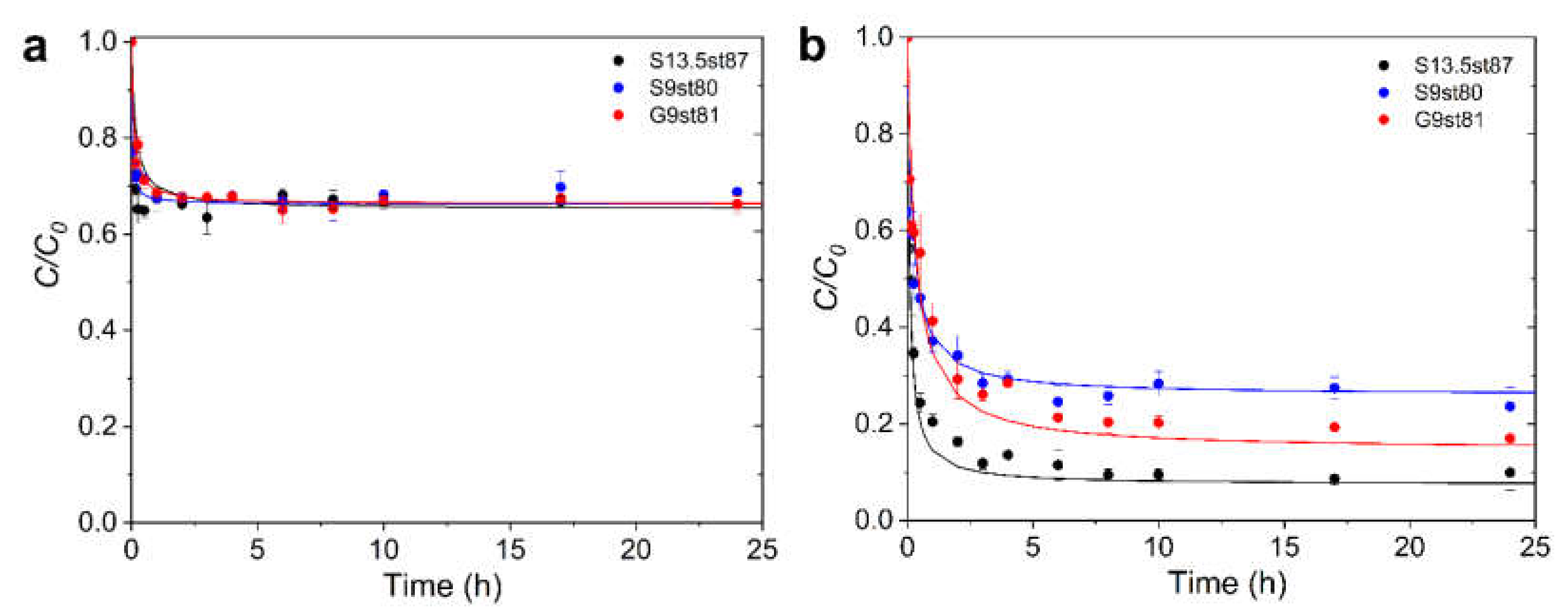
3.2. Ibuprofen and Iopamidol Adsorption onto Carbon Adsorbents
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- POSEIDON (June 2004). Assessment of Technologies for the Removal of Pharmaceuticals and Personal Care Products in Sewage and Drinking Water Facilities to Improve the Indirect Potable Water Reuse. Available online: https://cordis.europa.eu/project/id/EVK1-CT-2000-00047 (accessed on 5 September 2022).
- Carvalho, A.P.; Mestre, A.S.; Andrade, M.; Ania, C.O. Ibuprofen in the aquatic environment: Occurrence, ecotoxicity and water remediation technologies. In Ibuprofen: Clinical Pharmacology, Medical Uses and Adverse Effects; Carter, W.C., Brown, B.R., Eds.; Nova Science Publishers, Inc.: New York, NY, USA, 2013; pp. 1–84. ISBN1 978-162618659-0. Available online: http://www.scopus.com/inward/record.url?eid=2-s2.0-84890844077&partnerID=MN8TOARS (accessed on 15 September 2022)ISBN2 978-162618659-0.
- Rizzo, L.; Malato, S.; Antakyali, D.; Beretsou, V.G.; Đolić, M.B.; Gernjak, W.; Heath, E.; Ivancev-Tumbas, I.; Karaolia, P.; Lado Ribeiro, A.R.; et al. Consolidated vs new advanced treatment methods for the removal of contaminants of emerging concern from urban wastewater. Sci. Total Environ. 2019, 655, 986–1008. [Google Scholar] [CrossRef] [PubMed]
- Campinas, M.; Viegas, R.M.C.; Almeida, C.M.M.; Martins, A.; Silva, C.; Mesquita, E.; Coelho, M.R.; Silva, S.; Cardoso, V.V.; Benoliel, M.J.; et al. Powdered activated carbon full-scale addition to the activated sludge reactor of a municipal wastewater treatment plant: Pharmaceutical compounds control and overall impact on the process. J. Water Process Eng. 2022, 49, 102975. [Google Scholar] [CrossRef]
- Pi, Z.; Hou, K.; Yao, F.; He, L.; Chen, S.; Tao, Z.; Zhou, P.; Wang, D.; Li, X.; Yang, Q. In-situ regeneration of tetracycline-saturated hierarchical porous carbon by peroxydisulfate oxidation process: Performance, mechanism, and application. Chem. Eng. J. 2022, 427, 131749. [Google Scholar] [CrossRef]
- Lladó, J.; Lao-Luque, C.; Solé-Sardans, M.; Montemurro, N.; Pérez, S.; Fuente, E.; Ruiz, B. Elimination of persistent anthropogenic pollutants by micro-mesoporous carbon xerogels. Natural organic matter on surface water and textural properties influences. J. Environ. Chem. Eng. 2021, 9, 104885. [Google Scholar] [CrossRef]
- Labuto, G.; Carvalho, A.P.; Mestre, A.S.; dos Santos, M.S.; Modesto, H.R.; Martins, T.D.; Lemos, S.G.; da Silva, H.D.T.; Carrilho, E.N.V.M.; Carvalho, W.A. Individual and competitive adsorption of ibuprofen and caffeine from primary sewage effluent by yeast-based activated carbon and magnetic carbon nanocomposite. Sustain. Chem. Pharm. 2022, 28, 100703. [Google Scholar] [CrossRef]
- Mestre, A.S.; Viegas, R.M.C.; Mesquita, E.; Rosa, M.J.; Carvalho, A.P. Engineered pine nut shell derived activated carbons for improved removal of recalcitrant pharmaceuticals in urban wastewater treatment. J. Hazard. Mater. 2022, 437, 129319. [Google Scholar] [CrossRef] [PubMed]
- Viegas, R.M.C.; Mestre, A.S.; Mesquita, E.; Campinas, M.; Andrade, M.A.; Carvalho, A.P.; Rosa, M.J. Assessing the applicability of a new carob waste-derived powdered activated carbon to control pharmaceutical compounds in wastewater treatment. Sci. Total Environ. 2020, 743, 140791. [Google Scholar] [CrossRef]
- Mestre, A.S.; Campinas, M.; Viegas, R.M.C.; Mesquita, E.; Carvalho, A.P.; Rosa, M.J. Activated carbons in full-scale advanced wastewater treatment. In Advanced Materials for Sustainable Environmental Remediation: Terrestrial and Aquatic Environments; Giannakoudakis, D.A., Meili, L., Anastopoulos, I., Eds.; Elsevier: Boston, MA, USA, 2022; pp. 433–475. [Google Scholar] [CrossRef]
- Zwickenpflug, B.; Böhler, M.; Siegrist, H.; Gujer, W.; Behl, M.; Neuenschwander, S.; Joss, A.; Sterkele, B.; Dorusch, F.; Hollender, J. Einsatz von Pulveraktivkohle zur Elimination von Ikroverunreinigungen aus KOMMUNALEM Abwasser, Abschlussbericht (Use of Powdered Activated Carbon for the Elimination of Micropollutants from Municipal Wastewater, Final Report, in German). 2010. Available online: www.micropoll.ch (accessed on 5 September 2022).
- Mestre, A.S.; Pires, R.A.; Aroso, I.; Fernandes, E.M.; Pinto, M.L.; Reis, R.L.; Andrade, M.A.; Pires, J.; Silva, S.P.; Carvalho, A.P. Activated carbons prepared from industrial pre-treated cork: Sustainable adsorbents for pharmaceutical compounds removal. Chem. Eng. J. 2014, 253, 408–417. [Google Scholar] [CrossRef]
- Marsh, H.; Rodríguez-Reinoso, F. Activated Carbon; Elsevier: Oxford, UK, 2006. [Google Scholar]
- Mestre, A.S.; Carvalho, A.P. Nanoporous carbons synthesis: An old story with exciting new chapters. In Porosity—Process, Technologies and Applications; Ghrib, T., Ed.; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Wang, Q.; Li, H.; Chen, L.; Huang, X. Monodispersed hard carbon spherules with uniform nanopores. Carbon 2001, 39, 2211–2214. [Google Scholar] [CrossRef]
- Carvalho, A.P.; Mestre, A.S. Acid-chars-versatile materials for adsorption and catalysis. Boletín Grupo Español Carbón 2019, 54, 33–38. [Google Scholar]
- Chen, Z.; Wang, X.; Xue, B.; Li, W.; Ding, Z.; Yang, X.; Qiu, J.; Wang, Z. Rice husk-based hierarchical porous carbon for high performance supercapacitors: The structure-performance relationship. Carbon 2020, 161, 432–444. [Google Scholar] [CrossRef]
- Wang, L.; Guo, Y.; Zhu, Y.; Li, Y.; Qu, Y.; Rong, C.; Ma, X.; Wang, Z. A new route for preparation of hydrochars from rice husk. Bioresour. Technol. 2010, 101, 9807–9810. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Guo, Y.; Zou, B.; Rong, C.; Ma, X.; Qu, Y.; Li, Y.; Wang, Z. High surface area porous carbons prepared from hydrochars by phosphoric acid activation. Bioresour. Technol. 2011, 102, 1947–1950. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Atkinson, J.D. Tailored activated carbon from glycerol: Role of acid dehydrator on physiochemical characteristics and adsorption performance. J Mater. Chem. A 2017, 5, 16812–16821. [Google Scholar] [CrossRef]
- Mestre, A.S.; Hesse, F.; Freire, C.; Ania, C.O.; Carvalho, A.P. Chemically activated high grade nanoporous carbons from low density renewable biomass (Agave sisalana) for the removal of pharmaceuticals. J. Colloid Interface Sci. 2019, 536, 681–693. [Google Scholar] [CrossRef]
- Mestre, A.S.; Freire, C.; Pires, J.; Carvalho, A.P.; Pinto, M.L. High performance microspherical activated carbons for methane storage and landfill gas or biogas upgrade. J. Mater. Chem. A 2014, 2, 15337–15344. [Google Scholar] [CrossRef]
- Mestre, A.S.; Tyszko, E.; Andrade, M.A.; Galhetas, M.; Freire, C.; Carvalho, A.P. Sustainable activated carbons prepared from a sucrose-derived hydrochar: Remarkable adsorbents for pharmaceutical compounds. RSC Adv. 2015, 5, 19696–19707. [Google Scholar] [CrossRef]
- Wei, L.; Sevilla, M.; Fuertes, A.B.; Mokaya, R.; Yushin, G. Hydrothermal carbonization of abundant renewable natural organic chemicals for high-performance supercapacitor electrodes. Adv. Energy Mater. 2011, 1, 356–361. [Google Scholar] [CrossRef] [Green Version]
- Sevilla, M.; Fuertes, A.B.; Mokaya, R. High density hydrogen storage in superactivated carbons from hydrothermally carbonized renewable organic materials. Energy Environ. Sci. 2011, 4, 1400–1410. [Google Scholar] [CrossRef]
- Sevilla, M.; Fuertes, A.B. Sustainable porous carbons with a superior performance for CO2 capture. Energy Environ. Sci. 2011, 4, 1765–1771. [Google Scholar] [CrossRef] [Green Version]
- Titirici, M.-M.; White, R.J.; Falco, C.; Sevilla, M. Black perspectives for a green future: Hydrothermal carbons for environment protection and energy storage. Energy Environ. Sci. 2012, 5, 6796–6822. [Google Scholar] [CrossRef]
- Noh, J.S.; Schwarz, J.A. Estimation of the point of zero charge of simple oxides by mass titration. J. Colloid Interface Sci. 1989, 130, 157–164. [Google Scholar] [CrossRef]
- CEFIC. Test Methods for Activated Carbon; Conseil Européen des Fédérations de l’Industrie Chimique (European Council of Chemical Manufacturers’ Federation): Brussels, Belgium, 1986. [Google Scholar]
- AWWA. Standard for Powdered Activated Carbon; American Water Works Association: Denver, CO, USA, 1996. [Google Scholar]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Rouquerol, J.; Llewellyn, P.; Rouquerol, F. Is the bet equation applicable to microporous adsorbents. In Studies in Surface Science and Catalysis; Llewellyn, P.L., Rodriquez-Reinoso, F., Rouquerol, J., Seaton, N., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 49–56. [Google Scholar] [CrossRef]
- Rodriguez-Reinoso, F.; Martin-Martinez, J.M.; Prado-Burguete, C.; McEnaney, B. A standard adsorption isotherm for the characterization of activated carbons. J. Phys. Chem. 1987, 91, 515–516. [Google Scholar] [CrossRef]
- Rouquerol, F.; Rouquerol, J.; Sing, K. Adsorption by Powders and Porous Solids—Principles, Methodology and Applications; Academic Press: San Diego, CA, USA, 1999. [Google Scholar]
- Sevilla, M.; Ferrero, G.A.; Fuertes, A.B. Beyond KOH activation for the synthesis of superactivated carbons from hydrochar. Carbon 2017, 114, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Fuertes, A.B.; Ferrero, G.A.; Diez, N.; Sevilla, M. A green route to high-surface area carbons by chemical activation of biomass-based products with sodium thiosulfate. ACS Sustain. Chem. Eng. 2018, 6, 16323–16331. [Google Scholar] [CrossRef] [Green Version]
- Pinto, M.L.; Mestre, A.S.; Carvalho, A.P.; Pires, J. Comparison of methods to obtain micropore size distributions of carbonaceous materials from CO2 adsorption based on the Dubinin-Radushkevich isotherm. Ind. Eng. Chem. Res. 2010, 49, 4726–4730. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R.; Freitas, M.M.A.; Órfão, J.J.M. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Cody, G.D.; Alexander, C.M.O.D. NMR studies of chemical structural variation of insoluble organic matter from different carbonaceous chondrite groups. Geochim. Cosmochim. Acta 2005, 69, 1085–1097. [Google Scholar] [CrossRef]
- Haenel, M.W. Recent progress in coal structure research. Fuel 1992, 71, 1211–1223. [Google Scholar] [CrossRef]
- Yu, H.; Niu, S.; Lu, C.; Li, J.; Yang, Y. Sulfonated coal-based solid acid catalyst synthesis and esterification intensification under ultrasound irradiation. Fuel 2017, 208, 101–110. [Google Scholar] [CrossRef]
- Fanning, P.E.; Vannice, M.A. A DRIFTS study of the formation of surface groups on carbon by oxidation. Carbon 1993, 31, 721–730. [Google Scholar] [CrossRef]
- Biniak, S.; Szymanski, G.; Siedlewski, J.; Swiatkowski, A. The characterization of activated carbons with oxygen and nitrogen surface groups. Carbon 1997, 35, 1799–1810. [Google Scholar] [CrossRef]
- Mestre, A.S.; Pires, J.; Nogueira, J.M.F.; Carvalho, A.P. Activated carbons for the adsorption of ibuprofen. Carbon 2007, 45, 1979–1988. [Google Scholar] [CrossRef]
- Ho, Y.S. Citation review of Lagergren kinetic rate equation on adsorption reactions. Scientometrics 2004, 59, 171–177. [Google Scholar] [CrossRef]
- Ho, Y.S.; McKay, G. Pseudo-second order model for sorption processes. Process Biochem. 1999, 34, 451–465. [Google Scholar] [CrossRef]
- Mestre, A.S.; Machuqueiro, M.; Silva, M.; Freire, R.; Fonseca, I.M.; Santos, M.; Calhorda, M.J.; Carvalho, A.P. Influence of activated carbons porous structure on iopamidol adsorption. Carbon 2014, 77, 607–615. [Google Scholar] [CrossRef]
- Langmuir, I. The adsorption of gases on plane surfaces of glass, mica and platinum. J. Am. Chem. Soc. 1918, 40, 1361–1403. [Google Scholar] [CrossRef]
- Freundlich, H.M.F. Over the adsorption in solution. J. Phys. Chem. 1906, 57, 385–470. [Google Scholar] [CrossRef]
- Ho, Y.-S. Selection of optimum sorption isotherm. Carbon 2004, 42, 2115–2116. [Google Scholar] [CrossRef]
- Viegas, R.M.C.; Mestre, A.S.; Mesquita, E.; Machuqueiro, M.; Andrade, M.A.; Carvalho, A.P.; Rosa, M.J. Key factors for activated carbon adsorption of pharmaceutical compounds from wastewaters: A multivariate modelling approach. Water 2022, 14, 166. [Google Scholar] [CrossRef]
- Sevilla, M.; Falco, C.; Titirici, M.M.; Fuertes, A.B. High-performance CO2 sorbents from algae. RSC Adv. 2012, 2, 12792–12797. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | C (wt.%) | H (wt.%) | N (wt.%) | S (wt.%) | O (wt.%) | pHPZC |
|---|---|---|---|---|---|---|
| S13.5 | 60.43 | 3.91 | 0.39 | 1.17 | 34.10 | 2.2 |
| S12 | 55.21 | 3.31 | 0.47 | 3.05 | 37.96 | 2.3 |
| S9 | 60.38 | 4.14 | 0.39 | 1.16 | 33.98 | 2.3 |
| G13.5 | 62.00 | 3.53 | 0.46 | 0.95 | 33.06 | - |
| G12 | 57.13 | 3.53 | 0.26 | 2.59 | 36.49 | - |
| G9 | 57.98 | 4.00 | 0.26 | 1.60 | 36.16 | 2.5 |
| S13.5st87 | - | - | - | - | - | 6.1 |
| S12st82 | 89.89 | 0.58 | 1.05 | 0.27 | 8.21 | 6.2 |
| S9st80 | 93.91 | 0.10 | 0.96 | 0.31 | 4.72 | 5.7 |
| G9st81 | 92.39 | 0.85 | 2.18 | 1.43 | 3.15 | 6.0 |
| Sample | ABET (m2 g−1) | Vtotal (cm3 g−1) | Vmeso (cm3 g−1) | αs Method | ||
|---|---|---|---|---|---|---|
| Vα total (cm3 g−1) | Vα ultra (cm3 g−1) | Vα super (cm3 g−1) | ||||
| Sisal-derived activated carbons | ||||||
| S13.5st87 | 1987 | 0.96 | 0.09 | 0.87 | 0.00 | 0.87 |
| S13.5st80 | 1756 | 0.78 | 0.05 | 0.73 | 0.03 | 0.70 |
| S13.5st76 | 1520 | 0.65 | 0.03 | 0.62 | 0.16 | 0.46 |
| S13.5st71 | 1232 | 0.50 | 0.01 | 0.49 | 0.26 | 0.23 |
| S13.5st69 | 1149 | 0.47 | 0.01 | 0.46 | 0.25 | 0.21 |
| S12st82 | 1827 | 0.83 | 0.08 | 0.75 | 0.01 | 0.74 |
| S12st75 | 1430 | 0.60 | 0.03 | 0.57 | 0.20 | 0.37 |
| S12st71 | 1109 | 0.46 | 0.02 | 0.44 | 0.23 | 0.21 |
| S9st80 | 1670 | 0.82 | 0.16 | 0.66 | 0.05 | 0.61 |
| S9st75 | 1382 | 0.64 | 0.10 | 0.54 | 0.16 | 0.38 |
| S9st71 | 1275 | 0.54 | 0.04 | 0.50 | 0.27 | 0.23 |
| Glucose-derived activated carbons | ||||||
| G13.5st83 | 1910 | 0.87 | 0.08 | 0.79 | 0.00 | 0.79 |
| G12st86 | 1899 | 0.89 | 0.09 | 0.80 | 0.00 | 0.80 |
| G12st81 | 1787 | 0.78 | 0.04 | 0.74 | 0.01 | 0.73 |
| G12st73 | 1158 | 0.47 | 0.01 | 0.46 | 0.23 | 0.23 |
| G9st81 | 1697 | 0.91 | 0.28 | 0.63 | 0.05 | 0.58 |
| G9st71 | 1240 | 0.58 | 0.11 | 0.47 | 0.24 | 0.23 |
| Ibuprofen (IBU) | Iopamidol (IOP) | |
| Chemical structure |  |  |
| pKa | - | 10.7 |
| Water solubility (mg mL−1) | 100 | >200,000 |
| log Ko/w | 3.97 | 2.42 |
| Molecular dimensions (nm) | 1.32 (length) × 0.72 (width) × 0.72 (thickness) | Monomer 1.50 (length) × 1.50 (width) × 0.60 (thickness) Dimer 1.50 (length) × 1.50 (width) × 1.20 (thickness) Trimer 1.50 (length) × 1.50 (width) × 1.80 (thickness) |
| Sample | k2 (g mg−1 h−1) | R2 | h (mg g−1 h−1) | t1/2 (h) | Calculated Values | Experimental Results | |||
|---|---|---|---|---|---|---|---|---|---|
| qe,calc | Ce,calc (mg L−1) | qe,exp (mg g−1) | Ce,exp (mg L−1) | ||||||
| (mg g−1) | (mmol g−1) * | ||||||||
| Ibuprofen | |||||||||
| S13.5st87 | 0.021 | 0.9967 | 2000 | 0.16 | 313 | 1.53 | 118 | 327 | 117 |
| S9st80 | 0.109 | 0.9975 | 10000 | 0.03 | 303 | 1.48 | 119 | 299 | 121 |
| G9st81 | 0.036 | 0.9990 | 3333 | 0.09 | 303 | 1.48 | 119 | 306 | 118 |
| Iopamidol | |||||||||
| S13.5st87 | 0.014 | 0.9999 | 10000 | 0.08 | 833 | 1.07 | 13 | 834 | 8 |
| S9st80 | 0.008 | 0.9990 | 3333 | 0.20 | 667 | 0.86 | 47 | 706 | 38 |
| G9st81 | 0.004 | 0.9996 | 2500 | 0.31 | 769 | 0.99 | 26 | 745 | 31 |
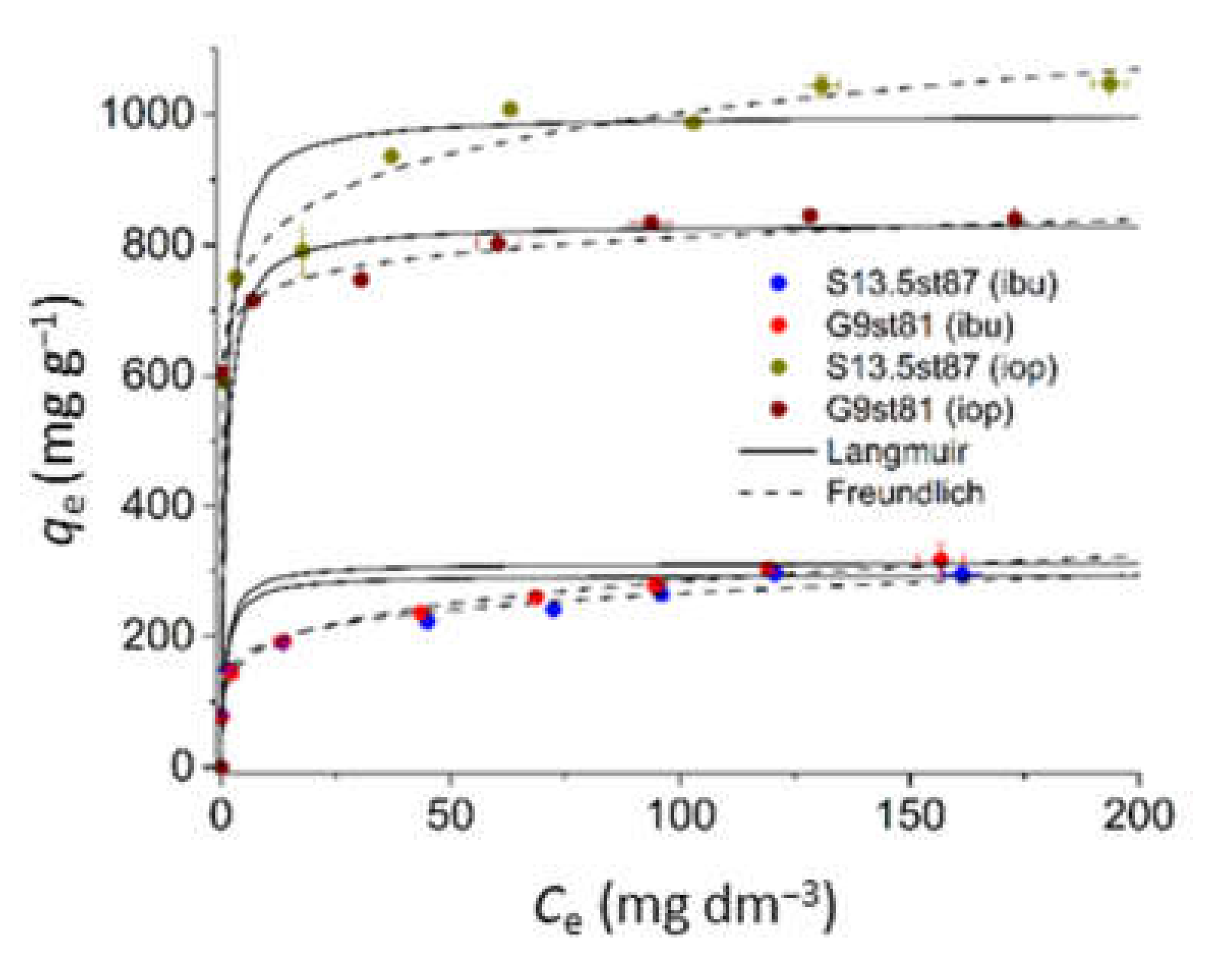
| Parameters | S13.5st87 | G9st81 | ||
|---|---|---|---|---|
| IOP | IBU | IOP | IBU | |
| Langmuir equation | ||||
| qm (mg g−1) | 1111 | 294 | 833 | 323 |
| KL (dm3 mg−1) | 0.29 | 0.16 | 3.00 | 0.13 |
| R2 | 0.9987 | 0.9850 | 0.9988 | 0.9879 |
| χ2 | 33.22 | 53.46 | 6.33 | 83.83 |
| Freundlich equation | ||||
| 1/nF | 0.0928 | 0.2063 | 0.0587 | 0.0823 |
| KF (mg1−1/n(L)1/n g−1) | 654 | 105 | 619 | 179 |
| R2 | 0.9639 | 0.8740 | 0.9244 | 0.9027 |
| χ2 | 9.01 | 4.32 | 3.99 | 1.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hubetska, T.S.; Mestre, A.S.; Kobylinska, N.G.; Carvalho, A.P. Steam Activation of Acid-Chars for Enhanced Textural Properties and Pharmaceuticals Removal. Nanomaterials 2022, 12, 3480. https://doi.org/10.3390/nano12193480
Hubetska TS, Mestre AS, Kobylinska NG, Carvalho AP. Steam Activation of Acid-Chars for Enhanced Textural Properties and Pharmaceuticals Removal. Nanomaterials. 2022; 12(19):3480. https://doi.org/10.3390/nano12193480
Chicago/Turabian StyleHubetska, Tetiana S., Ana S. Mestre, Natalia G. Kobylinska, and Ana P. Carvalho. 2022. "Steam Activation of Acid-Chars for Enhanced Textural Properties and Pharmaceuticals Removal" Nanomaterials 12, no. 19: 3480. https://doi.org/10.3390/nano12193480